Receptor Heterodimerization and Co-Receptor Engagement in TLR2 Activation Induced by MIC1 and MIC4 from Toxoplasma gondii

 , , ,
, , , 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
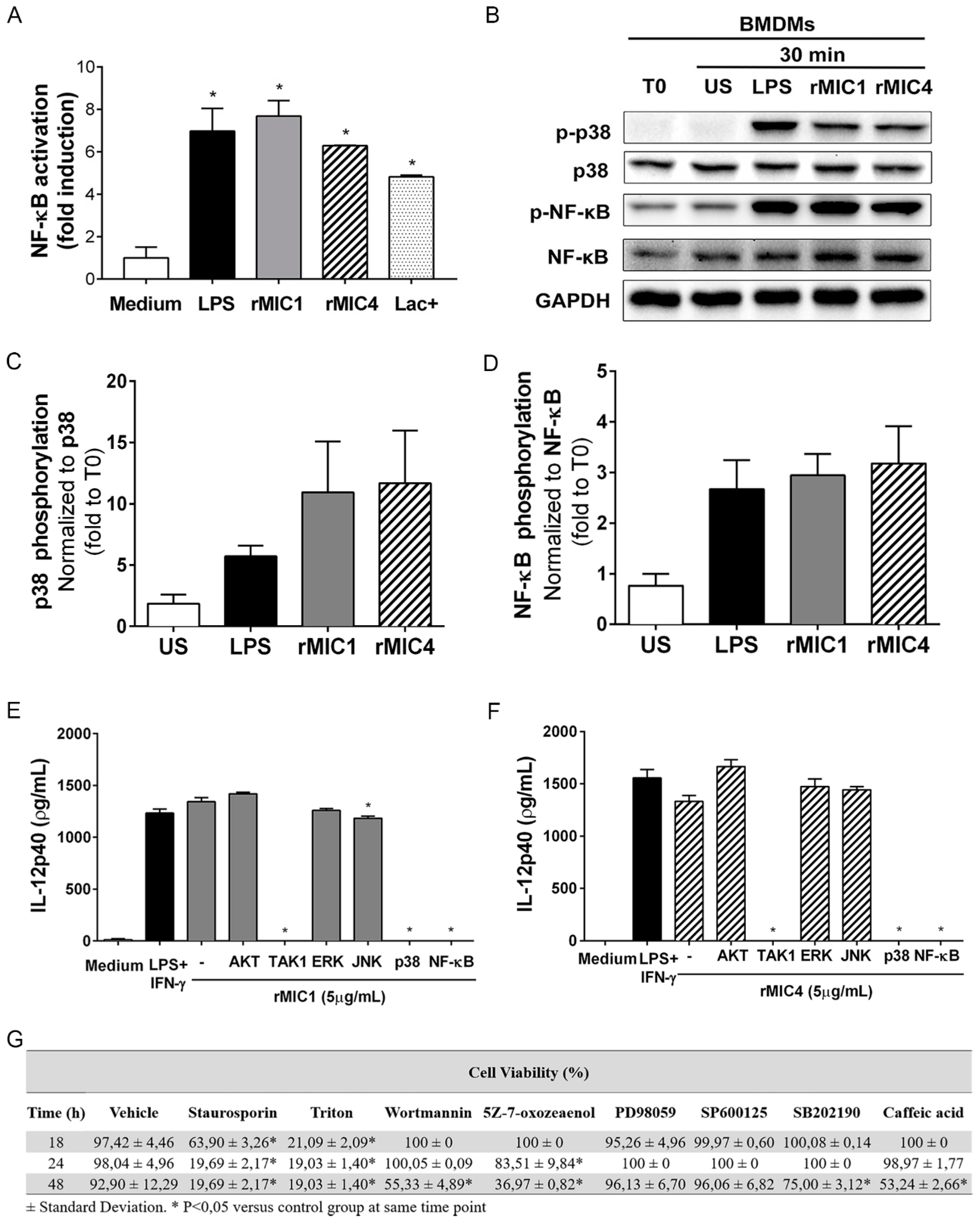
2.1. IL-12 Production in rMIC1- or rMIC4-Stimulated Macrophages Is Dependent on TAK1, p38, and NF-κB Phosphorylation
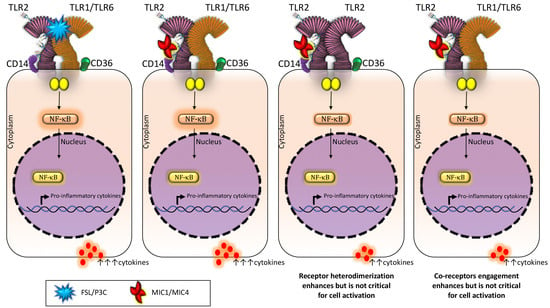
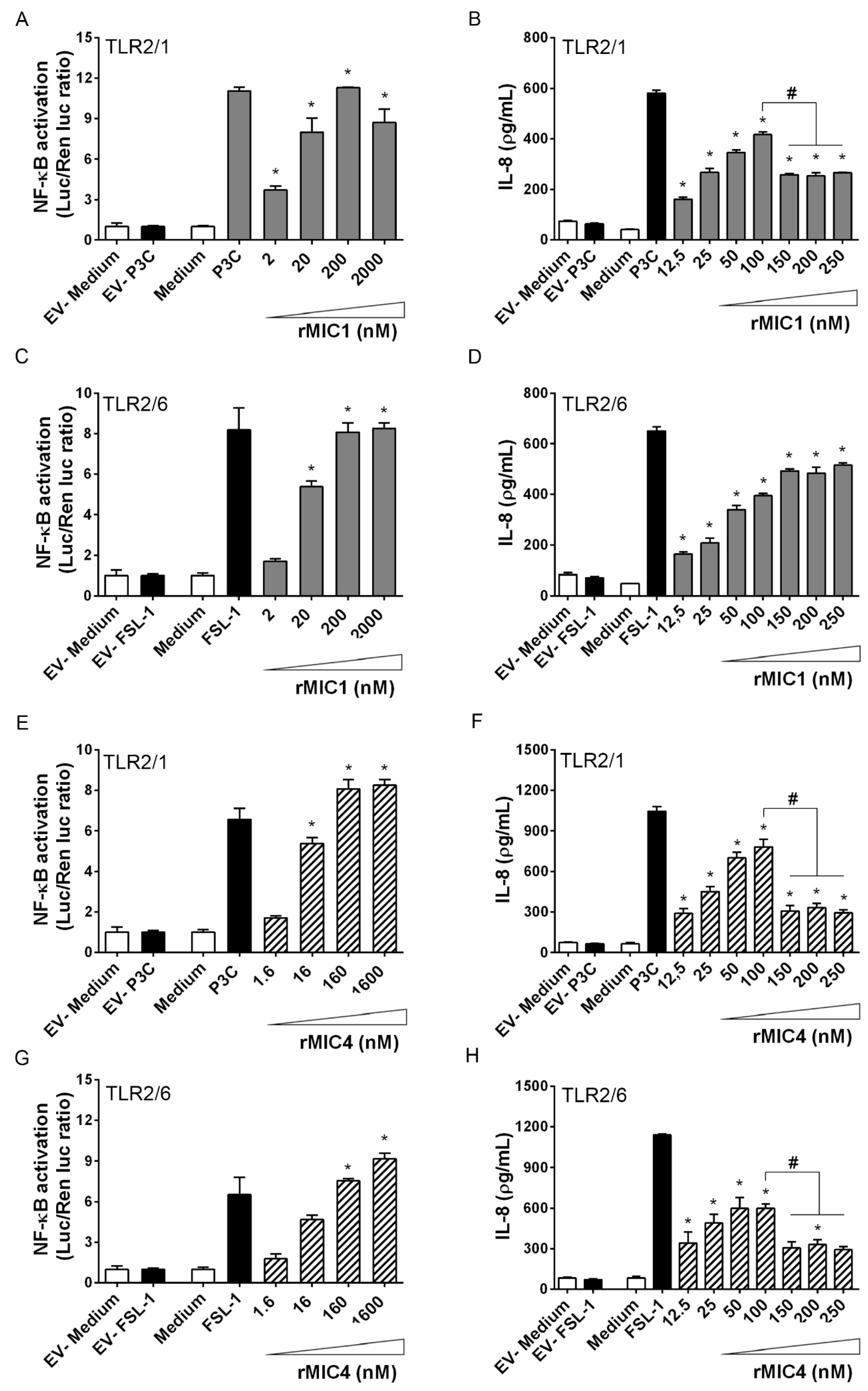
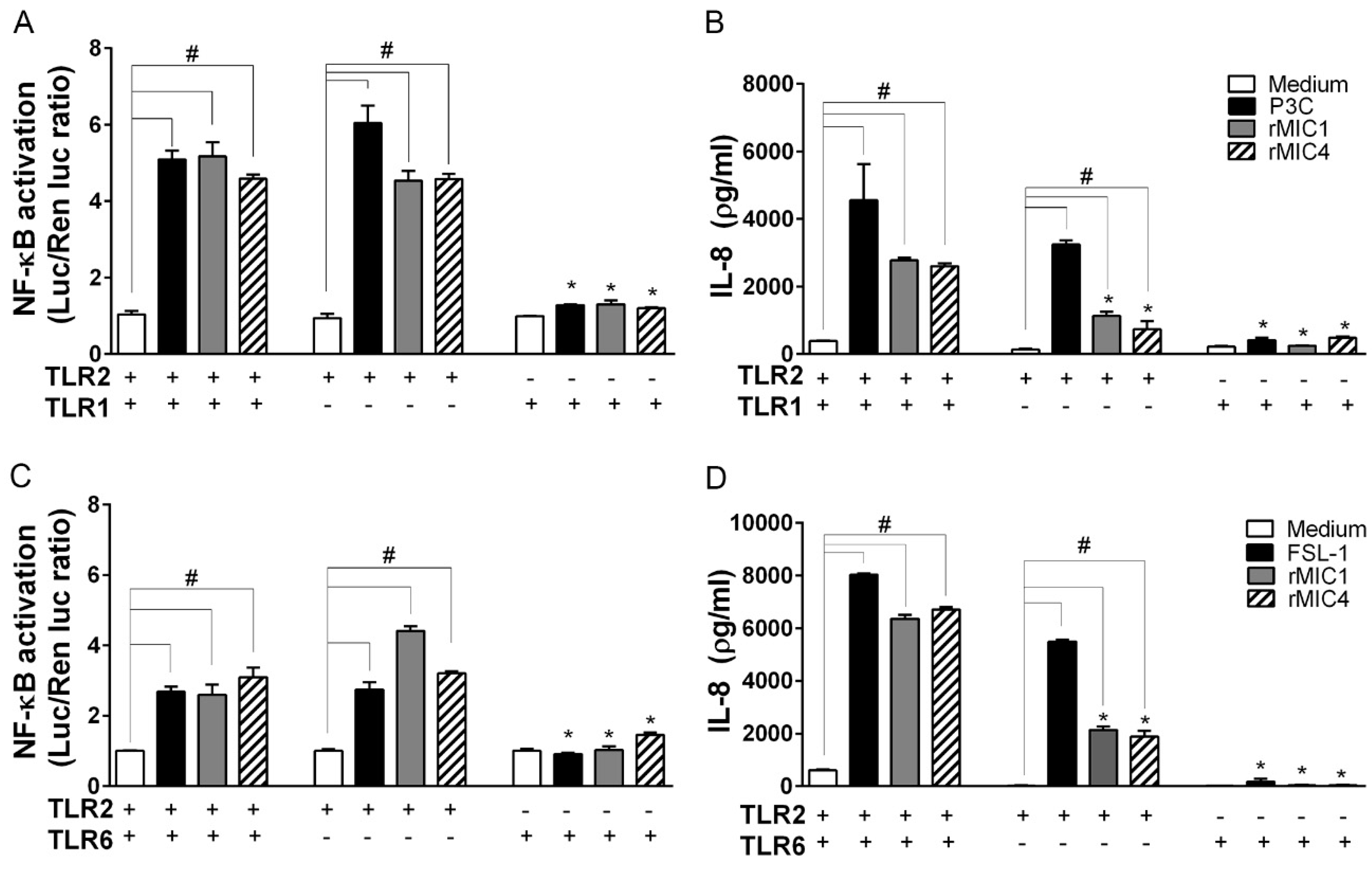
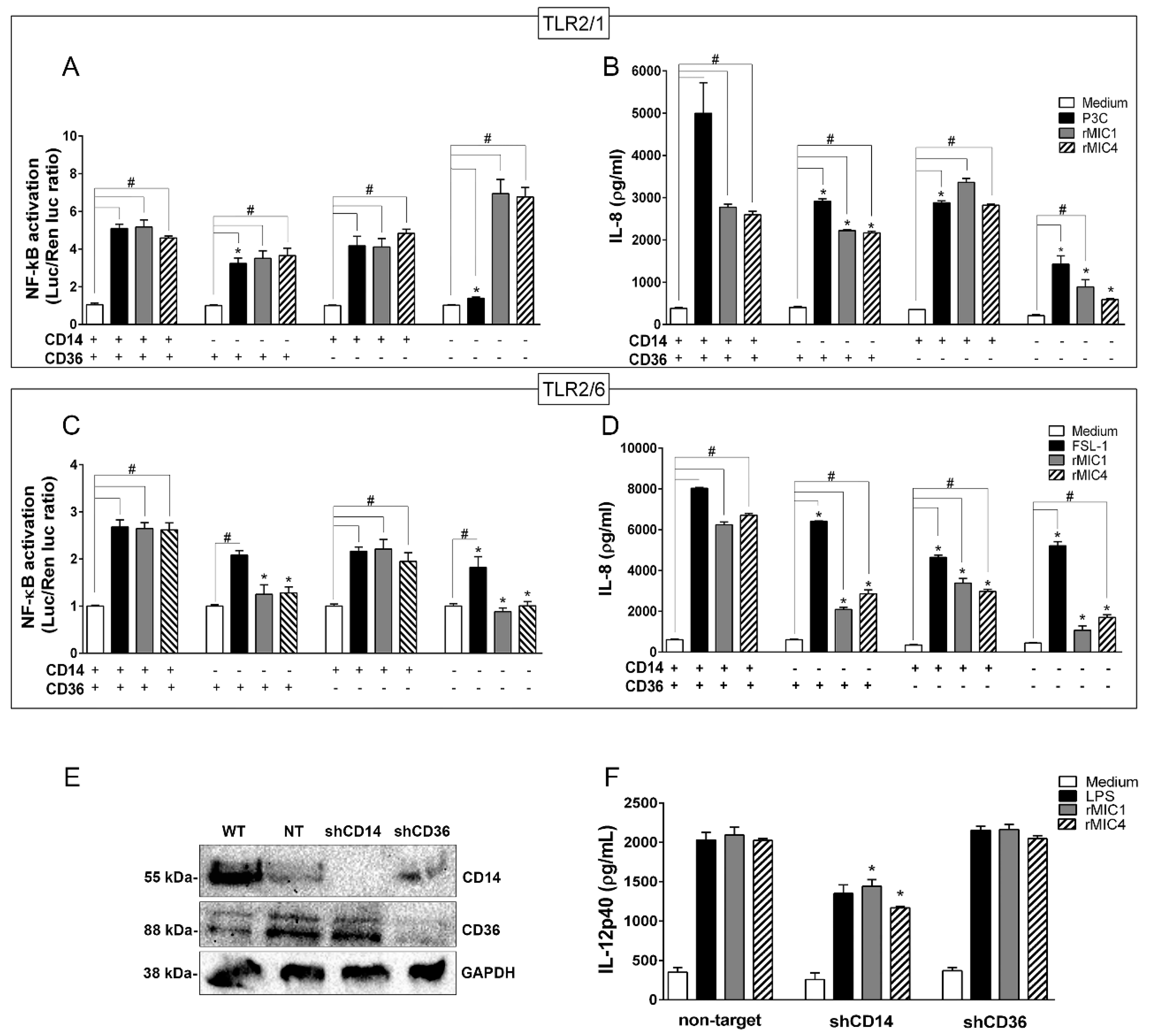
2.2. TLR2 Is Required for rMIC1- or rMIC4-Triggered Cell Activation, and Heterodimer Formation and Co-Receptor Engagement Enhance the Cell Response
3. Discussion
4. Materials and Methods
4.1. Animal Care and Ethics Statement
4.2. Preparation of the Lac+ Fraction from T. gondii and Recombinant MIC1 and MIC4
4.3. RAW264.7-Luc Cell Culture
4.4. Preparation of Bone Marrow-Derived Macrophages
4.5. Cell Signaling Inhibition Assay
4.6. Western Blotting
4.7. Transfection of HEK293T Cells with TLR2 Complexes
4.8. Luciferase Reporter Assays
4.9. Cd14 and Cd36 Knockdown by shRNA
4.10. Measurement of Cytokines
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sullivan, W.J.; Jeffers, V. Mechanisms of Toxoplasma gondii persistence and latency. FEMS Microbiol. Rev. 2012, 36, 717–733. [Google Scholar] [CrossRef] [PubMed]
- Montoya, J.G.; Liesenfeld, O. Toxoplasmosis. Lancet 2004, 363, 1965–1976. [Google Scholar] [CrossRef]
- Robert-Gangneux, F.; Dardé, M.-L. Epidemiology of and Diagnostic Strategies for Toxoplasmosis. Clin. Microbiol. Rev. 2012, 25, 264–296. [Google Scholar] [CrossRef]
- Kodym, P.; Malý, M.; Beran, O.; Jilich, D.; Rozsypal, H.; Machala, L.; Holub, M. Incidence, immunological and clinical characteristics of reactivation of latent Toxoplasma gondii infection in HIV-infected patients. Epidemiol. Infect. 2014, 143, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Carruthers, V.B.; Sibley, L.D. Sequential protein secretion from three distinct organelles of Toxoplasma gondii accompanies invasion of human fibroblasts. Eur. J. Cell Biol. 1997, 73, 114–123. [Google Scholar] [PubMed]
- Brecht, S.; Carruthers, V.B.; Ferguson, D.J.P.; Giddings, O.K.; Wang, G.; Jäkle, U.; Harper, J.M.; Sibley, L.D.; Soldati, D. The Toxoplasma micronemal protein MIC4 is an adhesin composed of six conserved apple domains. J. Biol. Chem. 2001, 276, 4119–4127. [Google Scholar] [CrossRef] [PubMed]
- Reiss, M.; Viebig, N.; Brecht, S.; Fourmaux, M.-N.; Soete, M.; Di Cristina, M.; Dubremetz, J.F.; Soldati, D. Identification and characterization of an escorter for two secretory adhesins in Toxoplasma gondii. J. Cell Biol. 2001, 152, 563–578. [Google Scholar] [CrossRef] [PubMed]
- Blumenschein, T.M.A.; Friedrich, N.; Childs, R.A.; Saouros, S.; Carpenter, E.P.; Campanero-Rhodes, M.A.; Simpson, P.; Chai, W.; Koutroukides, T.; Blackman, M.J. Atomic resolution insight into host cell recognition by Toxoplasma gondii. EMBO J. 2007, 26, 2808–2820. [Google Scholar] [CrossRef]
- Carruthers, V.B.; Håkansson, S.; Giddings, O.K.; Sibley, L.D. Toxoplasma gondii uses sulfated proteoglycans for substrate and host cell attachment. Infect. Immun. 2000, 68, 4005–4011. [Google Scholar] [CrossRef]
- Monteiro, V.G.; Soares, C.P.; De Souza, W. Host cell surface sialic acid residues are involved on the process of penetration of Toxoplasma gondii into mammalian cells. FEMS Microbiol. Lett. 1998, 164, 323–327. [Google Scholar] [CrossRef]
- Ortega-Barria, E.; Boothroyd, J.C. A Toxoplasma lectin-like activity specific for sulfated polysaccharides is involved in host cell infection. J. Biol. Chem. 1999, 274, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Lourenço, E.V.; Pereira, S.R.; Faça, V.M.; Coelho-Castelo, A.A.; Mineo, J.R.; Roque-Barreira, M.C.; Greene, L.J.; Panunto-Castelo, A. Toxoplasma gondii micronemal protein MIC1 is a lactose-binding lectin. Glycobiology 2001, 11, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, N.; Santos, J.M.; Liu, Y.; Palma, A.S.; Leon, E.; Saouros, S.; Kiso, M.; Blackman, M.J.; Matthews, S.; Feizi, T.; et al. Members of a novel protein family containing microneme adhesive repeat domains act as sialic acid-binding lectins during host cell invasion by apicomplexan parasites. J. Biol. Chem. 2010, 285, 2064–2076. [Google Scholar] [CrossRef] [PubMed]
- Marchant, J.; Cowper, B.; Liu, Y.; Lai, L.; Pinzan, C.; Marq, J.B.; Friedrich, N.; Sawmynaden, K.; Liew, L.; Chai, W.; et al. Galactose recognition by the apicomplexan parasite Toxoplasma gondii. J. Biol. Chem. 2012, 287, 16720–16733. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, N.; Matthews, S.; Soldati-Favre, D. Sialic acids: Key determinants for invasion by the Apicomplexa. Int. J. Parasitol. 2010, 40, 1145–1154. [Google Scholar] [CrossRef]
- Sardinha-Silva, A.; Mendonça-Natividade, F.C.; Pinzan, C.F.; Lopes, C.D.; Costa, D.L.; Jacot, D.; Fernandes, F.F.; Zorzetto-Fernandes, A.L.V.; Gay, N.J.; Sher, A.; et al. The lectin-specific activity of Toxoplasma gondii microneme proteins 1 and 4 binds Toll-like receptor 2 and 4 N-glycans to regulate innate immune priming. PLOS Pathog. 2019, 15, e1007871. [Google Scholar] [CrossRef] [PubMed]
- Lourenço, E.V.; Bernardes, E.S.; Silva, N.M.; Mineo, J.R.; Panunto-Castelo, A.; Roque-Barreira, M.C. Immunization with MIC1 and MIC4 induces protective immunity against Toxoplasma gondii. Microbes Infect. 2006, 8, 1244–1251. [Google Scholar] [CrossRef] [PubMed]
- Pinzan, C.F.; Sardinha-Silva, A.; Almeida, F.; Lai, L.; Lopes, C.D.; Lourenço, E.V.; Panunto-Castelo, A.; Matthews, S.; Roque-Barreira, M.C. Vaccination with recombinant microneme proteins confers protection against experimental toxoplasmosis in mice. PLoS ONE 2015, 10, e0143087. [Google Scholar] [CrossRef]
- Janeway, C.A. Approaching the asymptote? Evolution and revolution in immunology. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989; Volume 54, pp. 1–13. [Google Scholar]
- Gay, N.J.; Symmons, M.F.; Gangloff, M.; Bryant, C.E. Assembly and localization of Toll-like receptor signalling complexes. Nat. Rev. Immunol. 2014, 14, 546–558. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Golenbock, D.; Bowie, A.G. The history of Toll-like receptors-redefining innate immunity. Nat. Rev. Immunol. 2013, 13, 453–460. [Google Scholar] [CrossRef]
- Gay, N.J.; Gangloff, M.; Neill, L.A.J.O. What the Myddosome structure tells us about the initiation of innate immunity. Trends Immunol. 2011, 32, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Yarovinsky, F. Toll-like receptors and their role in host resistance to Toxoplasma gondii. Immunol. Lett. 2008, 119, 17–21. [Google Scholar] [CrossRef]
- Andrade, W.A.; do Carmo Souza, M.; Ramos-Martinez, E.; Nagpal, K.; Dutra, M.S.; Melo, M.B.; Bartholomeu, D.C.; Ghosh, S.; Golenbock, D.T.; Gazzinelli, R.T. Combined action of nucleic acid-sensing Toll-like receptors and TLR11/TLR12 heterodimers imparts resistance to Toxoplasma gondii in mice. Cell Host Microbe 2013, 13, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Yarovinsky, F.; Zhang, D.; Andersen, J.F.; Bannenberg, G.L.; Serhan, C.N.; Hayden, M.S.; Hieny, S.; Sutterwala, F.S.; Flavell, R.A.; Ghosh, S. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science 2005, 308, 1626–1629. [Google Scholar] [CrossRef] [PubMed]
- Koblansky, A.A.; Jankovic, D.; Oh, H.; Hieny, S.; Sungnak, W.; Mathur, R.; Hayden, M.S.; Akira, S.; Sher, A.; Ghosh, S. Recognition of profilin by Toll-like receptor 12 is critical for host resistance to Toxoplasma gondii. Immunity 2013, 38, 119–130. [Google Scholar] [CrossRef]
- Salazar Gonzalez, R.M.; Shehata, H.; O’connell, M.J.; Yang, Y.; Moreno-Fernandez, M.E.; Chougnet, C.A.; Aliberti, J. Toxoplasma gondii-derived profilin triggers human toll-like receptor 5-dependent cytokine production. J. Innate Immun. 2014, 6, 685–694. [Google Scholar] [CrossRef]
- Debierre-Grockiego, F.; Campos, M.A.; Azzouz, N.; Schmidt, J.; Bieker, U.; Resende, M.G.; Mansur, D.S.; Weingart, R.; Schmidt, R.R.; Golenbock, D.T. Activation of TLR2 and TLR4 by glycosylphosphatidylinositols derived from Toxoplasma gondii. J. Immunol. 2007, 179, 1129–1137. [Google Scholar] [CrossRef]
- Yarovinsky, F. Innate immunity to Toxoplasma gondii infection. Nat. Rev. Immunol. 2014, 14, 109–121. [Google Scholar] [CrossRef]
- Ozinsky, A.; Underhill, D.M.; Fontenot, J.D.; Hajjar, A.M.; Smith, K.D.; Wilson, C.B.; Schroeder, L.; Aderem, A. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc. Natl. Acad. Sci. USA 2000, 97, 13766–13771. [Google Scholar] [CrossRef]
- Nilsen, N.J.; Deininger, S.; Nonstad, U.; Skjeldal, F.; Husebye, H.; Rodionov, D.; von Aulock, S.; Hartung, T.; Lien, E.; Bakke, O.; et al. Cellular trafficking of lipoteichoic acid and Toll-like receptor 2 in relation to signaling: Role of CD14 and CD36. J. Leukoc. Biol. 2008, 84, 280–291. [Google Scholar] [CrossRef]
- Kirschning, C.J.; Schumann, R.R. TLR2: Cellular sensor for microbial and endogenous molecular patterns. In Toll-Like Receptor Family Members and Their Ligands; Springer: Berlin/Heidelberg, Germany, 2002; pp. 121–144. [Google Scholar]
- Triantafilou, M.; Manukyan, M.; Mackie, A.; Morath, S.; Hartung, T.; Heine, H.; Triantafilou, K. Lipoteichoic acid and toll-like receptor 2 internalization and targeting to the Golgi are lipid raft-dependent. J. Biol. Chem. 2004, 279, 40882–40889. [Google Scholar] [CrossRef] [PubMed]
- Marim, F.M.; Silveira, T.N.; Lima, D.S., Jr.; Zamboni, D.S. A Method for Generation of Bone Marrow-Derived Macrophages from Cryopreserved Mouse Bone Marrow Cells. PLoS ONE 2010, 5, e15263. [Google Scholar] [CrossRef] [PubMed]
- Joiner, K.A.; Roos, D.S. Secretory traffic in the eukaryotic parasite Toxoplasma gondii: Less is more. J. Cell Biol. 2002, 157, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Cérède, O.; Dubremetz, J.F.; Soête, M.; Deslée, D.; Vial, H.; Bout, D.; Lebrun, M. Synergistic role of micronemal proteins in Toxoplasma gondii virulence. J. Exp. Med. 2005, 201, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Paing, M.M.; Tolia, N.H. Multimeric assembly of host-pathogen adhesion complexes involved in apicomplexan invasion. PLoS Pathog. 2014, 10, e1004120. [Google Scholar] [CrossRef] [PubMed]
- Saouros, S.; Edwards-Jones, B.; Reiss, M.; Sawmynaden, K.; Cota, E.; Simpson, P.; Dowse, T.J.; Jäkle, U.; Ramboarina, S.; Shivarattan, T.; et al. A novel galectin-like domain from Toxoplasma gondii micronemal protein 1 assists the folding, assembly, and transport of a cell adhesion complex. J. Biol. Chem. 2005, 280, 38583–38591. [Google Scholar] [CrossRef] [PubMed]
- Takabatake, N.; Okamura, M.; Yokoyama, N.; Ikehara, Y.; Akimitsu, N.; Arimitsu, N.; Hamamoto, H.; Sekimizu, K.; Suzuki, H.; Igarashi, I. Glycophorin A-knockout mice, which lost sialoglycoproteins from the red blood cell membrane, are resistant to lethal infection of Babesia rodhaini. Vet. Parasitol. 2007. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, P.M.; Assis, R.R.; Torrecilhas, A.C.; Saraiva, E.M.; Pessoa, N.L.; Campos, M.A.; Marialva, E.F.; Ríos-Velasquez, C.M.; Pessoa, F.A.; Secundino, N.F.; et al. Lipophosphoglycans from Leishmania amazonensis Strains Display Immunomodulatory Properties via TLR4 and Do Not Affect Sand Fly Infection. PLoS Negl. Trop. Dis. 2016. [Google Scholar] [CrossRef]
- Favila, M.A.; Geraci, N.S.; Jayakumar, A.; Hickerson, S.; Mostrom, J.; Turco, S.J.; Beverley, S.M.; McDowell, M.A. Differential Impact of LPG-and PG-Deficient Leishmania major Mutants on the Immune Response of Human Dendritic Cells. PLoS Negl. Trop. Dis. 2015. [Google Scholar] [CrossRef]
- Persson, K.E.M.; McCallum, F.J.; Reiling, L.; Lister, N.A.; Stubbs, J.; Cowman, A.F.; Marsh, K.; Beeson, J.G. Variation in use of erythrocyte invasion pathways by Plasmodium falciparum mediates evasion of human inhibitory antibodies. J. Clin. Investig. 2008. [Google Scholar] [CrossRef]
- Jankovic, D.; Kullberg, M.C.; Hieny, S.; Caspar, P.; Collazo, C.M.; Sher, A. In the absence of IL-12, CD4+T cell responses to intracellular pathogens fail to default to a Th2 pattern and are host protective in an IL-10-/-setting. Immunity 2002. [Google Scholar] [CrossRef]
- Pifer, R.; Yarovinsky, F. Innate responses to Toxoplasma gondii in mice and humans. Trends Parasitol. 2011, 27, 388–393. [Google Scholar] [CrossRef]
- Wang, Y.; Yin, H. Research advances in microneme protein 3 of Toxoplasma gondii. Parasit. Vectors 2015, 8, 384. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.-H.; Yuan, Z.-G.; Zhou, D.-H.; He, X.-H.; Liu, M.-M.; Yan, C.; Yin, C.-C.; He, Y.; Lin, R.-Q.; Zhu, X.-Q. Toxoplasma gondii microneme protein 6 (MIC6) is a potential vaccine candidate against toxoplasmosis in mice. Vaccine 2009, 27, 6570–6574. [Google Scholar] [CrossRef]
- Liu, M.M.; Yuan, Z.G.; Peng, G.H.; Zhou, D.H.; He, X.H.; Yan, C.; Yin, C.C.; He, Y.; Lin, R.Q.; Song, H.Q. Toxoplasma gondii microneme protein 8 (MIC8) is a potential vaccine candidate against toxoplasmosis. Parasitol. Res. 2010, 106, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Tao, Q.; Fang, R.; Zhang, W.; Wang, Y.; Cheng, J.; Li, Y.; Fang, K.; Khan, M.K.; Hu, M.; Zhou, Y. Protective immunity induced by a DNA vaccine-encoding Toxoplasma gondii microneme protein 11 against acute toxoplasmosis in BALB/c mice. Parasitol. Res. 2013, 112, 2871–2877. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.-G.; Ren, D.; Zhou, D.-H.; Zhang, X.-X.; Petersen, E.; Li, X.-Z.; Zhou, Y.; Yang, G.-L.; Zhu, X.-Q. Evaluation of protective effect of pVAX-TgMIC13 plasmid against acute and chronic Toxoplasma gondii infection in a murine model. Vaccine 2013, 31, 3135–3139. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Avalos, A.M.; Ploegh, H.L. Accessory molecules for Toll-like receptors and their function. Nat. Rev. Immunol. 2012, 12, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.Y.; Lee, J.-O. Structural biology of the toll-like receptor family. Annu. Rev. Biochem. 2011, 80, 917–941. [Google Scholar] [CrossRef]
- Weber, A.N.R.; Morse, M.A.; Gay, N.J. Four N-linked glycosylation sites in human toll-like receptor 2 cooperate to direct efficient biosynthesis and secretion. J. Biol. Chem. 2004, 279, 34589–34594. [Google Scholar] [CrossRef]
- Da Silva Correia, J.; Ulevitch, R.J. MD-2 and TLR4 N-linked glycosylations are important for a functional lipopolysaccharide receptor. J. Biol. Chem. 2002, 277, 1845–1854. [Google Scholar] [CrossRef] [PubMed]
- Alegre-Maller, A.C.P.; Mendonça, F.C.; da Silva, T.A.; Oliveira, A.F.; Freitas, M.S.; Hanna, E.S.; Almeida, I.C.; Gay, N.J.; Roque-Barreira, M.C. Therapeutic Administration of Recombinant Paracoccin Confers Protection against Paracoccidioides brasiliensis Infection: Involvement of TLRs. PLoS Negl. Trop. Dis. 2014, 8, e3317. [Google Scholar] [CrossRef] [PubMed]
- Mariano, V.S.; Zorzetto-Fernandes, A.L.; da Silva, T.A.; Ruas, L.P.; Nohara, L.L.; de Almeida, I.C.; Roque-Barreira, M.C. Recognition of TLR2 N-Glycans: Critical Role in ArtinM Immunomodulatory Activity. PLoS ONE 2014, 9, e98512. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Azevedo, R.; Roque-Barreira, M.-C.; Gay, N.J. Targeting and Recognition of Toll-Like Receptors by Plant and Pathogen Lectins. Front. Immunol. 2017, 8, 1820. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, A.; Xu, M.; Chen, Z.J. Ubiquitin-mediated activation of TAK1 and IKK. Oncogene 2007. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef]
- Li, J.Y.; Liu, Y.; Gao, X.X.; Gao, X.; Cai, H. TLR2 and TLR4 signaling pathways are required for recombinant Brucella abortus BCSP31-induced cytokine production, functional upregulation of mouse macrophages, and the Th1 immune response in vivo and in vitro. Cell. Mol. Immunol. 2014, 11, 477–494. [Google Scholar] [CrossRef]
- Sun, S.C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373. [Google Scholar] [CrossRef]
- Ajibade, A.A.; Wang, H.Y.; Wang, R.-F. Cell type-specific function of TAK1 in innate immune signaling. Trends Immunol. 2013, 34, 307–316. [Google Scholar] [CrossRef]
- Dias-Baruffi, M.; Sakamoto, M.; Rossetto, S.; Vozári-Hampe, M.M.; Roque-Barreira, M.C. Neutrophil migration and aggregation induced by euphorbin, a lectin from the latex of Euphorbia milii, var. milii. Inflamm. Res. 2000. [Google Scholar] [CrossRef]
- Santos-de-Oliveira, R.; Dias-Baruffi, M.; Thomaz, S.M.; Beltramini, L.M.; Roque-Barreira, M.C. A neutrophil migration-inducing lectin from Artocarpus integrifolia. J. Immunol. 1994, 153, 1798–1807. [Google Scholar] [PubMed]
- Panunto-Castelo, A.; Souza, M.A.; Roque-Barreira, M.C.; Silva, J.S. KM+, a lectin from Artocarpus integrifolia, induces IL-12 p40 production by macrophages and switches from type 2 to type 1 cell-mediated immunity against Leishmania major antigens, resulting in BALB/c mice resistance to infection. Glycobiology 2001. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Sugiura, T.; Toyohira, Y.; Yoshida, Y.; Yanagihara, N.; Karasaki, Y. Stimulation of IFN-γ production by garlic lectin in mouse spleen cells: Involvement of IL-12 via activation of p38 MAPK and ERK in macrophages. Phytomedicine 2011. [Google Scholar] [CrossRef] [PubMed]
- Hajtó, T.; Fodor, K.; Perjési, P.; Németh, P. Difficulties and Perspectives of Immunomodulatory Therapy with Mistletoe Lectins and Standardized Mistletoe Extracts in Evidence-Based Medicine. Evid.-Based Complement. Altern. Med. 2011, 2011, 298972. [Google Scholar]
- Krutzik, S.R.; Ochoa, M.T.; Sieling, P.A.; Uematsu, S.; Ng, Y.W.; Legaspi, A.; Liu, P.T.; Cole, S.T.; Godowski, P.J.; Maeda, Y. Activation and regulation of Toll-like receptors 2 and 1 in human leprosy. Nat. Med. 2003, 9, 525–532. [Google Scholar] [CrossRef]
- Jin, M.S.; Kim, S.E.; Heo, J.Y.; Lee, M.E.; Kim, H.M.; Paik, S.-G.; Lee, H.; Lee, J.-O. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 2007, 130, 1071–1082. [Google Scholar] [CrossRef]
- Takeuchi, O.; Kawai, T.; Mühlradt, P.F.; Morr, M.; Radolf, J.D.; Zychlinsky, A.; Takeda, K.; Akira, S. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int. Immunol. 2001, 13, 933–940. [Google Scholar] [CrossRef]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; Van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2009, 11, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Botos, I.; Wang, Y.; Leonard, J.N.; Shiloach, J.; Segal, D.M.; Davies, D.R. Structural basis of toll-like receptor 3 signaling with double-stranded RNA. Science 2008, 320, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Miguel, R.N.; Wong, J.; Westoll, J.F.; Brooks, H.J.; O’Neill, L.A.J.; Gay, N.J.; Bryant, C.E.; Monie, T.P. A dimer of the toll-like receptor 4 cytoplasmic domain provides a specific scaffold for the recruitment of signalling adaptor proteins. PLoS ONE 2007, 2, e788. [Google Scholar]
- Yoon, S.; Kurnasov, O.; Natarajan, V.; Hong, M.; Gudkov, A.V.; Osterman, A.L.; Wilson, I.A. Structural basis of TLR5-flagellin recognition and signaling. Science 2012, 335, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Muta, T.; Takeshige, K. Essential roles of CD14 and lipopolysaccharide-binding protein for activation of toll-like receptor (TLR) 2 as well as TLR4. Eur. J. Biochem. 2001, 268, 4580–4589. [Google Scholar] [CrossRef] [PubMed]
- Van Bergenhenegouwen, J.; Plantinga, T.S.; Joosten, L.A.; Netea, M.G.; Folkerts, G.; Kraneveld, A.D.; Garssen, J.; Vos, A.P. TLR2 & Co: A critical analysis of the complex interactions between TLR2 and coreceptors. J. Leukoc. Biol. 2013, 94, 885–902. [Google Scholar] [PubMed]
- Nakata, T.; Yasuda, M.; Fujita, M.; Kataoka, H.; Kiura, K.; Sano, H.; Shibata, K. CD14 directly binds to triacylated lipopeptides and facilitates recognition of the lipopeptides by the receptor complex of Toll-like receptors 2 and 1 without binding to the complex. Cell. Microbiol. 2006, 8, 1899–1909. [Google Scholar] [CrossRef]
- Hoebe, K.; Georgel, P.; Rutschmann, S.; Du, X.; Mudd, S.; Crozat, K.; Sovath, S.; Shamel, L.; Hartung, T.; Zahringer, U.; et al. CD36 is a sensor of diacylglycerides. Nature 2005, 433, 523–527. [Google Scholar] [CrossRef]
- Triantafilou, M.; Gamper, F.G.J.; Haston, R.M.; Mouratis, M.A.; Morath, S.; Hartung, T.; Triantafilou, K. Membrane sorting of toll-like receptor (TLR)-2/6 and TLR2/1 heterodimers at the cell surface determines heterotypic associations with CD36 and intracellular targeting. J. Biol. Chem. 2006, 281, 31002–31011. [Google Scholar] [CrossRef]
- Rapsinski, G.J.; Newman, T.N.; Oppong, G.O.; van Putten, J.P.M.; Tükel, Ç. CD14 protein acts as an adaptor molecule for the immune recognition of Salmonella curli fibers. J. Biol. Chem. 2013, 288, 14178–14188. [Google Scholar] [CrossRef]
- Birch, H.L.; Alderwick, L.J.; Appelmelk, B.J.; Maaskant, J.; Bhatt, A.; Singh, A.; Nigou, J.; Eggeling, L.; Geurtsen, J.; Besra, G.S. A truncated lipoglycan from mycobacteria with altered immunological properties. Proc. Natl. Acad. Sci. USA 2010, 107, 2634–2639. [Google Scholar] [CrossRef]
- Jimenez-Dalmaroni, M.J.; Xiao, N.; Corper, A.L.; Verdino, P.; Ainge, G.D.; Larsen, D.S.; Painter, G.F.; Rudd, P.M.; Dwek, R.A.; Hoebe, K.; et al. Soluble CD36 ectodomain binds negatively charged diacylglycerol ligands and acts as a co-receptor for TLR2. PLoS ONE 2009, 4, e7411. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, T.A.; Zorzetto-Fernandes, A.L.V.; Cecílio, N.T.; Sardinha-Silva, A.; Fernandes, F.F.; Roque-Barreira, M.C. CD14 is critical for TLR2-mediated M1 macrophage activation triggered by N-glycan recognition. Sci. Rep. 2017, 7, 7083. [Google Scholar] [CrossRef] [PubMed]
- Di Gioia, M.; Zanoni, I. Toll-like receptor co-receptors as master regulators of the immune response. Mol. Immunol. 2015, 63, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Vasta, G.R. Lectins as Innate Immune Recognition Factors: Structural, Functional, and Evolutionary Aspects. Evol. Immune Syst. 2016, 205–224. [Google Scholar]
- Vasta, G.R.; Ahmed, H.; Tasumi, S.; Odom, E.W.; Saito, K. Biological roles of lectins in innate immunity: Molecular and structural basis for diversity in self/non-self recognition. In Current Topics in Innate Immunity; Springer: New York, NY, USA, 2007. [Google Scholar]
- Mooibroek, T.J.; Crump, M.P.; Davis, A.P. Synthesis and evaluation of a desymmetrised synthetic lectin: An approach to carbohydrate receptors with improved versatility. Org. Biomol. Chem. 2016. [Google Scholar] [CrossRef]
- Davis, A.P. Synthetic lectins. Org. Biomol. Chem. 2009. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, J.; Audfray, A.; Imberty, A. Binding sugars: From natural lectins to synthetic receptors and engineered neolectins. Chem. Soc. Rev. 2013. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, A.B.; Iaciura, B.M.F.; Nohara, L.L.; Lopes, C.D.; Veas, E.M.C.; Mariano, V.S.; Bozza, P.T.; Lopes, U.G.; Atella, G.C.; Almeida, I.C.; et al. Lysophosphatidylcholine Triggers TLR2- and TLR4-Mediated Signaling Pathways but Counteracts LPS-Induced NO Synthesis in Peritoneal Macrophages by Inhibiting NF-??B Translocation and MAPK/ERK Phosphorylation. PLoS ONE 2013, 8, e76233. [Google Scholar] [CrossRef]
- Kurt-Jones, E.A.; Popova, L.; Kwinn, L.; Haynes, L.M.; Jones, L.P.; Tripp, R.A.; Walsh, E.E.; Freeman, M.W.; Golenbock, D.T.; Anderson, L.J.; et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 2000, 1, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Lien, E.; Sellati, T.J.; Yoshimura, A.; Flo, T.H.; Rawadi, G.; Finberg, R.W.; Carroll, J.D.; Espevik, T.; Ingalls, R.R.; Radolf, J.D.; et al. Toll-like receptor 2 functions as a pattern recognition receptor for diverse bacterial products. J. Biol. Chem. 1999, 274, 33419–33425. [Google Scholar] [CrossRef]
- Young, L.; Sung, J.; Stacey, G.; Masters, J.R. Detection of Mycoplasma in cell cultures. Nat. Protoc. 2010, 5, 929–934. [Google Scholar]
- Medvedev, A.E.; Kopydlowski, K.M.; Vogel, S.N. Inhibition of Lipopolysaccharide-Induced Signal Transduction in Endotoxin-Tolerized Mouse Macrophages: Dysregulation of Cytokine, Chemokine, and Toll-Like Receptor 2 and 4 Gene Expression. J. Immunol. 2000, 164, 5564–5574. [Google Scholar]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa Mendonça-Natividade, F.; Duque Lopes, C.; Ricci-Azevedo, R.; Sardinha-Silva, A.; Figueiredo Pinzan, C.; Paiva Alegre-Maller, A.C.; L. Nohara, L.; B. Carneiro, A.; Panunto-Castelo, A.; C. Almeida, I.; et al. Receptor Heterodimerization and Co-Receptor Engagement in TLR2 Activation Induced by MIC1 and MIC4 from Toxoplasma gondii. Int. J. Mol. Sci. 2019, 20, 5001. https://doi.org/10.3390/ijms20205001
Costa Mendonça-Natividade F, Duque Lopes C, Ricci-Azevedo R, Sardinha-Silva A, Figueiredo Pinzan C, Paiva Alegre-Maller AC, L. Nohara L, B. Carneiro A, Panunto-Castelo A, C. Almeida I, et al. Receptor Heterodimerization and Co-Receptor Engagement in TLR2 Activation Induced by MIC1 and MIC4 from Toxoplasma gondii. International Journal of Molecular Sciences. 2019; 20(20):5001. https://doi.org/10.3390/ijms20205001
Chicago/Turabian StyleCosta Mendonça-Natividade, Flávia, Carla Duque Lopes, Rafael Ricci-Azevedo, Aline Sardinha-Silva, Camila Figueiredo Pinzan, Ana Claudia Paiva Alegre-Maller, Lilian L. Nohara, Alan B. Carneiro, Ademilson Panunto-Castelo, Igor C. Almeida, and et al. 2019. "Receptor Heterodimerization and Co-Receptor Engagement in TLR2 Activation Induced by MIC1 and MIC4 from Toxoplasma gondii" International Journal of Molecular Sciences 20, no. 20: 5001. https://doi.org/10.3390/ijms20205001
APA StyleCosta Mendonça-Natividade, F., Duque Lopes, C., Ricci-Azevedo, R., Sardinha-Silva, A., Figueiredo Pinzan, C., Paiva Alegre-Maller, A. C., L. Nohara, L., B. Carneiro, A., Panunto-Castelo, A., C. Almeida, I., & Roque-Barreira, M. C. (2019). Receptor Heterodimerization and Co-Receptor Engagement in TLR2 Activation Induced by MIC1 and MIC4 from Toxoplasma gondii. International Journal of Molecular Sciences, 20(20), 5001. https://doi.org/10.3390/ijms20205001