1. Introduction
Articular hyaline cartilage has alow repair ability; for this reason, when a lesion advances, it extends to the underlying subchondral bone, generating an osteochondral defect (OC), which leads to a complex therapy [
1].
The articular cartilage has an architecture defined not only by the type of proteins of the extracellular matrix ECM but by its arrangement, and it varies from the surface to the subchondral bone; this is known as an osteochondral unit. The histological structure of the osteochondral unit is divided into 4 regions defined according to the content of the proteoglycans, as well as the density of the chondrocytes as follows: (1) Superficial or tangential; fine region aligned parallel to the articular surface, composed mainly of type II collagen fibers; (2) Intermediate or transitional; it has a low density of chondrocytes with a spherical morphology, rich in proteoglycans and thick collagen II fibers; (3) Deep or radial, which is formed by chondrocytes organized with a columnar orientation; and finally, (4) a calcified layer of hypertrophic chondrocytes capable of attaching the cartilage to the bone by anchoring the collagen fibers with the subchondral bone [
2,
3].
When a defect with a critical size occurs in the osteochondral unit, degenerative changes are induced both in the cartilage and in the bone surrounding the lesion; these changes generate a mechanical destabilization that translates into a decrease in the load properties [
4]. Consequently, due to the complex architecture of the chondral tissue, the repair of osteochondral defects requires an approach based on tissue engineering.
Strategies based on tissue engineering offer a promising therapy for the restoration of articular cartilage, and thus, different designs have been proposed. Its principle involves the combination of stem or differentiated cells, scaffolding materials and bioactive factors that facilitate the repair of cartilage. This seems to be convenient for a direct use of the cells on the site of the lesion, in relation to the problem of cell delivery. However, a challenge on which efforts should be invested consists in improving the integration of the implant with the surrounding tissue, as well as the generation of a new tissue that mimics the osteochondral hierarchical structure.
In recent years, the use of biphasic scaffolds has been intensified because they positively influence the hierarchical organization of osteochondral tissue; additionally, there is increasing evidence that these designs favor the integration of the implant with the surrounding tissue [
5,
6].
Concerning biomaterials of a protein nature, silkworm fibroin is an ideal candidate. Fibroin is a natural biopolymer that is highly biocompatible and biodegradable, which allows it to be used not only in a wide variety of biomedical devices but in an interesting way in the development of new regeneration technologies [
7].
In its semicrystalline structure, fibroin has a highly ordered phase of β-antiparallel sheets that give it resistance and toughness; on the other hand, interleaved separators of less-ordered β sheets are placed, contributing to the flexibility and elasticity of the material [
8].
Among the biological properties of fibroin, we highlight the ability to sustain the proliferation and differentiation of various cell types, including the chondrogenic lineage; This makes it an attractive biomaterial in regenerative cartilage medicine [
9,
10].
The use of silk fibroin has been expanded due to its versatility in generating different types of scaffolding, ranging from films to scaffolds. The selection of manufacturing methods and even the use of different solvents in the solubilization of fibroin influence induction capacities toward specific cell lineages such as chondrogenic [
11].
For the functionality of a biomaterial within a complex structural context such as the osteochondral one, it is necessary to adapt the properties of the biomaterials to the needs of the chondral or bone tissue. To overcome this issue, biofunctionalization is presented as a valuable procedure.
In this sense, biofunctionalization with an ECM derived from several tissues and decellularized organs has become more attractive and is used for the regeneration of tissues, such as skeletal muscle [
12], blood vessels [
13], nerves [
14], and cartilage [
15,
16]. The ECM provides informative signals and a unique composition that mimics the natural tissue environment and leads to proper tissue regeneration. In articular cartilage, chondrocytes are surrounded by a highly hydrated ECM consisting of type II collagen (Col II), proteoglycans, and several other proteins, which play a crucial role in chondrogenesis [
17,
18].
In this study, the design of a biphasic scaffold based on the assembly of a cartilage phase consisting of silk fibroin, biofunctionalized with a bovine cartilage matrix, cellularized with differentiated pre-chondrocytes from adipose tissue stem cells (autologous) and well-attached to the bone phase (decellularized bovine bone) is described and evaluated in terms of mechanical and chondroinductive features. Furthermore, we evaluated its ability to generate well-organized hyaline cartilage and subchondral bone, 4 months after its implantation in an osteochondral lesion in a porcine model.
3. Discussion
The aim of this study was to examine the effect of a new biphasic scaffold on the integration with the host and on the tissue organization during osteochondral defect repair.
The biphasic scaffold, based on biofunctionalized fibroin with BCM attached to a bone phase and cellularized only in the chondral phase, showed not only an appropriate integration with the host tissue but also the ideal replacement of the damaged area, with cartilage that maintained the hierarchical structure of the chondral tissue.
Hydrogels, based on various polymeric biomaterials such as PLGA [Poly (lactide-co-glycolide)] and PEG [Poly (ethylene glycol)] or natural materials such as silk fibroin (both capable of generating a three-dimensional network structure), offer multiple advantages in terms of cartilage tissue engineering; these include: (1) a simple and highly reproducible manufacturing methodology; (2) the ability to integrate different types of cells; (3) an adequate biodegradability and biocompatibility to allow cartilage regeneration; and (4) the modulation of mechanical properties [
21,
22,
23].
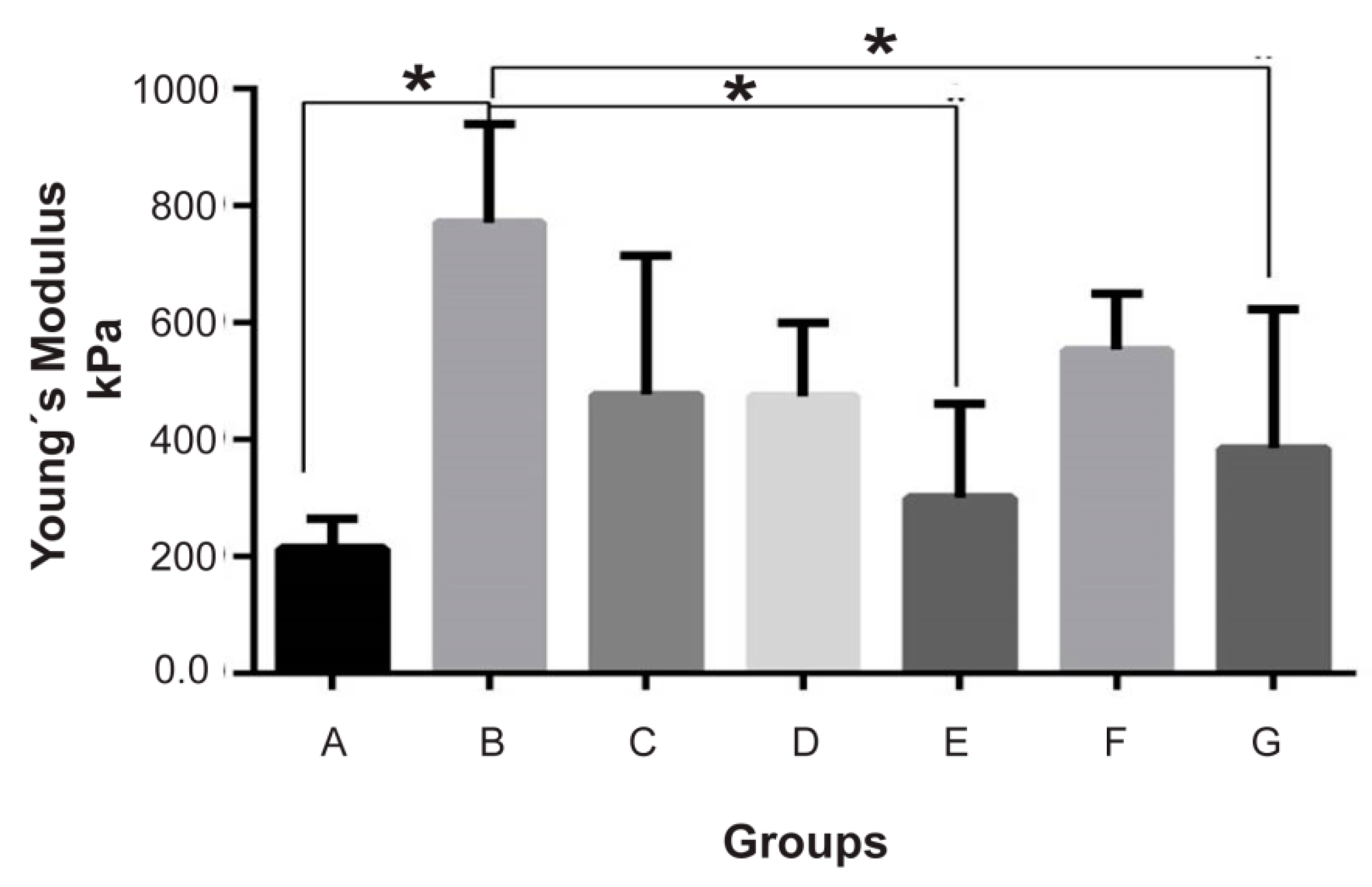
In this sense, the resistance and tenacity of the fibroinmimic the characteristics of the cartilage, which is not the case for the elastic modulus that, as shown in
Figure 2, is naturally inferior to that required by the cartilage to carry out its function.
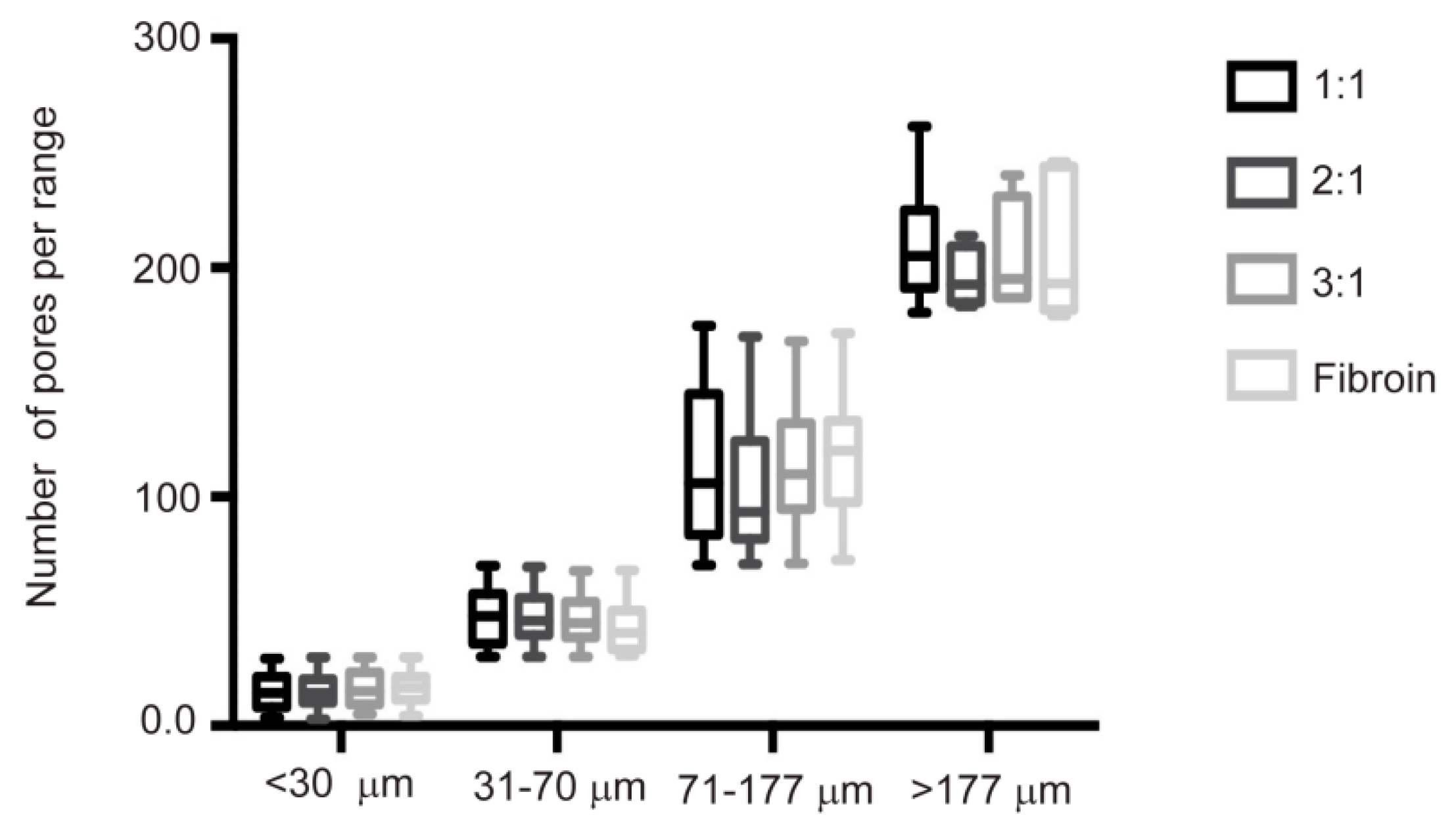
The biphasic design described here presents a cartilage phase based on fibroin manufactured by crystallization with methanol. On the other hand, the porosity was generated by the leaching method with NaCl as porogen with a particle size between 77 and 177 micrometers, in order to obtain a uniform porous and interconnected structure with pore sizes ranging between 400 and 500 micrometers [
24,
25,
26]. Although, via this method, we obtained a chondral scaffold with an interconnected pore ultrastructure (
Figure 1 and
Figure 3) that favors adhesion and cell-cell contact, the resulting mechanical properties were still insufficient for the solicitation of the chondral tissue.
To overcome mechanical tissue demands, a strategy used by our group [
16] and others has been to incorporate decellularized ECM processed in the form of micronized particles.
Even though the effect of the particle size on the properties of proliferation and differentiation toward adipocytes [
27], osteoblasts [
28] and chondrocytes [
29] has been studied, its impact on mechanical properties and especially on elasticity has been rarely addressed.
In this work, the biofunctionalization of silk fibroin was not only used to recreate the microenvironment of the hyaline cartilage [
30] but also to modulate its mechanical properties. Regarding the mechanical features; as shown in
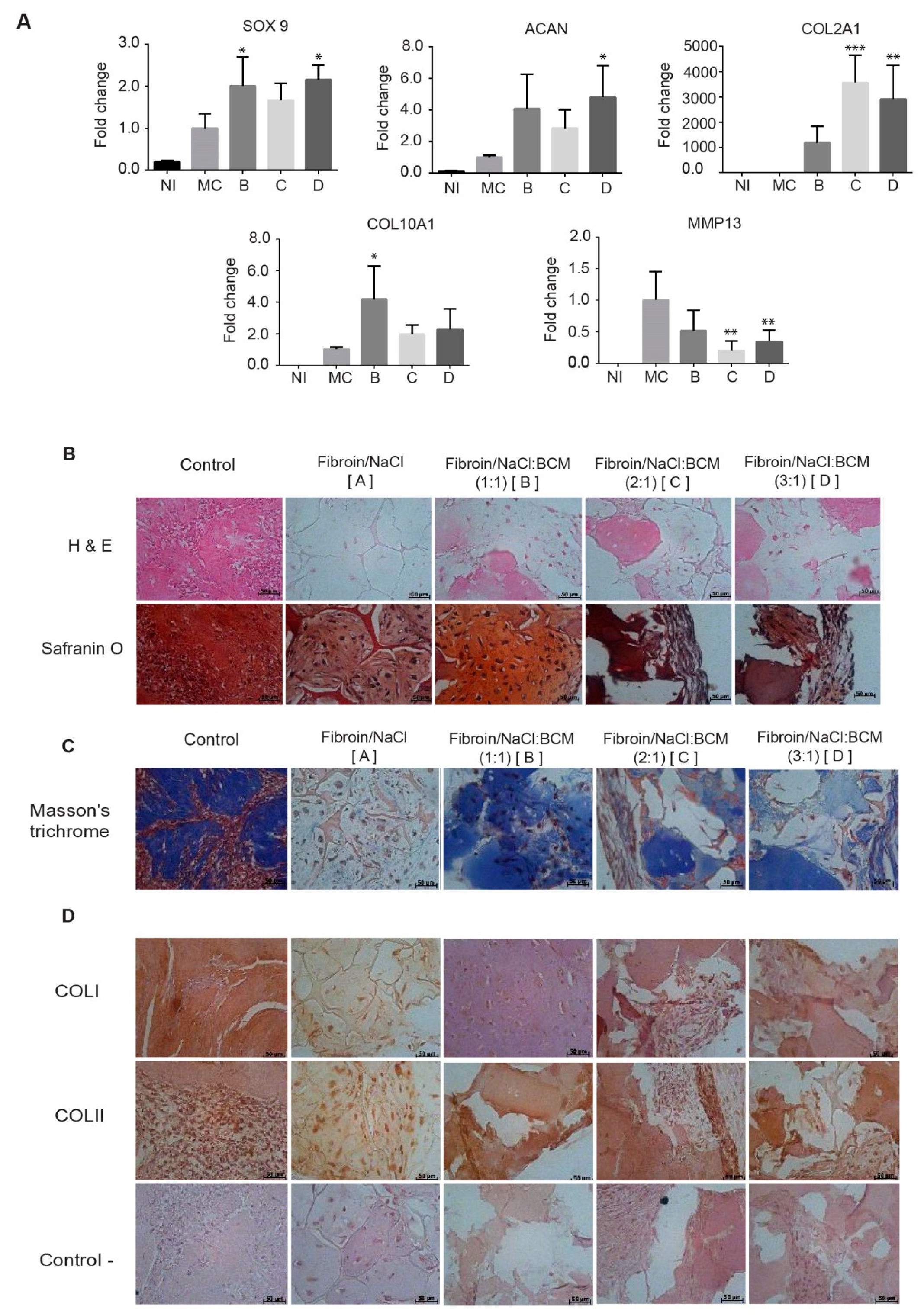
Figure 2, the addition of BCM clearly increased Young’s modulus by 2- to 4-fold, and the magnitude of the increase depended on the size of the matrix particle, contributing to a better elastic module than the one that was assembled with a BCM between 2 and 100 µm.
To our knowledge, this is the first research that analyzes the ECM in two different size ranges to evaluate its influence on mechanical properties.
Kun et al. reported that 6% of porcine cartilage matrix nanofibers prepared via the freeze-dried method, with a particle size ranging from 50 to 500 nm, have an elastic modulus of 40.208 ± 5.097 kPa [
31]; this is insufficient to cover the requirements of the chondral tissue.
Unlike Kun et al. we compared particle sizes above the range of nanoparticles; and interestingly we observed that implants constructed with a larger particle size (20–100 µm) showed improved mechanics that were even similar to native articular cartilage (
Figure 2).
In this research, in addition to studying the effect of the particle size of BCM on Young’s modulus, the effect of three different proportions with respect to the fibroin hydrogel system obtained by the NaCl leaching method was also analyzed.
Rowland et al. studied the effect of different concentrations (between 7% and 11%) of a porcine cartilage matrix, with a particle size no greater than 97 μm, on the mechanical properties of a reticulated collagen system. They observed that only a concentration of 11% is able to resist the deformation of the scaffold (better compression module); however, this affected the porosity and therefore the cellular infiltration [
32]. These data confirm that an increase in the concentration of the cartilage matrix does not necessarily lead to a scaffold with good mechanical and biological properties.
In our system, in contrast to the data reported by Rowland, different proportions of the matrix did not exert a statistically significant effect on the mechanical properties of the scaffold (
Figure 2).
These discrepancies may be due to differences in the nature of the scaffolding. The manufacturing leaching process provides a controlled porosity for the resulting scaffolding.
Although there were no differences in the Young elastic modulus with respect to the BCM proportions that were analyzed, we certainly do observe a considerable influence of the microenvironment created in different proportions on the behavior of the chondrocytes cultivated in the scaffolds.
It has been shown that the mechanical features of articular cartilage are closely related to two of the main components of the hyaline cartilage matrix: sulfated proteoglycans and collagens [
33].
According to Muiznieks et.al., type II, IX and XI collagens are responsible for the shape, tensile stiffness and strength of cartilage tissue [
34], while proteoglycans are responsible for their compressive strength [
35].
The data presented in
Figure 2 and
Figure 5 are consistent with this assertion, where the highest content of proteoglycans in Group II conferred a greater elastic modulus. Via safranin O, the presence of chondrocytes surrounded by sulfated proteoglycans was demonstrated within a properly organized matrix. Moreover, Masson’s trichrome stain confirmed the presence of abundant well-organized collagen fibers in the newly formed matrix, where a type II collagen expression predominated, generating a Col II:Col I ratio typical for hyaline cartilage (
Figure 5D).
We postulate that this increase of the matrix synthesis in the chondral phase was a combined effect of a homogeneous and interconnected porosity, and the presence of the BCM rich in chondro-inductive signals in a proportion (1:1 ratio of fibroin/NaCl:BCM) that allowed for themaintenance of the scaffolding microstructure.
Although the in vitro models offer valuable information about the behavior of an implant under a controlled environment, they do not give evidence of two aspects that determine the clinical application of an implant: the integration with the surrounding tissue and the organization of the tissue in the area of repair of an osteochondral defect. To date, the important challenge of integrating and maintaining the hierarchical structure of the osteochondral tissue has been poorly addressed [
36,
37].
In order to generate a scaffold to guide the synthesis and hierarchical organization of the newly formed ECM in the context of osteochondral damage, a biphasic scaffold was designed by tissue engineering and was evaluated in a porcine osteochondral damage model.
In the strategy described here, the pre-chondrocytes (Groups II and III) and the osteoblasts (Group III) were included in scaffolds of a defined size, shape and architecture. The biological similarity of the constructions with the native articular cartilage was demonstrated in
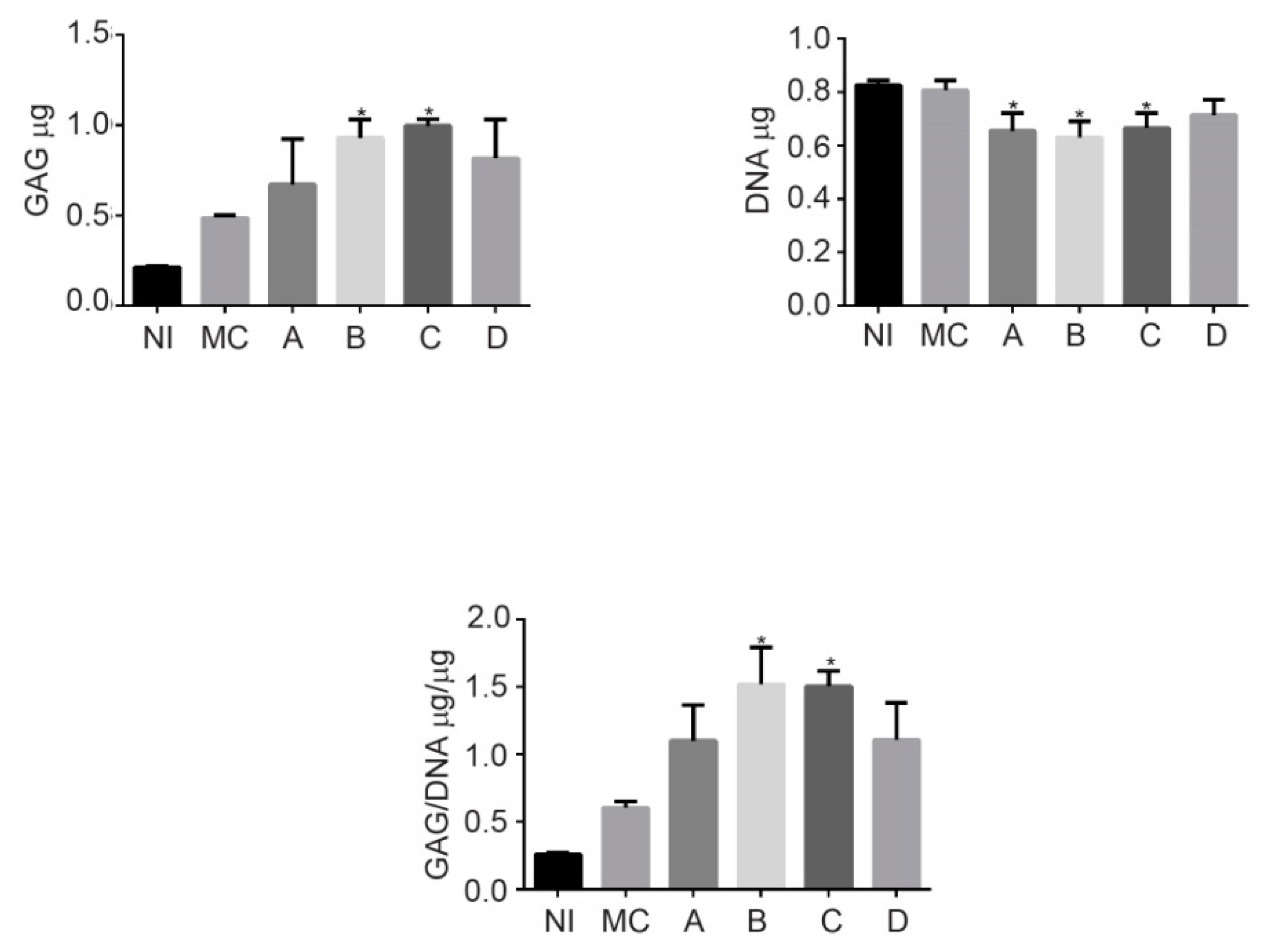
Figure 1C. The procedure involved a phase of in vitro maturation of the cellularized implant. This step allowed the embedded cells to begin the synthesis of the ECM under a controlled system, as the presence of abundant type II collagen and GAG demonstrated (
Figure 4; this is key, since it has been reported that this profile correlates with the mechanical quality of engineering hyaline cartilage [
38]. Moreover, it also encourages the platform on which the host cells are established to preserve the hierarchical organization in the neoformed ECM.
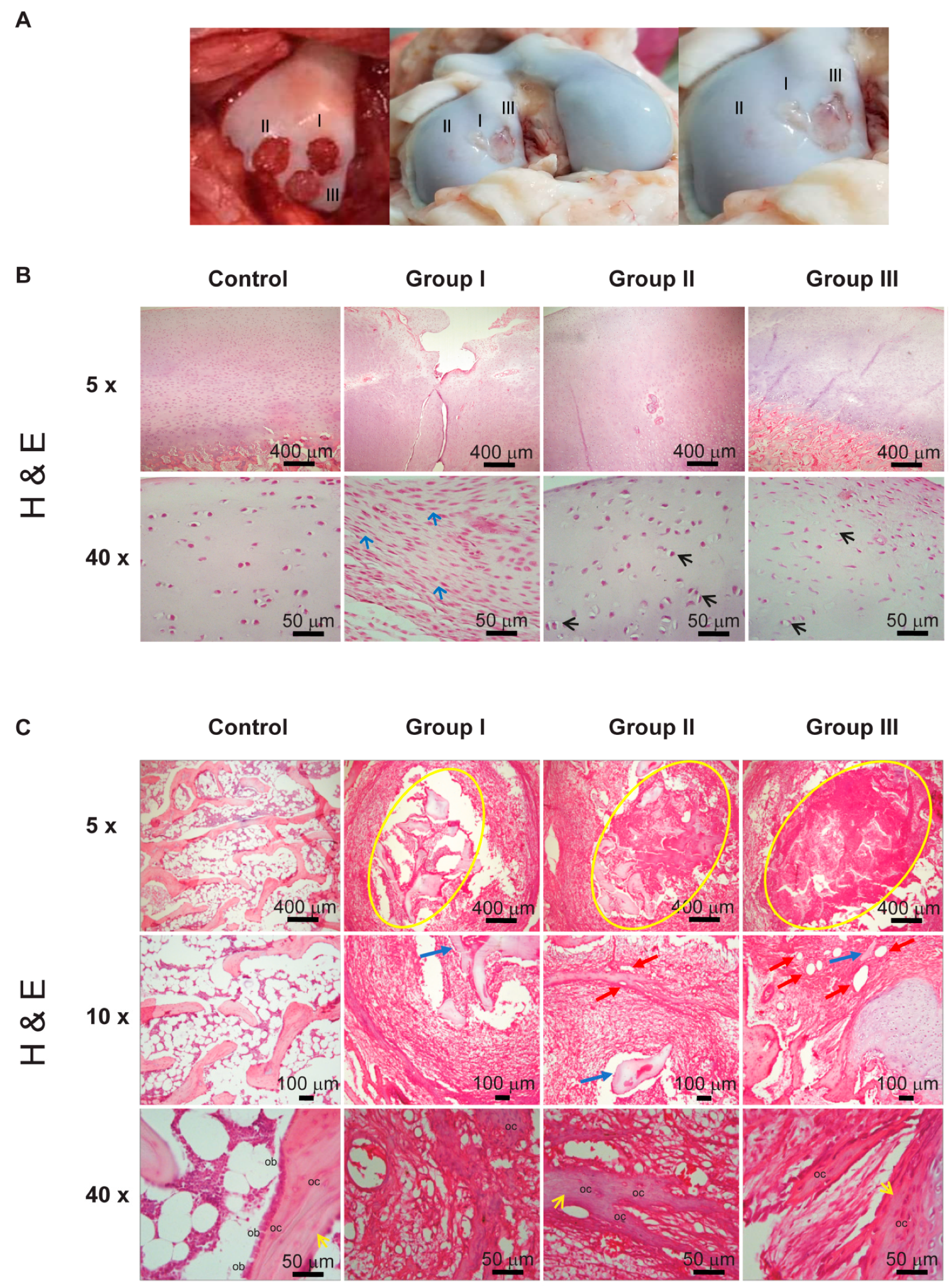
8 weeks after placing the implants, our histological data (
Figure 6) showed that only the cellularized biphasic implant in the chondral phase (Group II) had succeeded in establishing the architecture of the osteochondral tissue (
Figure 6B and
Figure 7A), constituted by an ECM moderately rich in GAGs (
Figure 7B) and presenting, toward the surface, a smooth texture (
Figure 6A).
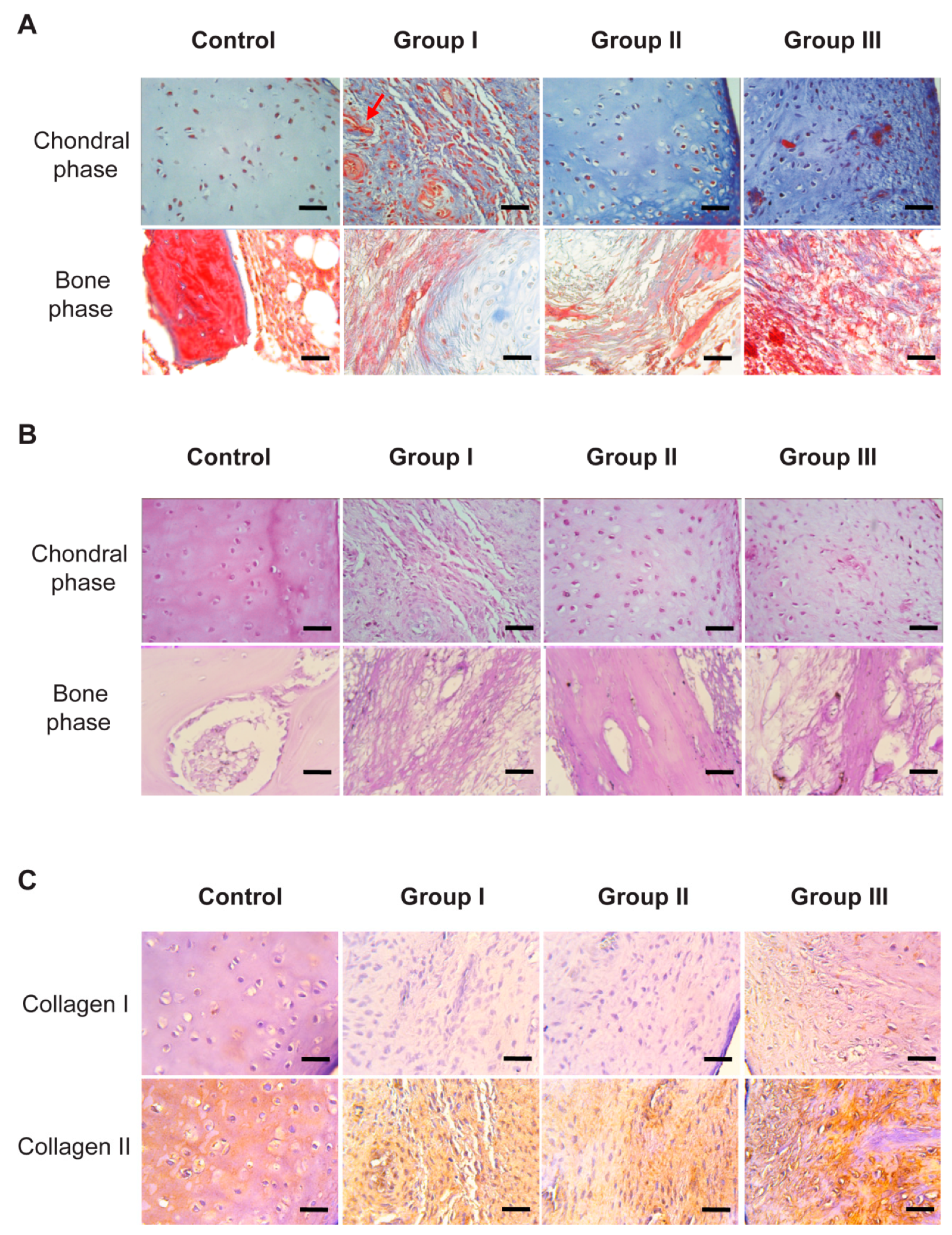
The tissue formation of hyaline cartilage was evident in Group II, as evidenced by the presence of lacunae containing cells embedded within the basophilic substance that was rich in GAG and type II collagen. Regarding Groups I and III, fibro-cartilage repair tissue was observed, as indicated by the presence of type I collagen, in addition to the poor secretion of proteoglycans compared to native articular cartilage.
It has already been described that a lesion on the articular surface generally causes the proliferation of chondrocytes in the proximal zone and increases the synthesis of matrix macromolecules. However, this anabolic response of the chondrocytes terminates a short time after the injury, which leads to an inadequate filling of the defect in the tissue [
39]. The influence of the biphasic implant, but not the monophasic one, could have supported the compensatory mechanisms of the chondrocytes under a scaffolding system that served as a guide for the hierarchical structure, leading to the production and proper structuring of a matrix rich in proteoglycans and collagens in repaired chondral tissue.
Regarding the bone phase, it has been reported that the created microenvironment is capable of activating the surrounding stem cells into responding, thereby promoting osteogenesis [
40,
41].
According to the immunohistochemical observation, both in Group II (non-cellularized in the bone phase) and in Group III (cellularized in vitro with osteoblasts), the expression of type I collagen was evidenced, which is an indicator of a good recovery of the osteoblastic tissue, and which also coincides with the expression of type II collagen in the chondral phase, suggesting an adequate repair of the osteochondral tissue as a whole [
42].
Consequently, what is the influence of the cellularization of this bone phase in terms of osteochondral tissue repair?
The different patterns of vascularization within the framework of the osteochondral tissue are a major issue. The bone is a highly vascularized tissue, while the cartilage is avascular. The avascularity of the cartilage and also its resistance to the invasion of the vascular networks are important for its function [
43]; otherwise, the cartilage can result in ossification of the deep and intermediate zone and consequently exacerbate the joint damage.
As indicated by the H&E staining, in the bone phase of Group II a greater number of nascent blood vessels were observed in comparison with Group III, which suggests that a non-cellularized scaffolding system allows the establishment of host cells for the promotion of an adequate ECM, as well as for the modulation of the vascularization degree. We also noted that the ECM of the cartilage, in this Group II, showed almost no angiogenesis, which benefited the repair of cartilage.
Poor integration has discarded many cartilage repair approaches, and this problem derives from the intrinsic metabolism of cartilage [
44].
The ability of the implants to integrate with the host tissue was histologically evaluated. In the case of Group II, the ECM that was produced was integrated with the surrounding tissue, which resulted in a significantly better macroscopic and microscopic healing of the cartilage defects when compared to Groups I and III. No signs of cleft, delamination and fissures were observed at the boundary between the host tissue and the repair tissue (
Figure 5).
We postulate that the integration observed in Group II is an additive effect resulting from a scaffolding system that serves as a guide for maintaining the hierarchical structure of osteochondral tissue, as a bone phase capable of promoting cell migration, as the generation of a newly formed tissue that entwines with the collagen fibers in the recipient site and as a vascularization profile that maintains the healthy state of the bone without invading the chondral tissue.
In conclusion, the biphasic implant, based on fibroin and biofunctionalized with BCM in a 1:1 ratio with respect to fibroin/NaCl, develops an ECM rich in sulfated proteoglycans and collagen fibers with an appropriate ratio of Col II:Col I, and with mechanical characteristics similar to native cartilage. The implant, when it is only cellularized in the cartilage phase, is able to repair a full-thickness osteochondral lesion, showing a bright white tissue surface after a period of 4 months. Repair tissue evidenced a hierarchical organization of chondrocytes and collagen fibers similar to that of native cartilage. Taking together, these data show that we can establish that the design described here represents a promising alternative for osteochondral tissue-engineering applications.
4. Materials and Methods
4.1. Preparation of Natural Bovine Cartilage Matrix (BCM)
Bovine knees explants (thickness = 2 mm) were obtained from six animals (aged <3 years.) in an aseptic environment. The joints were acquired from an abattoir with TIF certification within 24 h of sacrifice. The cartilage was collected from the area of the condyles, patella, and trochlea with a scalpel No. 24 (SensiMedical). The physicochemical decellularization consisted of five cycles of thermal shock in liquid nitrogen for 5 min followed by a wash in PBS for 10 min. Later, the matrix was crushed with a blender. Once crushed, the matrix was washed for 24 h in hypotonic buffer (10 mM TRIS-HCL, 2 mM EDTA, pH 8) supplemented with 100 mM KCl and 5 mM MgCl2. Next, hypotonic buffer supplemented with 100 mM KCl, MgCl2 and 0.5% SDS was added for 18 h. Finally, the matrix was washed with a hypotonic buffer with 0.5% SDS for 36 h. Following sterilePBS rinsing, the samples were immediately frozen at −80 °C. Before implantation, the samples were lyophilized for 24 h for the complete removal of interstitial fluid, followed by fine grinding. For the pulverization, a pulverizer mill K10 (Micron, Shanghai, China) and a Freezer/mill 6870 (SPEX(R) samplePrep, Metuchen, NJ, USA) were used, generating a particle size of 10–20 µm and 20–100 µm, respectively.
4.2. Preparation of Natural Bovine Bone Chips (BBC)
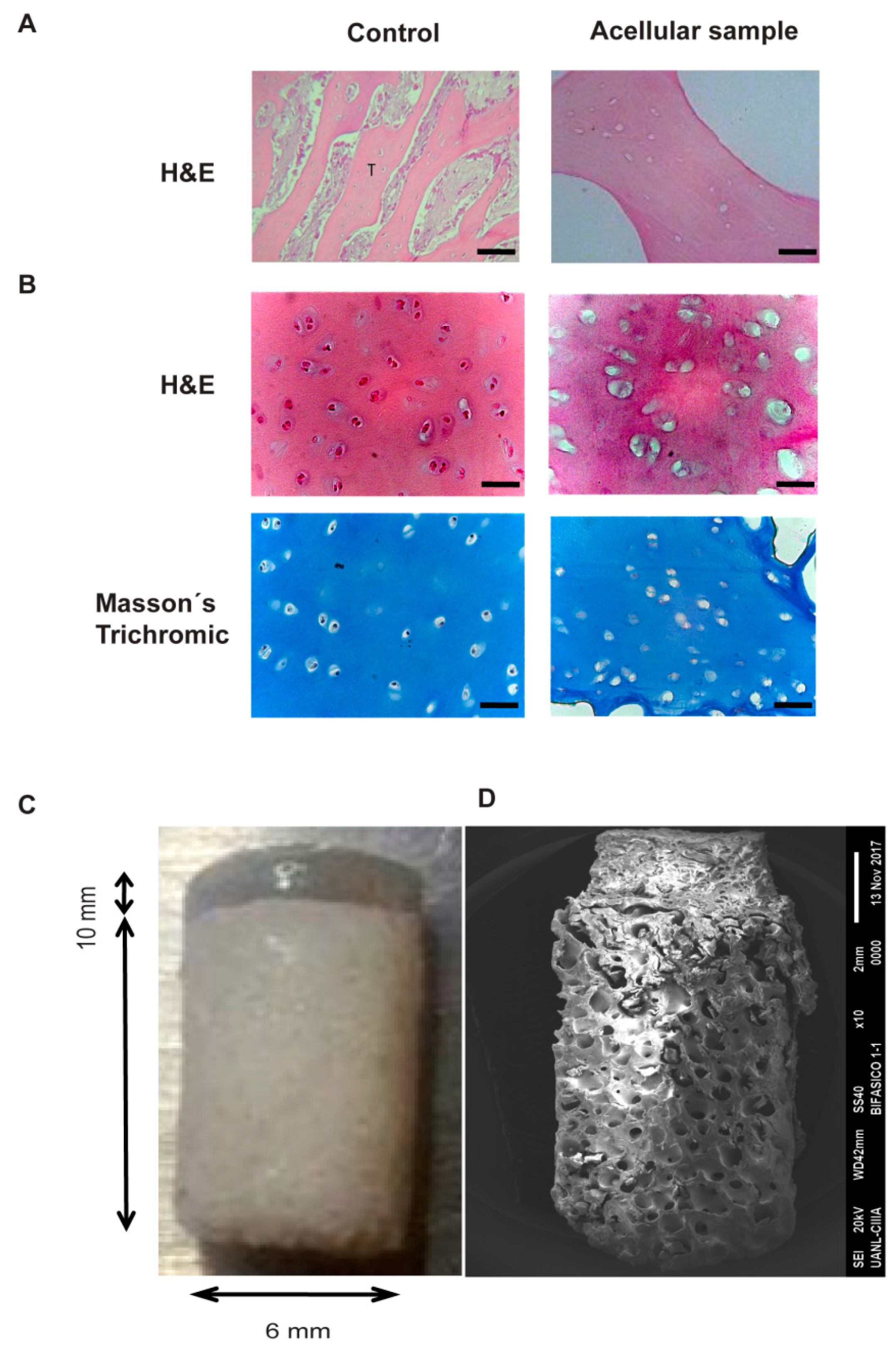
Subchondral bone was collected from bovine knees with trephine 6 mm in diameter. The decellularization consisted of 5 washing cycles in 0.1% SDS in PBS (Sigma-Aldrich, St. Louis, MO, USA) at 100 °C followed by incubation with 30% H2O2 (Sigma-Aldrich, St. Louis, MO, USA) for 5 h. Subsequently, the bone chips were washed with deionized water in order to remove the remains of SDS and H2O2. Finally, the decellularized bones were allowed to dry at room temperature and stored until use. Their decellularization by H&E was evaluated.
4.3. Biphasic Implant Assembly
For the chondral phase, the silk fibroin was dissolved in Hexafluoro-isopropanol (HFIP) (Sigma-Aldrich, St. Louis, MO, USA) to obtain an 8% solution. The chondral phase was manufactured with fibroin and NaCl (particle size from 74 to 177 µm) in a 1:1 ratio with or without BCM (pulverized with the K10 mill or the Freezer/thousand 6870) at different proportions (NaCl:BCM; 1:1, 2:1 and 3:1). The biphasic scaffold was prepared as follows. The NaCl with or without BCM was placed in cylindrical Teflon molds of 2 cm × 6 mm, and 140 μL of silk fibroin (SF) was added by scaffolding. Subsequently, the BBC (pre-soaked with 90% methanol) was placed into the mold, and 90% methanol (Sigma-Aldrich, St. Louis, MO, USA) was added to induce the crystallization of the SF. The scaffolds in the mold were allowed to stand for 24 h at room temperature. To remove the salt, the scaffolds were washed in ultrapure water for 3 days. Finally, the scaffolds were frozen at −80 °C and lyophilized for 24 h. The sterilization was carried out with ethylene oxide. Seven groups of implants were obtained; Groups B, C and D were manufactured with the pulverized matrix in the Freezer/mill 6870, while Groups E, F, G and H were manufactured with the BCM pulverized with the K10 mill. Group A: SF, Group B: SF/NaCl:BCM 1:1, Group C: SF/NaCl:BCM2:1, Group D: SF/NaCl:BCM3:1, Group E: SF/NaCl:BCM 1:1, Group F: SF/NaCl:BCM 2:1, and Group G: SF/NaCl:BCM 3:1.
4.4. Scanning Electron Microscopy
The samples were fixed in 4% glutaraldehyde, and then soaked in 0.1M sodium cacodylate for 10 min and washed three times. The samples were transferred into 1% osmium tetraoxide for 2 h for post-fixation and washed with 0.1M sodium cacodylate. The fixed samples were dehydrated in a graded series of acetone, placed into the critical point dryer (CPD Baltec-030) for 30 min, mounted onto a stub sputtered with gold coating in a Sputter Coater Polaron E-5100 scanning electron microscopy SEM Coating Unit and viewed under a JEOL JSM 6400 scanning electron microscope (JEOL Corporation Ltd., Tokyo, Japan).
4.5. Mechanical Testing
An unconfined compression assay of acellular scaffolds (n = 6 per group) was performed on circular discs of 3 mm diameter. The mechanical testing of the scaffolds was carried out with a mechanical tester (Bose Electroforce model 3230 SERIES II). The dimension of the scaffolds was measured with a calibrator, and then they were placed between the compressive motor and load cell and subjected to a 10% compression (0.2 mm) at a speed of 0.01 mm/s. The force on the displacement was graphed. Young’s modulus was calculated from the slope of the line. All testing groups in this study consisted of 6 individual samples, and the statistical significance between the groups was determined by a one-way ANOVA followed by the Tukey-Kramer HSD test. Samples with a p < 0.05 were determined to be statistically significant.
4.6. Porcine Adipose-Derived Stem Cell Isolation and Cell Cultivation
Porcine ADSCs were isolated from adipose tissues obtained from the subcutaneous fat from the caudal area of euthanized adult Yorkshire pigs of 70 kg (n = 4). The adipose tissue sample (0.5 g) was minced into small pieces and digested in 0.1% collagenase I (Gibco-Invitrogen, Carlsbad, CA, USA) at 37 °C for 2 h. The collagenase was inactivated by the addition of supplemented DMEM (Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum (FBS)) (HyClone, Logan, UT, USA), L-glutamine 10 mM and, penicillin (100 U/mL), streptomycin (100 μg/mL) and amphotericin B (0.25 mg/mL) (all from CORNING Cellgro, Manassas, VA, USA). Cells were cultured in a 25 cm2 flask at 37 °C and 5% CO2. After a week, the non-adherent cells were removed, and the adherent cells were further cultured in supplemented DMEM. The medium was changed every 2–3 days until the monolayer of adherent cells reached 80% confluence.
4.7. Cell Differentiation Assays
To confirm the multipotential differentiation ability of cultured porcine ADSCs, cells were cultivated with the different induction media for 14d. For the adipogenic differentiation, ADSCs were grown to 80% confluence in a 4-well chamber (Thermo Fischer Scientific Nunc, Langenselbold, Germany) in the DMEM supplemented with 10% FBS, 1 mM dexamethasone, 1 mg/mL insulin, 0.5 mM 3-isobutyl-1-methylxanthine, and 100 mM indomethacin. After induction, the cultures were fixed in 10% paraformaldehyde and stained with Oil-red-O solution to detect lipid droplets. For the chondrogenic differentiation, the ADSCs were plated at 2 × 103 cells/cm2 in a chondrogenic medium containing DMEM (GIBCO, Scotland, UK) supplemented with 0.010 mg/mL of ITS [insulin transferring selenium] (GIBCO, Scotland, UK), 100 nM of dexamethasone (Invitrogen, Carlsbad, CA, USA), 0.001 mg/mL of ascorbic acid and 10 ng/mL of TGFβ1. The cells were fixed with 3% paraformaldehyde and stained with 1% Alcian blue (Sigma-Aldrich, St. Louis, MO, USA) in 0.1 N HCl for 30 min. A blue coloration is indicative of the synthesis of proteoglycans by chondrocytes. To induce osteogenic differentiation, cells were plated at 2 × 103 cells/cm2 in an osteogenic medium that contained the DMEM (GIBCO, Scotland, UK) supplemented with 10% FBS, 10 mM β-glycerophosphate, 0.25 mM ascorbic acid, and 10−8 M dexamethasone (Sigma-Aldrich, St. Louis, MO, USA). The phenotype was confirmed using Alizarin red staining to evaluate calcium-rich deposits. Control cultures of ADSCs without differentiation medium were also maintained simultaneously (non-induced group).
4.8. Cellularization of Scaffolds and In Vitro Maturation
The ADSCs in passage 3 were cultured in a monolayer with supplemented DMEM at 37 °C and 5% CO
2 until reaching 90% confluence; subsequently, the cells were pre-differentiated in the chondrogenic medium during 5 d and then trypsinized with 0.25% trypsin (GIBCO, Scotland, UK). Scaffolds of 3 mm in diameter by 9 mm deep were cellularized after hydration with supplemented DMEM for 2 h. The chondral phase was cellularized by injection with 0.5 × 10
6 pre-chondrocytes in 10 μL of supplemented DMEM. The scaffolds were placed in low adhesion 48-well plates and incubated at 37 °C and 5% CO
2 for 2 h to allow the cells to adhere to the scaffold. This procedure was followed for the scaffolds of Groups I, II and III. For Group III, after chondral cellularization, the scaffolds were cellularized with osteoblasts. Previously, the ADSCs were differentiated in the osteogenic medium during 5 d and then trypsinized with 0.25% trypsin (GIBCO, Scotland, UK). The bone phase was cellularized by injection with 2.25 × 10
6 osteoblasts in 20 μL of osteogenic medium and incubated for 2 more hours to allow the cells to adhere to the scaffold. The ones from Groups I and II were grown in low adhesion 48-well plates in a chondrogenic medium, while those in Group III were incubated in a two-chamber bioreactor [
42], which allowed the chondral phase to be cultivated in a chondrogenic medium and the bone phase in an osteogenic medium, preventing the media from mixing. All groups were cultured for 12 days for maturation at 37 °C in 5% CO
2 before being implanted.
4.9. Quantification of GAGs, Normalized with the DNA Content
The content of sulfated GAGs was quantified using a Blyscan™ sGAG Assay Kit (B1000; Biocolor, Carrickfergus, UK). The samples were minced into small pieces and digested with the papain extraction reagent (Sigma-Aldrich, St. Louis, MO, USA), L-Cysteina-HCl (Sigma-Aldrich, St. Louis, MO, USA), sodium acetate (Sigma-Aldrich, St. Louis, MO, USA) and EDTA (Sigma-Aldrich, St. Louis, MO, USA) at 65 °C for 18 h. For the DNA quantification, the QuanticoTM PicoGreenTM dsDNA Assay Kit (Thermo Fischer Scientific Nunc, Langenselbold, Germany) was used. The fluorescence was read on a plate reader (Biotek) at an excitation length of 485 nm and an emission of 520 nm.
4.10. RNA Extraction and Quantitative PCR
RNA (100 ng) was isolated from the chondrocyte/scaffold composites after 28 days in culture using the TRIzol reagent (Life Technologies, Carlsbad, CA, USA). 1 μg cDNA was amplified in a PCR mixture including SYBR Green Realtime PCR Master Mix-Plus (Applied Biosystems, Foster City, CA, USA) and gene-specific primers (
Table 2), in accordance with the manufacturer’s information. The reaction was performed using a Step One Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). GAPDH was used as an endogenous control for the study. The relative gene expression profiles were normalized to a GAPDH expression and analyzed using the 2
−ΔΔCT approach (
n = 3).
4.11. Histology and Immunohistochemistry (IHC)
After decalcification with 10% formic acid (Sigma-Aldrich, St. Louis, MO, USA), the samples were processed via a conventional histological technique until their inclusion in paraffin blocks. Subsequently, histological sections of 4 μm were stained with hematoxylin and eosin (H&E) for the analysis of the general histology, with safranin O to detect the presence of sulphated proteoglycans, and with Masson’s trichrome for the staining of connective and muscular tissue components, as well as with the acid histochemical technique Periodic Schiff (PAS). This technique selectively identifies the complex polysaccharides (glycosaminoglycans, proteoglycans, and adhesion glycoproteins) present in the extracellular matrix. These samples were analyzed by light microscopy. An immunohistochemical analysis was performed on sections of 4 μm for the specific identification of type I and II collagen fibers in the chondral phase I and collagen I phase in the bone phase. Anti-col I monoclonal antibodies (1:400) and anti col II (1:400) were used. The mouse and rabbit specific detection system HRP/DAB (ABC) detection IHC kit (ab64264) was used. The antibodies and the detection system were purchased from Abcam®, (Cambridge, MA, USA). The positivity was identified with 3, 3′ diaminobenzidine (DAB), and the nuclei were contrasted with Gill’s hematoxylin. Samples of articular cartilage and trabecular bone tissue were used as positive controls of the technique, and the primary antibody was omitted as a negative control. The samples were analyzed and also photographed using an Olympus AX70 microscope (Olympus, Tokyo, Japan).
4.12. Intraarticular Implantation Surgery
In order to reduce differences in the quality of tissue repair due to the genetic background of the animal, three different implants were placed on the same knee. Each lesion was 3 mm in diameter by 9 mm in length, reaching the subchondral bone; the defects were created in the center of the distal weight-bearing surface. The animal was anesthetized with intravenous xylazine (0.1 mg/kg) and Ketamine (7 mg/kg), and then placed in a lateral recumbent position. For surgery, a medial parapatellar approach to the joint capsule in its left hind leg, continuing to dislocate the patella laterally and expose the femoral condyles, was performed. A circular drill bit with a 6 mm diameter was used to perform the lesions to the subchondral bone. Three treatments were applied per knee as follow: Group 1: chondral monophasic scaffold, cellularized (AM); Group 2: biphasic scaffold, cellularized only in the chondral phase; and Group 3: biphasic scaffolding, cellularized in both the chondral and bone phases. In order to hold the graft onto the surrounding tissue, two drops of glue tisuacryl (Tisuacryl®, BIOMAT) were placed on the surface. Finally, two internal sutures were performed with polyglycolic acid (PGA Atramat®, International Pharmaceutical SA de CV Mexico, DF) in the synovial capsule, muscle and tendons, and with outer nylon (Vicryl 3-0 Ethicon, Inc.; Nylon ™ Suture ETHILON 3-0, Ethicon, Inc. Somerville, NJ) on the skin. During the immediate postoperative period, the animals were maintained in cages with standard care. After this period of time, the motion of the knee was not restricted and full weight-bearing on the surgical joints was allowed.
4.13. Ethics Statement
The protocol with the identification code P218-00337 involving research on animals was revised and approved by the UANL School of Medicine & University Hospital Institutional Review Board (October 2018), and experiments were conducted following the Mexican standard for the treatment of experimental animals (Norma Oficial Mexicana 062-ZOO-1999).

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}