Determination of the Pathological Features of NPC1 Variants in a Cellular Complementation Test

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Results
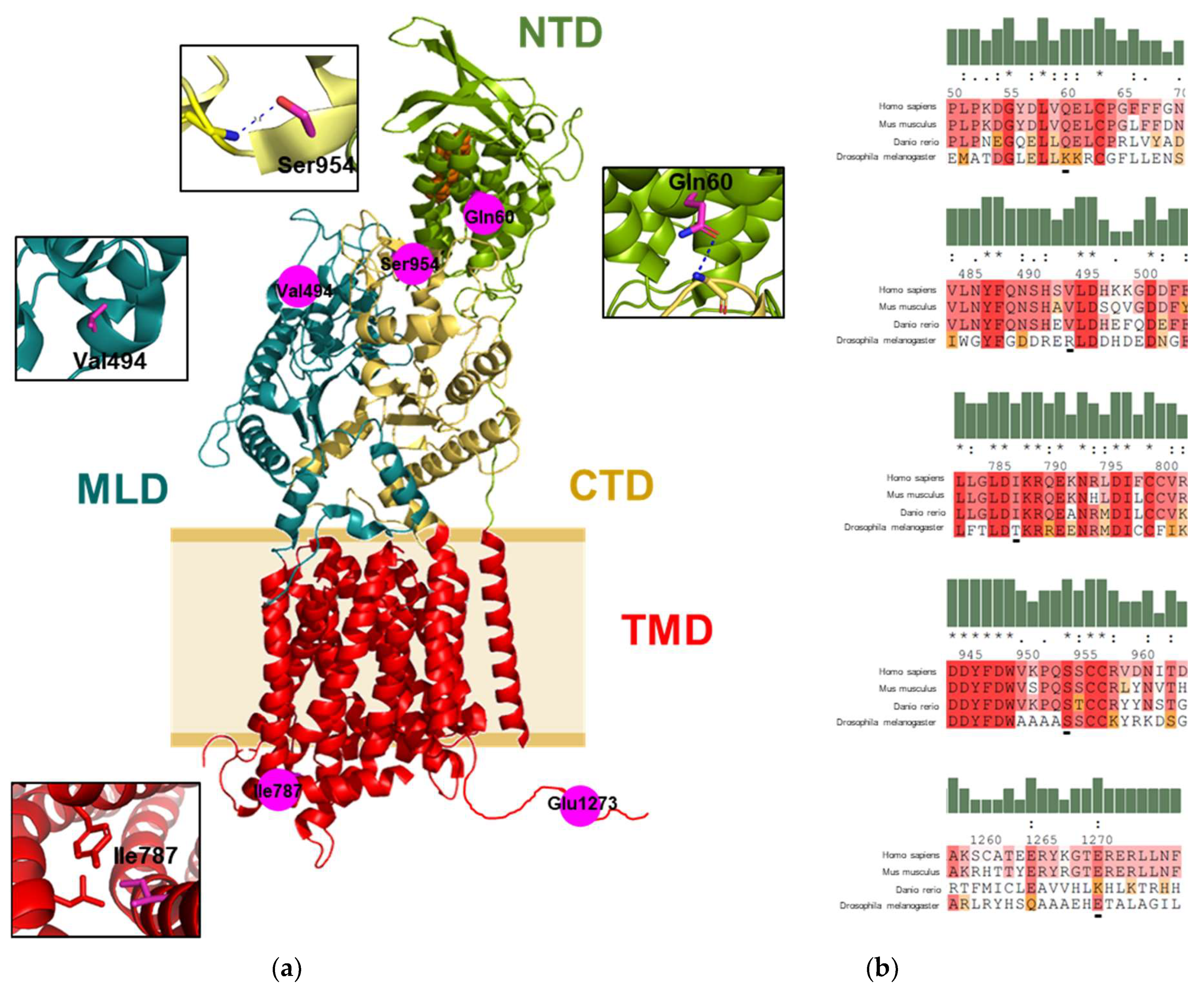
2.1. Mapping of the Gene Variants on the Protein Structure
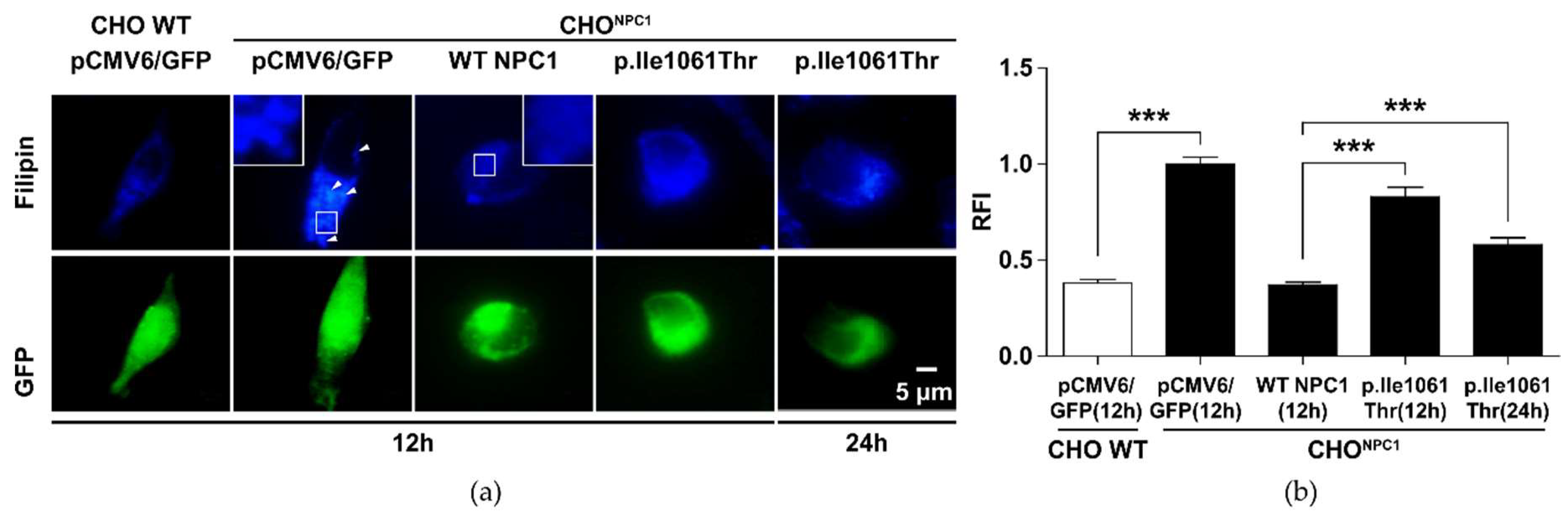
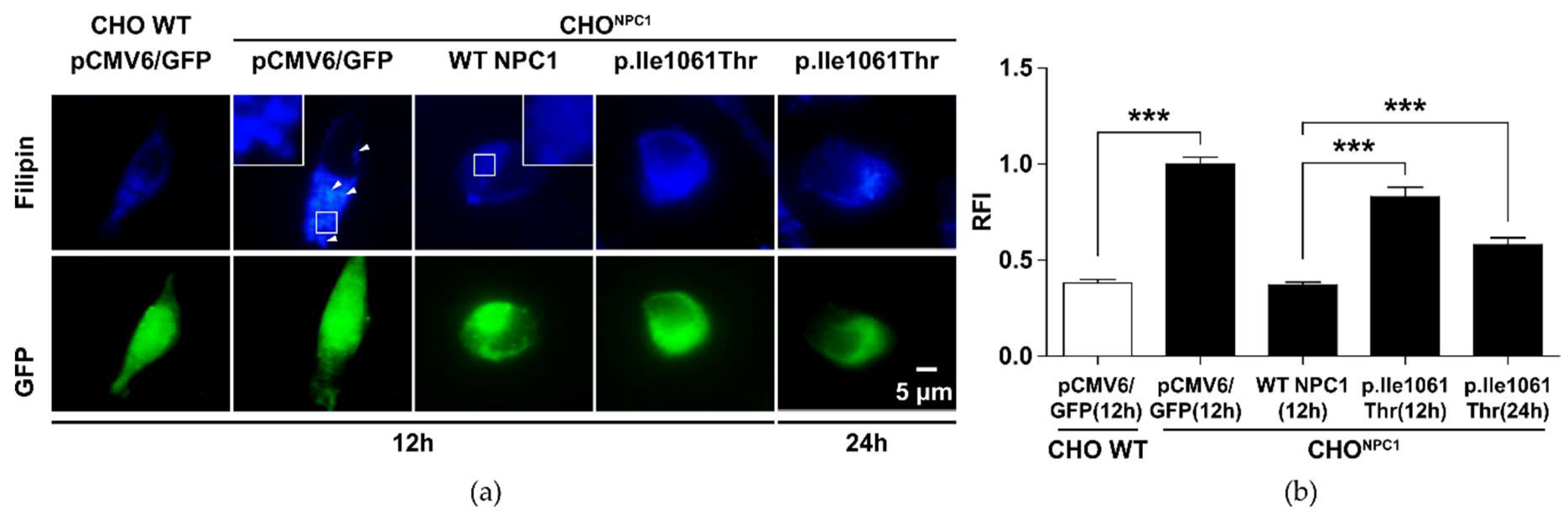
2.2. p.Ile1061Thr Has an Impaired Ability to Eliminate Cholesterol from LSOs
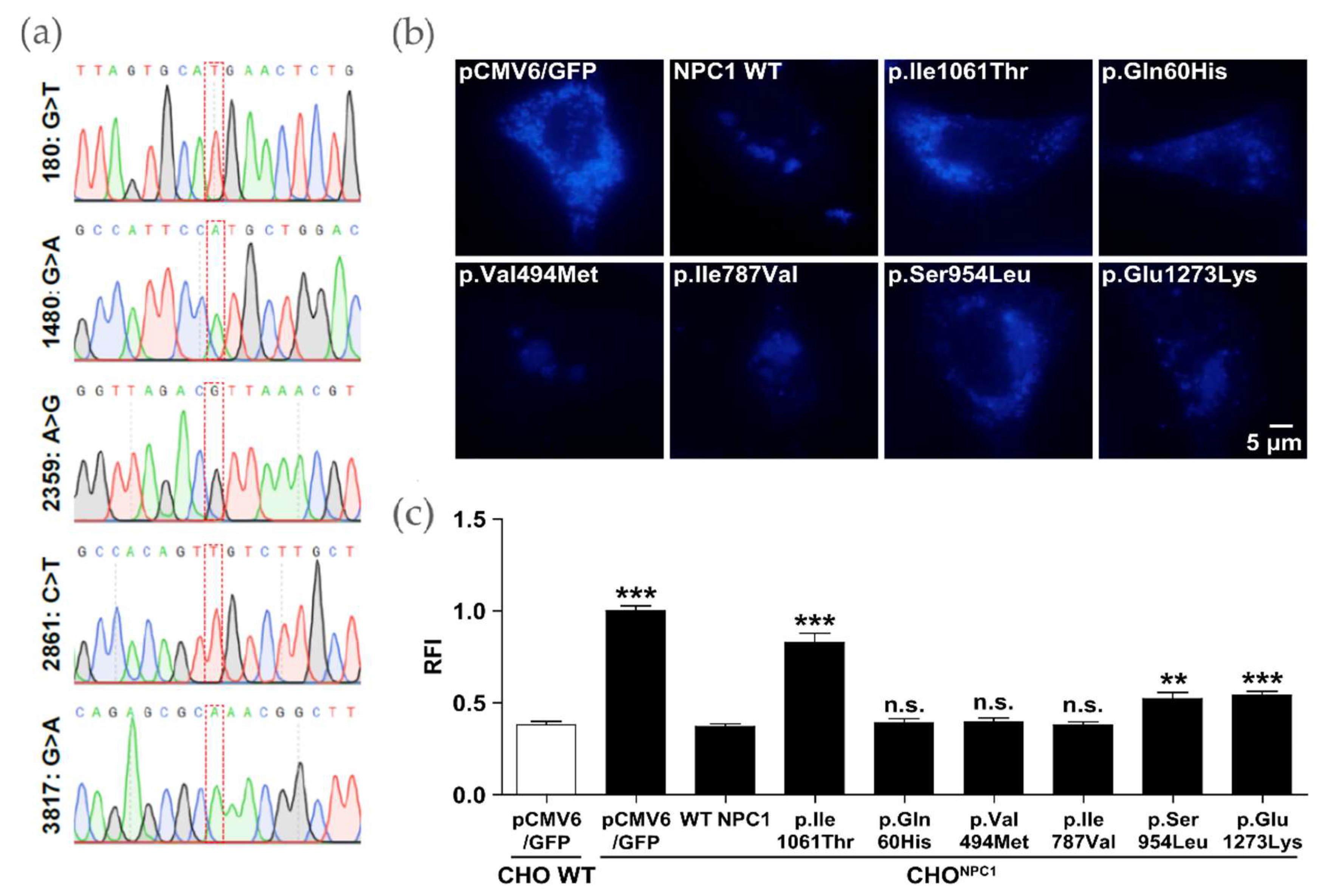
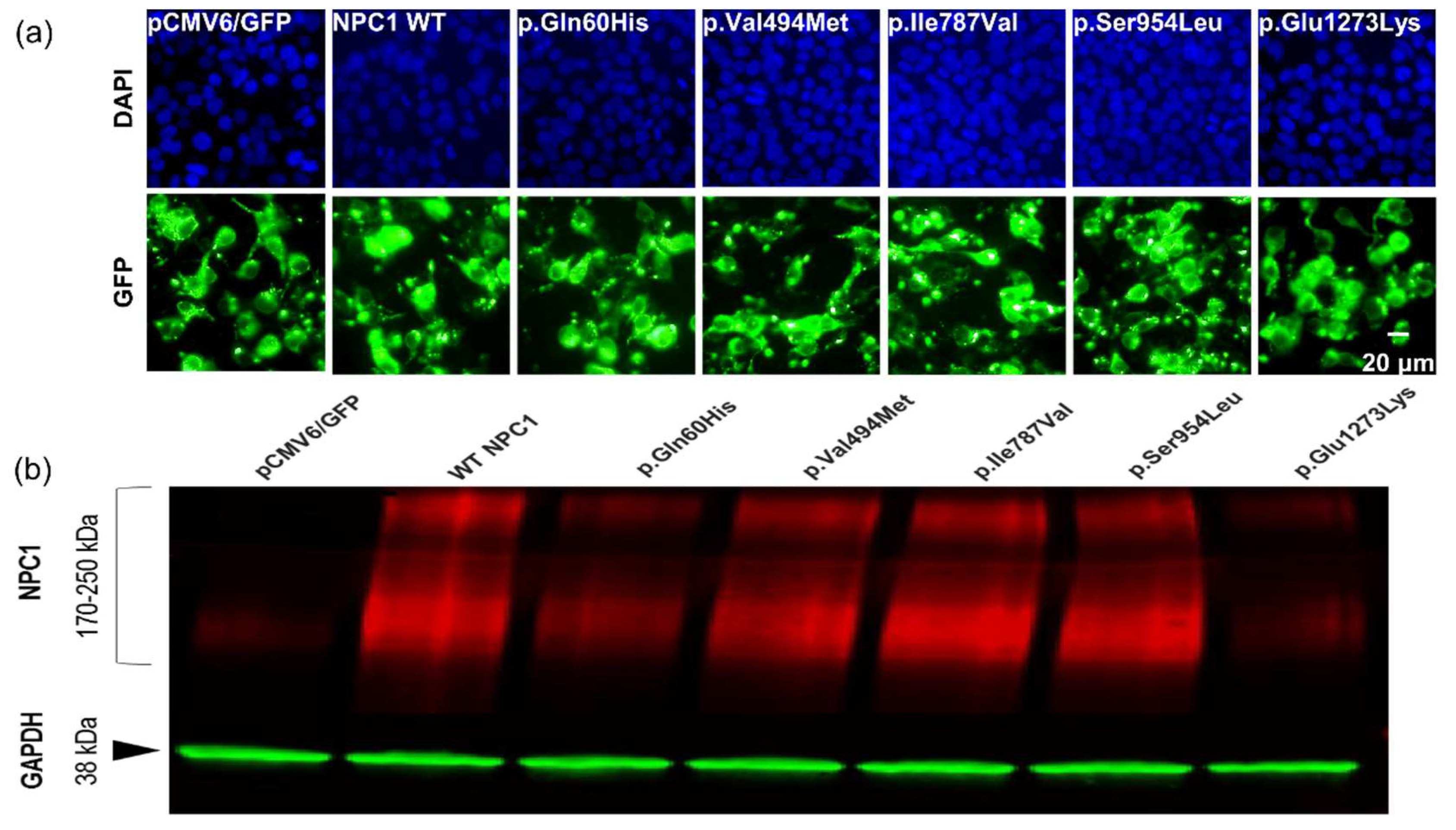
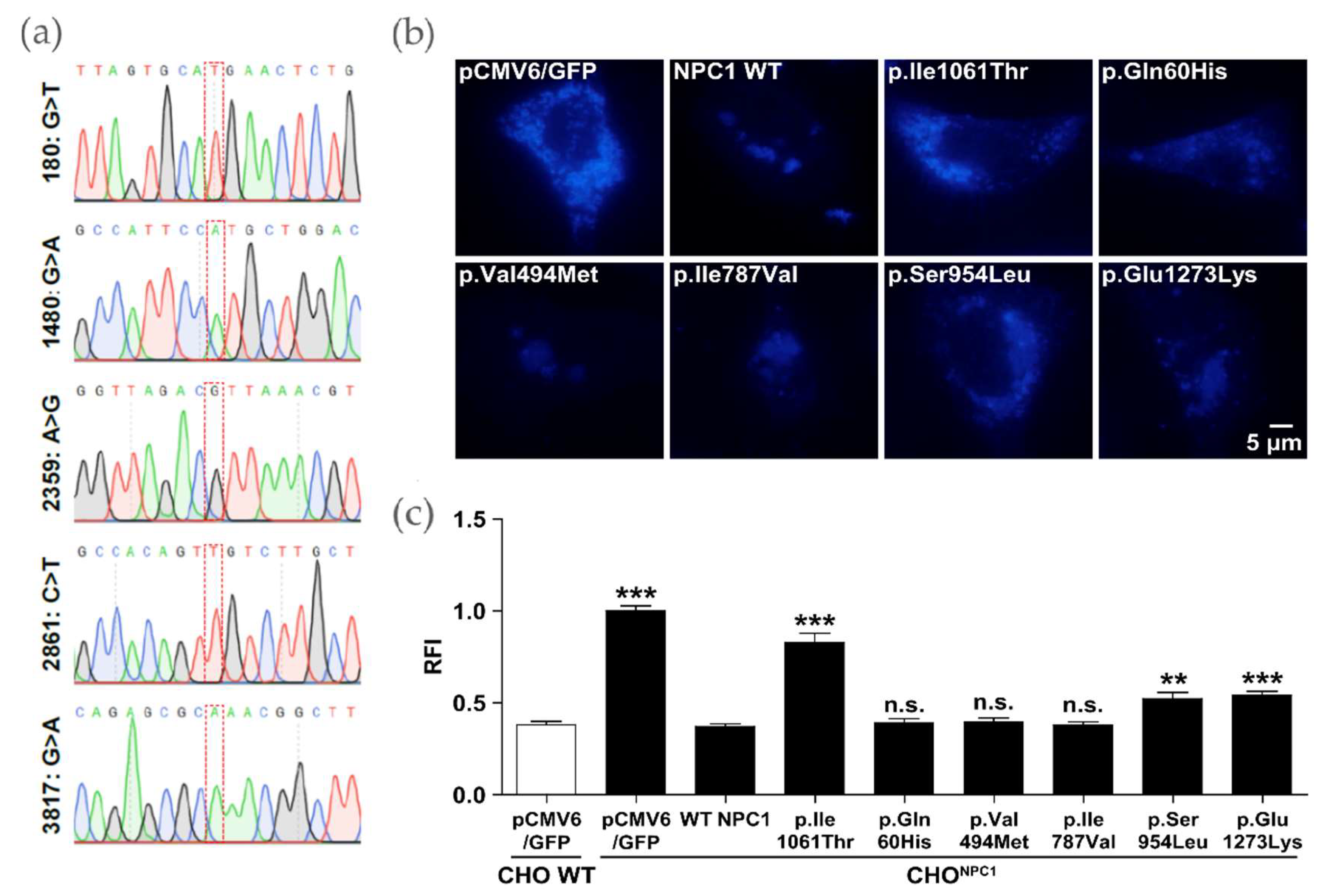
2.3. Certain NPC1 Variants Display Molecular Damage
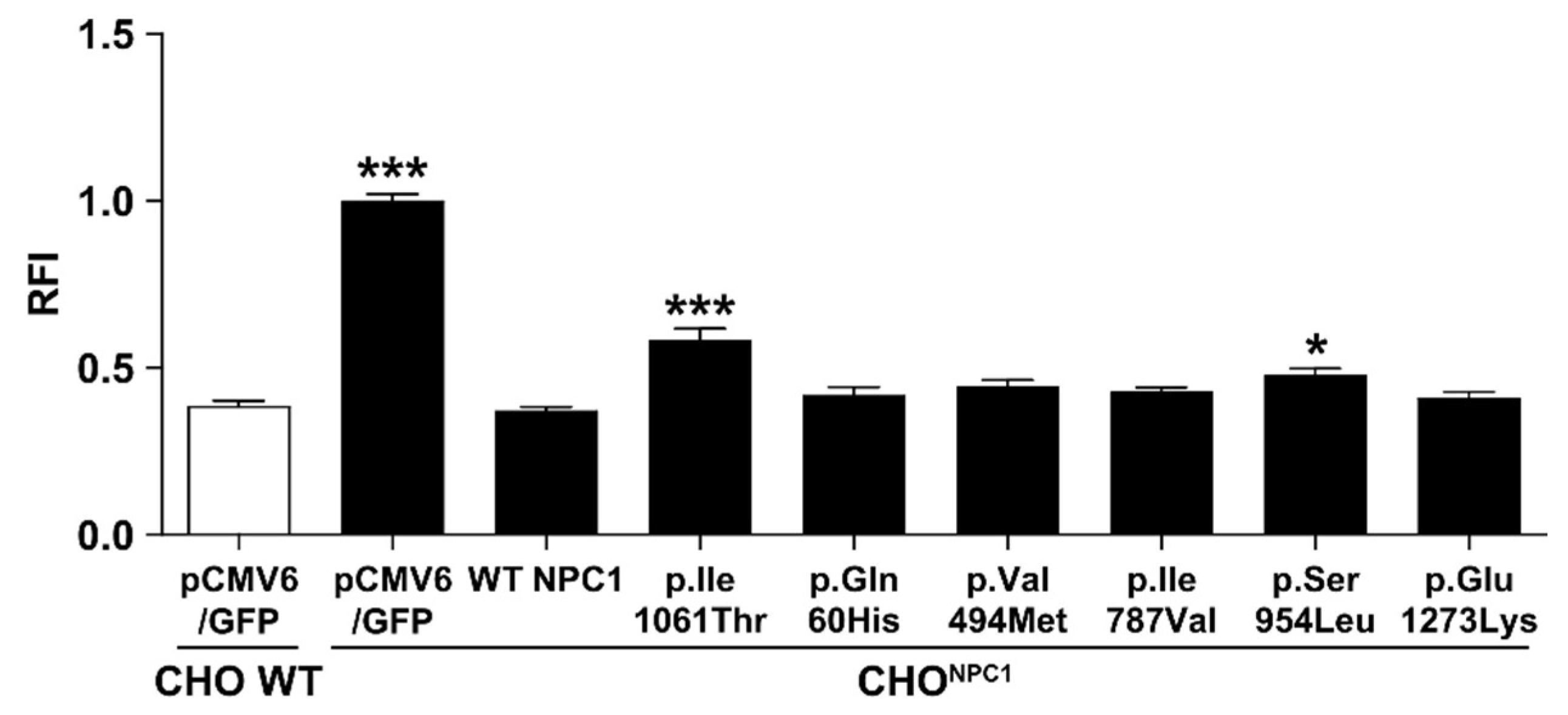
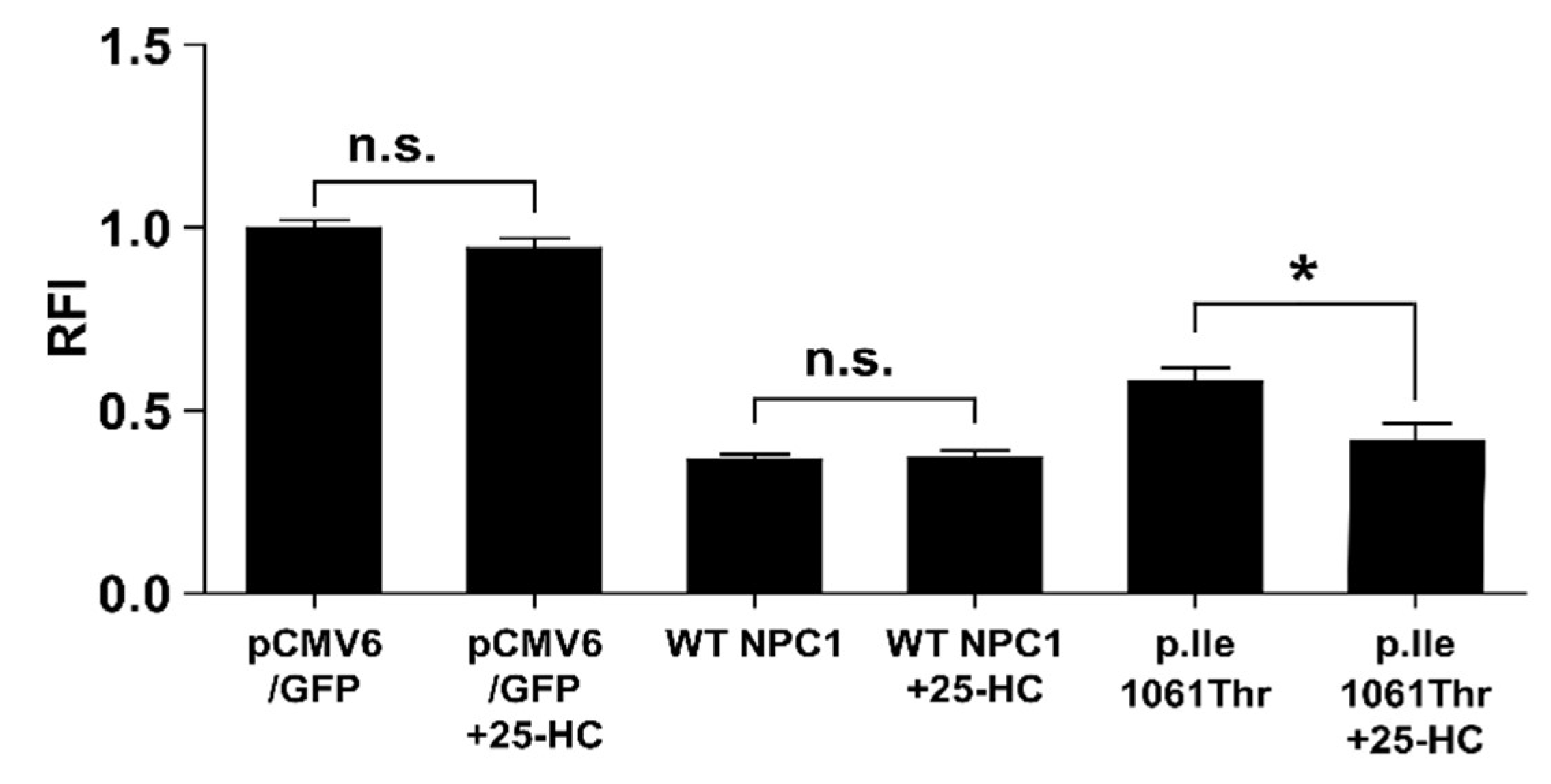
2.4. 25-Hydroxycholesterol Enhances Cholesterol Elimination from LSOs
3. Materials and Methods
3.1. Patients
3.2. Variant Selection
3.2.1. Whole Genome Sequencing and Variant Selection
3.2.2. Plasmid Generation
3.2.3. Cell Culture
3.2.4. Transfection
3.2.5. Western Blot
3.2.6. Filipin Staining and Fluorescence Microscopy
3.2.7. Statistical Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence (5′-3′) |
|---|---|
| NPC1_c.180G>T_fw | TGGATATGACTTAGTGCATGAACTCTGTCCAGGATTC |
| NPC1_c.180G>T_rev | GAATCCTGGACAGAGTTCATGCACTAAGTCATATCCA |
| NPC1_ c.1480G>A_fw | CCAGAACAGCCATTCCATGCTGGACCACAAGAA |
| NPC1_ c.1480G>A_rev | TTCTTGTGGTCCAGCATGGAATGGCTGTTCTGG |
| NPC1_ c.2359A>G_fw | GTGAGTCTCTTGGGGTTAGACGTTAAACGTCAAGAGAAAAATC |
| NPC1_ c.2359A>G_rev | GATTTTTCTCTTGACGTTTAACGTCTAACCCCAAGAGACTCAC |
| NPC1_c.2861C>T_fw | GGGTGAAGCCACAGTTGTCTTGCTGTCGAGT |
| NPC1_c.2861C>T_rev | ACTCGACAGCAAGACAACTGTGGCTTCACCC |
| NPC1_ c.3817G>A_fw | ACAAAGGAACAGAGCGCAAACGGCTTCTAAATTTC |
| NPC1_ c.3817G>A_rev | GAAATTTAGAAGCCGTTTGCGCTCTGTTCCTTTGT |
| NPC1_ c.3182T>C_fw | GAAGAAAGCCCGACTTACAGCCAGTAATGTCACCG |
| NPC1_ c.3182T>C_rev | CGGTGACATTACTGGCTGTAAGTCGGGCTTTCTTC |
| T7_Standard | TAATACGACTCACTATAGG |
| NPC1_cds_1 | GTGAAAGAGTTACAATACTACG |
| NPC1_cds_2 | CCCATCGATAGCAATATAGC |
| NPC1_cds_3 | CACCGTATAACACGAACTGC |
| NPC1_cds_4 | GCTGAAGATGGAACAAGCGT |
| NPC1_cds_5 | AGTGGTTGACCCTGCCTGCGTT |
| NPC1_cds_6 | TTGGAGTTATGTGGCTCTGG |
References
- Vanier, M.T. Niemann-Pick disease type C. Orphanet J. Rare Dis. 2010, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.M.; McNamara, J.; Seashore, M.R.; Mistry, P.K. Misdiagnosis of Niemann-Pick disease type C as Gaucher disease. J. Inherit. Metab. Dis. 2010, 33, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Connemann, B.J.; Gahr, M.; Schmid, M.; Runz, H.; Freudenmann, R.W. Low ceruloplasmin in a patient with Niemann-Pick Type C disease. J. Clin. Neurosci. 2012, 19, 620–621. [Google Scholar] [CrossRef] [PubMed]
- Bountouvi, E.; Papadopoulou, A.; Vanier, M.T.; Nyktari, G.; Kanellakis, S.; Michelakakis, H.; Dinopoulos, A. Novel NPC1 mutations with different segregation in two related Greek patients with Niemann-Pick type C disease: Molecular study in the extended pedigree and clinical correlations. BMC Med. Genet. 2017, 18, 51. [Google Scholar] [CrossRef]
- Patterson, M. Niemann-Pick Disease Type C; Margaret, P.A., Holly, H.A., Roberta, A.P., Stephanie, E.W., Lora, J.H.B., Karen, S., Anne, A., Eds.; GeneReviews: Seattle, WA, USA, 1993. [Google Scholar]
- Pipalia, N.H.; Cosner, C.C.; Huang, A.; Chatterjee, A.; Bourbon, P.; Farley, N.; Helquist, P.; Wiest, O.; Maxfield, F.R. Histone deacetylase inhibitor treatment dramatically reduces cholesterol accumulation in Niemann-Pick type C1 mutant human fibroblasts. Proc. Natl. Acad. Sci. USA 2011, 108, 5620–5625. [Google Scholar] [CrossRef] [Green Version]
- Frolov, A.; Zielinski, S.E.; Crowley, J.R.; Dudley-Rucker, N.; Schaffer, J.E.; Ory, D.S. NPC1 and NPC2 Regulate Cellular Cholesterol Homeostasis through Generation of Low Density Lipoprotein Cholesterol-derived Oxysterols. J. Boil. Chem. 2003, 278, 25517–25525. [Google Scholar] [CrossRef] [Green Version]
- Tängemo, C.; Weber, D.; Theiss, S.; Mengel, E.; Runz, H. Niemann-Pick Type C disease: Characterizing lipid levels in patients with variant lysosomal cholesterol storage. J. Lipid Res. 2011, 52, 813–825. [Google Scholar] [CrossRef]
- Jiang, X.; Sidhu, R.; Porter, F.D.; Yanjanin, N.M.; Speak, A.O.; te Vruchte, D.T.; Platt, F.M.; Fujiwara, H.; Scherrer, D.E.; Zhang, J.; et al. A sensitive and specific LC-MS/MS method for rapid diagnosis of Niemann-Pick C1 disease from human plasma. J. Lipid Res. 2011, 52, 1435–1445. [Google Scholar] [CrossRef] [Green Version]
- Giese, A.K.; Mascher, H.; Grittner, U.; Eichler, S.; Kramp, G.; Lukas, J.; te Vruchte, D.; Al Eisa, N.; Cortina-Borja, M.; Porter, F.D.; et al. A novel, highly sensitive and specific biomarker for Niemann-Pick type C1 disease. Orphanet J. Rare Dis. 2015, 10, 78. [Google Scholar] [CrossRef]
- Wittmann, J.; Karg, E.; Turi, S.; Legnini, E.; Wittmann, G.; Giese, A.-K.; Lukas, J.; Gölnitz, U.; Klingenhäger, M.; Bodamer, O.; et al. Newborn Screening for Lysosomal Storage Disorders in Hungary. JIMD Rep. 2012, 6, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Zech, M.; Nübling, G.; Castrop, F.; Jochim, A.; Schulte, E.C.; Mollenhauer, B.; Lichtner, P.; Peters, A.; Gieger, C.; Marquardt, T.; et al. Niemann-Pick C Disease Gene Mutations and Age-Related Neurodegenerative Disorders. PLoS ONE 2013, 8, e82879. [Google Scholar] [CrossRef] [PubMed]
- Synofzik, M.; Harmuth, F.; Stampfer, M.; Hagen, J.M.V.; Schöls, L.; Bauer, P. NPC1 is enriched in unexplained early onset ataxia: A targeted high-throughput screening. J. Neurol. 2015, 262, 2557–2563. [Google Scholar] [CrossRef] [PubMed]
- Higaki, K.; Ninomiya, H.; Sugimoto, Y.; Suzuki, T.; Taniguchi, M.; Niwa, H.; Pentchev, P.G.; Vanier, M.T.; Ohno, K. Isolation of NPC1-deficient Chinese hamster ovary cell mutants by gene trap mutagenesis. J. Biochem. 2001, 129, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, 980–985. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. ClinVar; [VCV000181457.3]. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000181457.3 (accessed on 1 October 2019).
- Millat, G.; Marcais, C.; Rafi, M.A.; Yamamoto, T.; Morris, J.A.; Pentchev, P.G.; Ohno, K.; Wenger, D.A.; Vanier, M.T. Niemann-Pick C1 Disease: The I1061T Substitution Is a Frequent Mutant Allele in Patients of Western European Descent and Correlates with a Classic Juvenile Phenotype. Am. J. Hum. Genet. 1999, 65, 1321–1329. [Google Scholar] [CrossRef] [Green Version]
- Gelsthorpe, M.E.; Baumann, N.; Millard, E.; Gale, S.E.; Langmade, S.J.; Schaffer, J.E.; Ory, D.S. Niemann-Pick type C1 I1061T mutant encodes a functional protein that is selected for endoplasmic reticulum-associated degradation due to protein misfolding. J. Boil. Chem. 2008, 283, 8229–8236. [Google Scholar] [CrossRef]
- Ohgane, K.; Karaki, F.; Dodo, K.; Hashimoto, Y. Discovery of Oxysterol-Derived Pharmacological Chaperones for NPC1: Implication for the Existence of Second Sterol-Binding Site. Chem. Boil. 2013, 20, 391–402. [Google Scholar] [CrossRef] [Green Version]
- Grabe, H.J.; Assel, H.; Bahls, T.; Dörr, M.; Endlich, K.; Endlich, N.; Erdmann, P.; Ewert, R.; Felix, S.B.; Fiene, B.; et al. Cohort profile: Greifswald approach to individualized medicine (GANI_MED). J. Transl. Med. 2014, 12, 144. [Google Scholar] [CrossRef]
- Trujillano, D.; Oprea, G.-E.; Schmitz, Y.; Bertoli-Avella, A.M.; Jamra, R.A.; Rolfs, A. A comprehensive global genotype–phenotype database for rare diseases. Mol. Genet. Genom. Med. 2016, 5, 66–75. [Google Scholar] [CrossRef]
- Yamamoto, T.; Ninomiya, H.; Matsumoto, M.; Ohta, Y.; Nanba, E.; Tsutsumi, Y.; Yamakawa, K.; Millat, G.; Vanier, M.T.; Pentchev, P.G.; et al. Genotype-phenotype relationship of Niemann-Pick disease type C: A possible correlation between clinical onset and levels of NPC1 protein in isolated skin fibroblasts. J. Med. Genet. 2000, 37, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Greer, W.L.; Dobson, M.J.; Girouard, G.S.; Byers, D.M.; Riddell, D.C.; Neumann, P.E. Mutations in NPC1 highlight a conserved NPC1-specific cysteine-rich domain. Am. J. Hum. Genet. 1999, 65, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, P.C. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Tang, H.; Thomas, P.D. Tools for Predicting the Functional Impact of Nonsynonymous Genetic Variation. Genetics 2016, 203, 635–647. [Google Scholar] [CrossRef] [Green Version]
- Gong, X.; Qian, H.; Zhou, X.; Wu, J.; Wan, T.; Cao, P.; Huang, W.; Zhao, X.; Wang, X.; Wang, P.; et al. Structural Insights into the Niemann-Pick C1 (NPC1)-Mediated Cholesterol Transfer and Ebola Infection. Cell 2016, 165, 1467–1478. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.J.; Abi-Mosleh, L.; Wang, M.L.; Deisenhofer, J.; Goldstein, J.L.; Brown, M.S.; Infante, R.E. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell 2009, 137, 1213–1224. [Google Scholar] [CrossRef]
- Snapp, E. Design and use of fluorescent fusion proteins in cell biology. Curr. Protoc. Cell Biol. 2005, 27, 21–24. [Google Scholar] [CrossRef]
- Lamri, A.; Pigeyre, M.; Garver, W.S.; Meyre, D. The Extending Spectrum of NPC1-Related Human Disorders: From Niemann–Pick C1 Disease to Obesity. Endocr. Rev. 2018, 39, 192–220. [Google Scholar] [CrossRef]
- Sun, X.; Marks, D.L.; Park, W.D.; Wheatley, C.L.; Puri, V.; O’Brien, J.F.; Kraft, D.L.; Lundquist, P.A.; Patterson, M.C.; Pagano, R.E.; et al. Niemann-Pick C Variant Detection by Altered Sphingolipid Trafficking and Correlation with Mutations within a Specific Domain of NPC1. Am. J. Hum. Genet. 2001, 68, 1361–1372. [Google Scholar] [CrossRef] [Green Version]
- Rujoi, M.; Pipalia, N.H.; Maxfield, F.R. Cholesterol pathways affected by small molecules that decrease sterol levels in Niemann-Pick type C mutant cells. PLoS ONE 2010, 5, e12788. [Google Scholar] [CrossRef] [PubMed]
| DNA | Protein | Exon | Prediction | Clinical Significance 1 | Allele Frequency (MAF) 2 | |
|---|---|---|---|---|---|---|
| PolyPhen2 | SIFT | |||||
| c.180G>T | p.Gln60His | 2 | possibly damaging | tolerated | no entry | 3.314 × 10−4 |
| c.1480G>A | p.Val494Met | 9 | benign | tolerated | GVUS | 1.657 × 10−4 |
| c.2359A>G | p.Ile787Val | 15 | benign | tolerated | no entry | 1.450 × 10−4 |
| c.2861C>T | p.Ser954Leu | 19 | possibly damaging | damaging | pathogenic | 8.292 × 10−5 |
| c.3817G>A | p.Glu1273Lys | 25 | benign | damaging | no entry | no data |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, X.; Cozma, C.; Pantoom, S.; Hund, C.; Iwanov, K.; Petters, J.; Völkner, C.; Bauer, C.; Vogel, F.; Bauer, P.; et al. Determination of the Pathological Features of NPC1 Variants in a Cellular Complementation Test. Int. J. Mol. Sci. 2019, 20, 5185. https://doi.org/10.3390/ijms20205185
Feng X, Cozma C, Pantoom S, Hund C, Iwanov K, Petters J, Völkner C, Bauer C, Vogel F, Bauer P, et al. Determination of the Pathological Features of NPC1 Variants in a Cellular Complementation Test. International Journal of Molecular Sciences. 2019; 20(20):5185. https://doi.org/10.3390/ijms20205185
Chicago/Turabian StyleFeng, Xiao, Claudia Cozma, Supansa Pantoom, Christina Hund, Katharina Iwanov, Janine Petters, Christin Völkner, Claudia Bauer, Florian Vogel, Peter Bauer, and et al. 2019. "Determination of the Pathological Features of NPC1 Variants in a Cellular Complementation Test" International Journal of Molecular Sciences 20, no. 20: 5185. https://doi.org/10.3390/ijms20205185
APA StyleFeng, X., Cozma, C., Pantoom, S., Hund, C., Iwanov, K., Petters, J., Völkner, C., Bauer, C., Vogel, F., Bauer, P., Weiss, F. U., Lerch, M. M., Knospe, A.-M., Hermann, A., Frech, M. J., Luo, J., Rolfs, A., & Lukas, J. (2019). Determination of the Pathological Features of NPC1 Variants in a Cellular Complementation Test. International Journal of Molecular Sciences, 20(20), 5185. https://doi.org/10.3390/ijms20205185