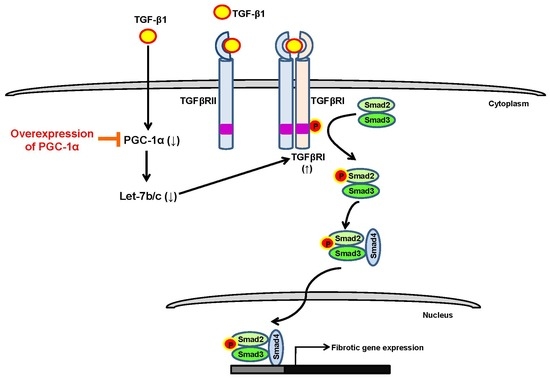
PGC-1α Suppresses the Activation of TGF-β/Smad Signaling via Targeting TGFβRI Downregulation by let-7b/c Upregulation

 , ,
, ,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Downregulation of PGC-1α in UUO-Induced Kidney Injury and TGF-β-treated HK-2 Cells
2.2. Protective Effects of PGC-1α on TGF-β-Induced Epithelial-Mesenchymal Transition (EMT)
2.3. Negative Regulation of TGF-β/Smad Signaling by PGC-1α
2.4. PGC-1α-Specific TGFβRI Downregulation
2.5. PGC-1α Regulates let-7b/c-Mediated TGFβRI Expression
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. UUO Model
4.3. Cell Culture and TGF-β Treatment
4.4. Transfection of siRNA
4.5. Quantification of mRNA and miRNA
4.6. Immunofluorescence (IF) Staining
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Meng, X.M.; Tang, P.M.; Li, J.; Lan, H.Y. TGF-beta/Smad signaling in renal fibrosis. Front. Physiol. 2015, 6, 82. [Google Scholar] [CrossRef]
- Bottinger, E.P.; Bitzer, M. TGF-beta signaling in renal disease. J. Am. Soc. Nephrol. 2002, 13, 2600–2610. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Isaka, Y. Targeting TGF-beta Signaling in Kidney Fibrosis. Int. J. Mol. Sci. 2018, 19, 2532. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Ward, W.F. PGC-1alpha: A key regulator of energy metabolism. Adv. Physiol Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.; Tam, D.; Bardia, A.; Bhasin, M.; Rowe, G.C.; Kher, A.; Zsengeller, Z.K.; Akhavan-Sharif, M.R.; Khankin, E.V.; Saintgeniez, M.; et al. PGC-1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. J. Clin. Invest. 2011, 121, 4003–4014. [Google Scholar] [CrossRef]
- Lynch, M.R.; Tran, M.T.; Parikh, S.M. PGC1alpha in the kidney. Am. J. Physiol. Renal Physiol 2018, 314, F1–F8. [Google Scholar] [CrossRef] [PubMed]
- Han, S.H.; Wu, M.Y.; Nam, B.Y.; Park, J.T.; Yoo, T.H.; Kang, S.W.; Park, J.; Chinga, F.; Li, S.Y.; Susztak, K. PGC-1alpha Protects from Notch-Induced Kidney Fibrosis Development. J. Am. Soc. Nephrol. 2017, 28, 3312–3322. [Google Scholar] [CrossRef]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Sharma, K.; Karl, B.; Mathew, A.V.; Gangoiti, J.A.; Wassel, C.L.; Saito, R.; Pu, M.; Sharma, S.; You, Y.H.; Wang, L.; et al. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J. Am. Soc. Nephrol. 2013, 24, 1901–1912. [Google Scholar] [CrossRef]
- Han, S.H.; Malaga-Dieguez, L.; Chinga, F.; Kang, H.M.; Tao, J.; Reidy, K.; Susztak, K. Deletion of Lkb1 in Renal Tubular Epithelial Cells Leads to CKD by Altering Metabolism. J. Am. Soc. Nephrol. 2016, 27, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Badal, S.S.; Ye, Z.; Wang, Y.; Ayanga, B.A.; Galvan, D.L.; Green, N.H.; Chang, B.H.; Overbeek, P.A.; Danesh, F.R. Long noncoding RNA Tug1 regulates mitochondrial bioenergetics in diabetic nephropathy. J. Clin. Invest. 2016, 126, 4205–4218. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.I.; Kim, H.J.; Park, J.S.; Kim, I.J.; Bae, E.H.; Ma, S.K.; Kim, S.W. PGC-1alpha attenuates hydrogen peroxide-induced apoptotic cell death by upregulating Nrf-2 via GSK3beta inactivation mediated by activated p38 in HK-2 Cells. Sci. Rep. 2017, 7, 4319. [Google Scholar] [CrossRef] [PubMed]
- Brennan, E.P.; Nolan, K.A.; Borgeson, E.; Gough, O.S.; McEvoy, C.M.; Docherty, N.G.; Higgins, D.F.; Murphy, M.; Sadlier, D.M.; Ali-Shah, S.T.; et al. Lipoxins attenuate renal fibrosis by inducing let-7c and suppressing TGFbetaR1. J. Am. Soc. Nephrol. 2013, 24, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Jha, J.C.; Hagiwara, S.; McClelland, A.D.; Jandeleit-Dahm, K.; Thomas, M.C.; Cooper, M.E.; Kantharidis, P. Transforming growth factor-beta1-mediated renal fibrosis is dependent on the regulation of transforming growth factor receptor 1 expression by let-7b. Kidney Int. 2014, 85, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Duann, P.; Lin, P.H. Mitochondria Damage and Kidney Disease. Adv. Exp. Med. Biol. 2017, 982, 529–551. [Google Scholar]
- Forbes, J.M. Mitochondria-Power Players in Kidney Function? Trends Endocrinol Metab. 2016, 27, 441–442. [Google Scholar] [CrossRef] [PubMed]
- Quadri, M.M.; Fatima, S.S.; Che, R.C.; Zhang, A.H. Mitochondria and Renal Fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 501–524. [Google Scholar]
- Leaf, I.A.; Duffield, J.S. What can target kidney fibrosis? Nephrol Dial. Transplant. 2017, 32, i89–i97. [Google Scholar] [CrossRef]
- Galvan, D.L.; Green, N.H.; Danesh, F.R. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int. 2017, 92, 1051–1057. [Google Scholar] [CrossRef]
- Luangmonkong, T.; Suriguga, S.; Bigaeva, E.; Boersema, M.; Oosterhuis, D.; de Jong, K.P.; Schuppan, D.; Mutsaers, H.A.M.; Olinga, P. Evaluating the antifibrotic potency of galunisertib in a human ex vivo model of liver fibrosis. Br. J. Pharmacol. 2017, 174, 3107–3117. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wu, H.; Tarr, P.T.; Zhang, C.Y.; Wu, Z.; Boss, O.; Michael, L.F.; Puigserver, P.; Isotani, E.; Olson, E.N.; et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 2002, 418, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Wang, Q.; Lou, T.; Qin, J.; Jung, S.; Shetty, V.; Li, F.; Wang, Y.; Feng, X.H.; Mitch, W.E.; et al. Myokine mediated muscle-kidney crosstalk suppresses metabolic reprogramming and fibrosis in damaged kidneys. Nat. Commun. 2017, 8, 1493. [Google Scholar] [CrossRef] [PubMed]
- Frost, R.J.; Olson, E.N. Control of glucose homeostasis and insulin sensitivity by the Let-7 family of microRNAs. Proc. Natl. Acad. Sci. USA 2011, 108, 21075–21080. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Han, S.; Kwon, C.S.; Lee, D. Biogenesis and regulation of the let-7 miRNAs and their functional implications. Protein Cell 2016, 7, 100–113. [Google Scholar] [CrossRef]
- Lv, W.; Fan, F.; Wang, Y.; Gonzalez-Fernandez, E.; Wang, C.; Yang, L.; Booz, G.W.; Roman, R.J. Therapeutic potential of microRNAs for the treatment of renal fibrosis and CKD. Physiol. Genomics 2018, 50, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Park, J.T.; Kato, M.; Lanting, L.; Castro, N.; Nam, B.Y.; Wang, M.; Kang, S.W.; Natarajan, R. Repression of let-7 by transforming growth factor-beta1-induced Lin28 upregulates collagen expression in glomerular mesangial cells under diabetic conditions. Am. J. Physiol. Renal Physiol. 2014, 307, F1390–F1403. [Google Scholar] [CrossRef]
- Serhan, C.N. Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot Essent Fatty Acids 2005, 73, 141–162. [Google Scholar] [CrossRef]
- Borgeson, E.; Docherty, N.G.; Murphy, M.; Rodgers, K.; Ryan, A.; O’Sullivan, T.P.; Guiry, P.J.; Goldschmeding, R.; Higgins, D.F.; Godson, C. Lipoxin A(4) and benzo-lipoxin A(4) attenuate experimental renal fibrosis. FASEB J. 2011, 25, 2967–2979. [Google Scholar] [CrossRef]
- Sato, Y.; Yanagita, M. Resident fibroblasts in the kidney: A major driver of fibrosis and inflammation. Inflamm. Regen 2017, 37, 17. [Google Scholar] [CrossRef]
- Lv, W.; Booz, G.W.; Wang, Y.; Fan, F.; Roman, R.J. Inflammation and renal fibrosis: Recent developments on key signaling molecules as potential therapeutic targets. Eur. J. Pharmacol. 2018, 820, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Fontecha-Barriuso, M.; Martin-Sanchez, D.; Martinez-Moreno, J.M.; Carrasco, S.; Ruiz-Andres, O.; Monsalve, M.; Sanchez-Ramos, C.; Gomez, M.J.; Ruiz-Ortega, M.; Sanchez-Nino, M.D.; et al. PGC-1alpha deficiency causes spontaneous kidney inflammation and increases the severity of nephrotoxic AKI. J. Pathol. 2019, 249, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Spiegelman, B.M. The role of exercise and PGC1alpha in inflammation and chronic disease. Nature 2008, 454, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.F.; Ku, H.C.; Lin, H. PGC-1alpha as a Pivotal Factor in Lipid and Metabolic Regulation. Int. J. Mol. Sci. 2018, 19, 3447. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, J.; Zhou, F.; Wang, W.; Chen, N. PGC-1alpha ameliorates kidney fibrosis in mice with diabetic kidney disease through an antioxidative mechanism. Mol. Med. Rep. 2018, 17, 4490–4498. [Google Scholar] [PubMed]
- Marmolino, D.; Manto, M.; Acquaviva, F.; Vergara, P.; Ravella, A.; Monticelli, A.; Pandolfo, M. PGC-1alpha down-regulation affects the antioxidant response in Friedreich’s ataxia. PLoS ONE 2010, 5, e10025. [Google Scholar] [CrossRef]
- Miglio, G.; Rosa, A.C.; Rattazzi, L.; Collino, M.; Lombardi, G.; Fantozzi, R. PPARgamma stimulation promotes mitochondrial biogenesis and prevents glucose deprivation-induced neuronal cell loss. Neurochem. Int. 2009, 55, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Wenz, T.; Diaz, F.; Spiegelman, B.M.; Moraes, C.T. Activation of the PPAR/PGC-1alpha pathway prevents a bioenergetic deficit and effectively improves a mitochondrial myopathy phenotype. Cell Metab. 2008, 8, 249–256. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, H.-I.; Park, J.S.; Kim, D.-H.; Kim, C.S.; Bae, E.H.; Ma, S.K.; Kim, S.W. PGC-1α Suppresses the Activation of TGF-β/Smad Signaling via Targeting TGFβRI Downregulation by let-7b/c Upregulation. Int. J. Mol. Sci. 2019, 20, 5084. https://doi.org/10.3390/ijms20205084
Choi H-I, Park JS, Kim D-H, Kim CS, Bae EH, Ma SK, Kim SW. PGC-1α Suppresses the Activation of TGF-β/Smad Signaling via Targeting TGFβRI Downregulation by let-7b/c Upregulation. International Journal of Molecular Sciences. 2019; 20(20):5084. https://doi.org/10.3390/ijms20205084
Chicago/Turabian StyleChoi, Hoon-In, Jung Sun Park, Dong-Hyun Kim, Chang Seong Kim, Eun Hui Bae, Seong Kwon Ma, and Soo Wan Kim. 2019. "PGC-1α Suppresses the Activation of TGF-β/Smad Signaling via Targeting TGFβRI Downregulation by let-7b/c Upregulation" International Journal of Molecular Sciences 20, no. 20: 5084. https://doi.org/10.3390/ijms20205084
APA StyleChoi, H.-I., Park, J. S., Kim, D.-H., Kim, C. S., Bae, E. H., Ma, S. K., & Kim, S. W. (2019). PGC-1α Suppresses the Activation of TGF-β/Smad Signaling via Targeting TGFβRI Downregulation by let-7b/c Upregulation. International Journal of Molecular Sciences, 20(20), 5084. https://doi.org/10.3390/ijms20205084