HDAC8 Inhibitor WK2-16 Therapeutically Targets Lipopolysaccharide-Induced Mouse Model of Neuroinflammation and Microglial Activation

 ,
,
Abstract
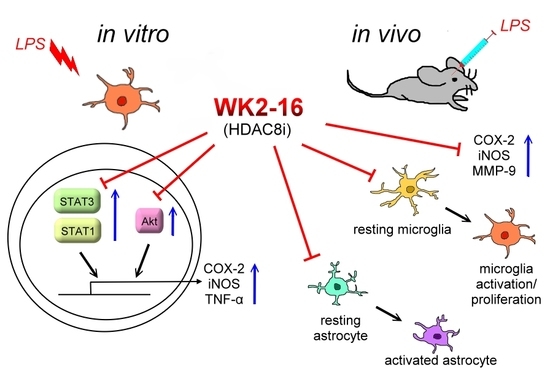
{kind=link}
{kind=link}
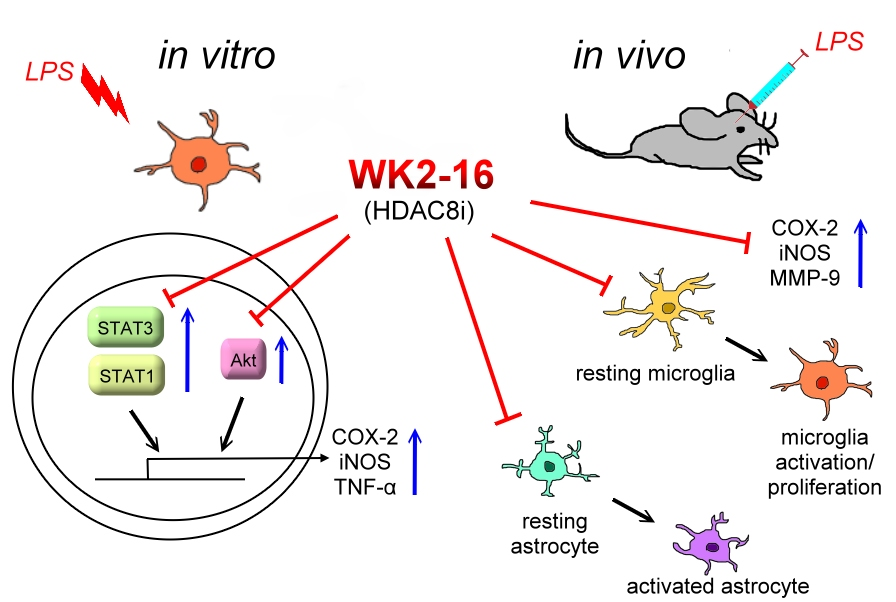
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. WK2-16 Attenuated the Inflammatory Responses and Improved Neurological Functions in LPS-Induced Neuroinflammation in C57BL6 Mice
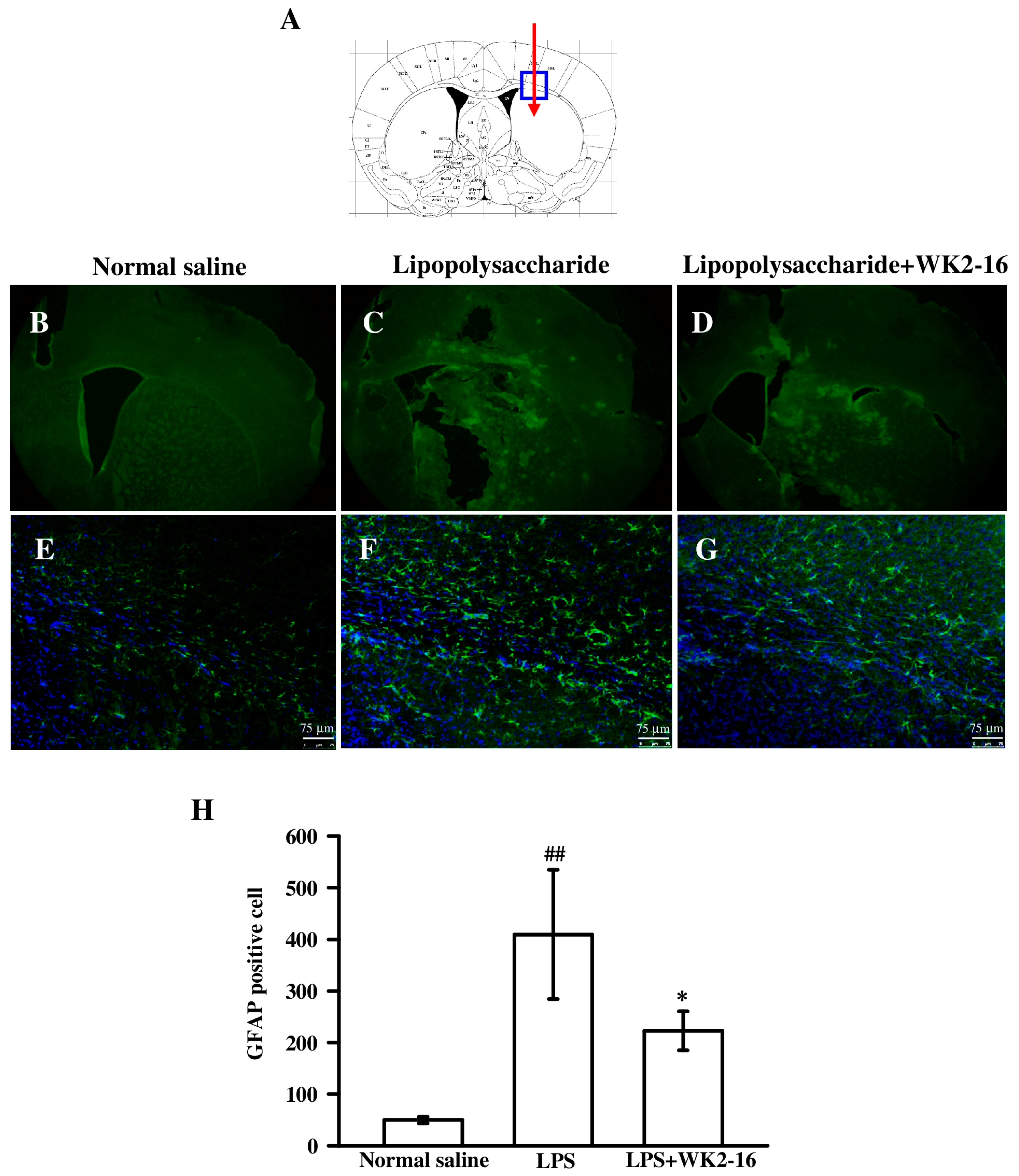
2.2. WK2-16 Inhibited Astrogliosis and Microglia Proliferation in LPS-Challenged C57BL6 Mice
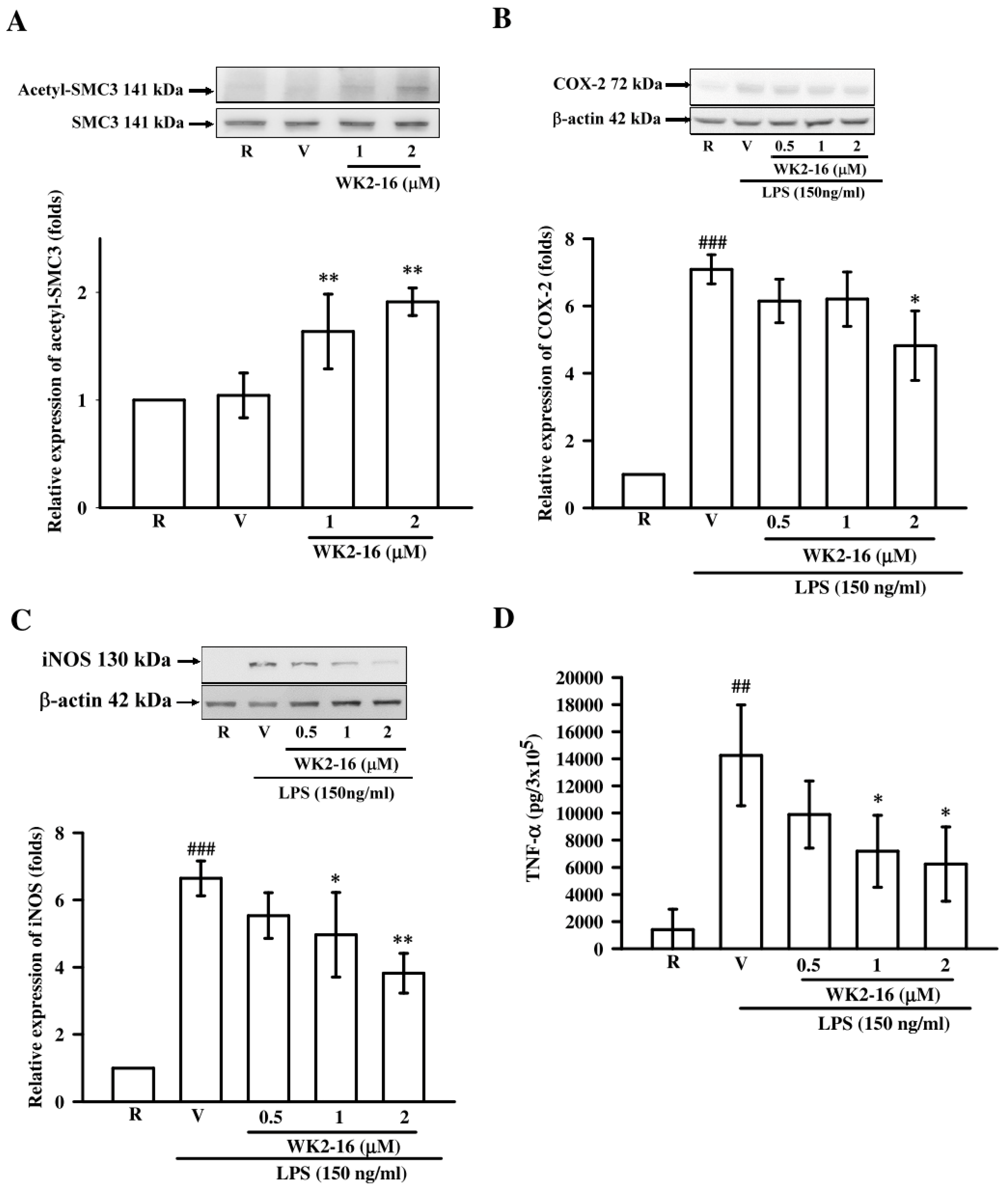
2.3. WK2-16 Inhibited Inflammatory Mediators and Cytokine in LPS-Stimulated Microglial BV2 Cells
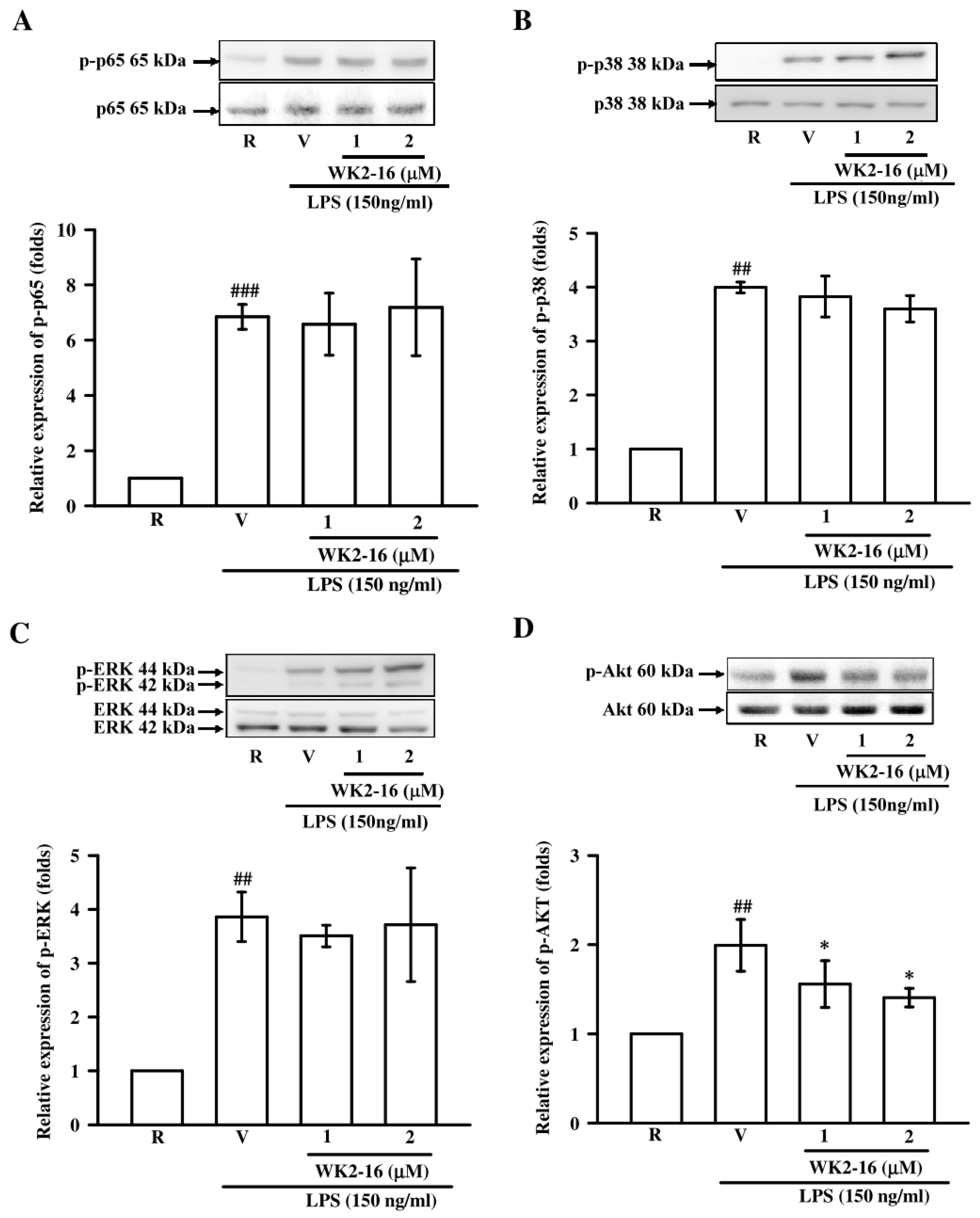
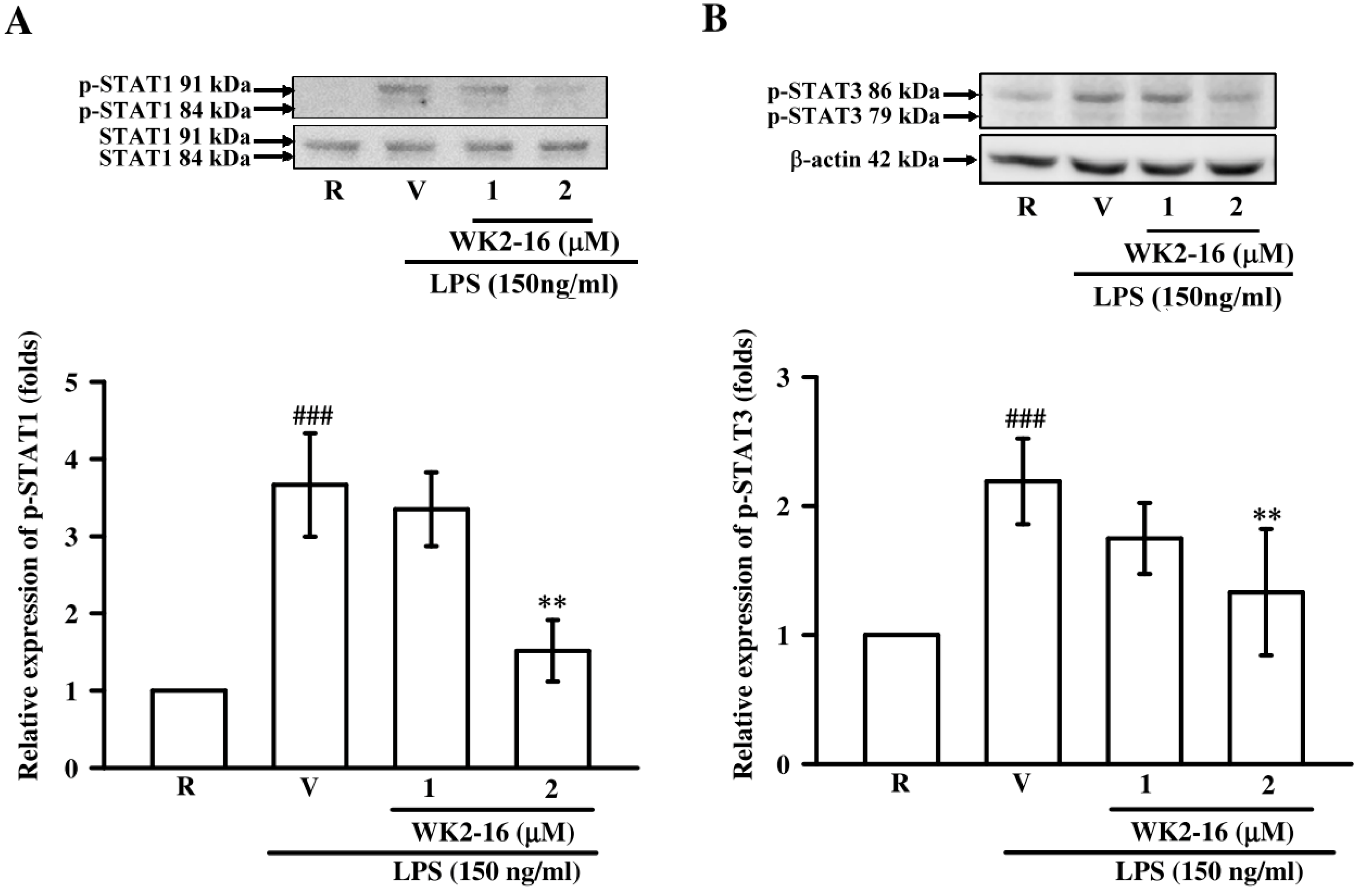
2.4. WK2-16 Decreased LPS-Stimulated Inflammatory Responses through STAT-1/-3 Signaling Pathway
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animals
4.3. Stereotactic Injections and WK2-16 Administration
4.4. Neurological Score
4.5. Western blot Analysis
4.6. Gelatin Zymography Analysis
4.7. Immunofluorescence
4.8. Microglial Cell Culture
4.9. MTT Assay
4.10. Sulforhodamine B (SRB) Assay
4.11. Enzyme-Linked Immunosorbent Assay (ELISA)
4.12. Statistical Analyses
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Davis, R.L.; Stevens, C.W.; Thomas Curtis, J. The opioid antagonist, beta-funaltrexamine, inhibits lipopolysaccharide-induced neuroinflammation and reduces sickness behavior in mice. Physiol. Behav. 2017, 173, 52–60. [Google Scholar] [CrossRef]
- Shastri, A.; Bonifati, D.M.; Kishore, U. Innate immunity and neuroinflammation. Mediat. Inflamm. 2013, 2013, 342931. [Google Scholar] [CrossRef]
- Lull, M.E.; Block, M.L. Microglial activation and chronic neurodegeneration. Neurotherapeutics 2010, 7, 354–365. [Google Scholar] [CrossRef]
- Czeh, M.; Gressens, P.; Kaindl, A.M. The yin and yang of microglia. Dev. Neurosci. 2011, 33, 199–209. [Google Scholar] [CrossRef]
- Liu, M.; Bing, G. Lipopolysaccharide animal models for Parkinson’s disease. Park. Dis. 2011, 2011, 327089. [Google Scholar] [CrossRef]
- Saijo, K.; Winner, B.; Carson, C.T.; Collier, J.G.; Boyer, L.; Rosenfeld, M.G.; Gage, F.H.; Glass, C.K. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell 2009, 137, 47–59. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, Y.H.; Park, G. Cucurbitacins attenuate microglial activation and protect from neuroinflammatory injury through Nrf2/ARE activation and STAT/NF-kappaB inhibition. Neurosci. Lett. 2015, 609, 129–136. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Kunnumakkara, A.B.; Harikumar, K.B.; Gupta, S.R.; Tharakan, S.T.; Koca, C.; Dey, S.; Sung, B. Signal transducer and activator of transcription-3, inflammation, and cancer: How intimate is the relationship? Ann. N. Y. Acad. Sci. 2009, 1171, 59–76. [Google Scholar] [CrossRef]
- Zeng, K.W.; Wang, S.; Dong, X.; Jiang, Y.; Tu, P.F. Sesquiterpene dimer (DSF-52) from Artemisia argyi inhibits microglia-mediated neuroinflammation via suppression of NF-kappaB, JNK/p38 MAPKs and Jak2/Stat3 signaling pathways. Phytomedicine 2014, 21, 298–306. [Google Scholar] [CrossRef]
- Park, S.Y.; Bae, Y.S.; Ko, M.J.; Lee, S.J.; Choi, Y.W. Comparison of anti-inflammatory potential of four different dibenzocyclooctadiene lignans in microglia; action via activation of PKA and Nrf-2 signaling and inhibition of MAPK/STAT/NF-kappaB pathways. Mol. Nutr. Food Res. 2014, 58, 738–748. [Google Scholar] [CrossRef]
- Lin, H.Y.; Tang, C.H.; Chen, Y.H.; Wei, I.H.; Chen, J.H.; Lai, C.H.; Lu, D.Y. Peptidoglycan enhances proinflammatory cytokine expression through the TLR2 receptor, MyD88, phosphatidylinositol 3-kinase/AKT and NF-kappaB pathways in BV-2 microglia. Int. Immunopharmacol. 2010, 10, 883–891. [Google Scholar] [CrossRef]
- Cui, Y.; Park, J.Y.; Wu, J.; Lee, J.H.; Yang, Y.S.; Kang, M.S.; Jung, S.C.; Park, J.M.; Yoo, E.S.; Kim, S.H.; et al. Dieckol Attenuates Microglia-mediated Neuronal Cell Death via ERK, Akt and NADPH Oxidase-mediated Pathways. Korean J. Physiol. Pharmacol. 2015, 19, 219–228. [Google Scholar] [CrossRef]
- Jung, J.S.; Choi, M.J.; Lee, Y.Y.; Moon, B.I.; Park, J.S.; Kim, H.S. Suppression of Lipopolysaccharide-Induced Neuroinflammation by Morin via MAPK, PI3K/Akt, and PKA/HO-1 Signaling Pathway Modulation. J. Agric. Food Chem. 2017, 65, 373–382. [Google Scholar] [CrossRef]
- Patnala, R.; Arumugam, T.V.; Gupta, N.; Dheen, S.T. HDAC Inhibitor Sodium Butyrate-Mediated Epigenetic Regulation Enhances Neuroprotective Function of Microglia During Ischemic Stroke. Mol. Neurobiol. 2017, 54, 6391–6411. [Google Scholar] [CrossRef]
- Dolinoy, D.C.; Weidman, J.R.; Jirtle, R.L. Epigenetic gene regulation: Linking early developmental environment to adult disease. Reprod. Toxicol. 2007, 23, 297–307. [Google Scholar] [CrossRef]
- Zhang, B.; West, E.J.; Van, K.C.; Gurkoff, G.G.; Zhou, J.; Zhang, X.M.; Kozikowski, A.P.; Lyeth, B.G. HDAC inhibitor increases histone H3 acetylation and reduces microglia inflammatory response following traumatic brain injury in rats. Brain Res. 2008, 1226, 181–191. [Google Scholar] [CrossRef]
- Kannan, V.; Brouwer, N.; Hanisch, U.K.; Regen, T.; Eggen, B.J.; Boddeke, H.W. Histone deacetylase inhibitors suppress immune activation in primary mouse microglia. J. Neurosci. Res. 2013, 91, 1133–1142. [Google Scholar] [CrossRef]
- Yamawaki, Y.; Yoshioka, N.; Nozaki, K.; Ito, H.; Oda, K.; Harada, K.; Shirawachi, S.; Asano, S.; Aizawa, H.; Yamawaki, S.; et al. Sodium butyrate abolishes lipopolysaccharide-induced depression-like behaviors and hippocampal microglial activation in mice. Brain Res. 2018, 1680, 13–38. [Google Scholar] [CrossRef]
- Li, S.; Fossati, G.; Marchetti, C.; Modena, D.; Pozzi, P.; Reznikov, L.L.; Moras, M.L.; Azam, T.; Abbate, A.; Mascagni, P.; et al. Specific inhibition of histone deacetylase 8 reduces gene expression and production of proinflammatory cytokines in vitro and in vivo. J. Biol. Chem. 2015, 290, 2368–2378. [Google Scholar] [CrossRef]
- Jan, J.S.; Chou, Y.C.; Cheng, Y.W.; Chen, C.K.; Huang, W.J.; Hsiao, G. The Novel HDAC8 Inhibitor WK2-16 Attenuates Lipopolysaccharide-Activated Matrix Metalloproteinase-9 Expression in Human Monocytic Cells and Improves Hypercytokinemia In Vivo. Int. J. Mol. Sci. 2017, 18, 1394. [Google Scholar] [CrossRef]
- Garcia, J.H.; Wagner, S.; Liu, K.F.; Hu, X.J. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke 1995, 26, 627–634. [Google Scholar] [CrossRef]
- Streit, W.J.; Mrak, R.E.; Griffin, W.S. Microglia and neuroinflammation: A pathological perspective. J. Neuroinflamm. 2004, 1, 14. [Google Scholar] [CrossRef][Green Version]
- Rettig, I.; Koeneke, E.; Trippel, F.; Mueller, W.C.; Burhenne, J.; Kopp-Schneider, A.; Fabian, J.; Schober, A.; Fernekorn, U.; von Deimling, A.; et al. Selective inhibition of HDAC8 decreases neuroblastoma growth in vitro and in vivo and enhances retinoic acid-mediated differentiation. Cell Death Dis. 2015, 6, e1657. [Google Scholar] [CrossRef]
- Lin, F.L.; Ho, J.D.; Cheng, Y.W.; Chiou, G.C.Y.; Yen, J.L.; Chang, H.M.; Lee, T.H.; Hsiao, G. Theissenolactone C Exhibited Ocular Protection of Endotoxin-Induced Uveitis by Attenuating Ocular Inflammatory Responses and Glial Activation. Front. Pharmacol. 2018, 9, 326. [Google Scholar] [CrossRef]
- Song, F.; Zeng, K.; Liao, L.; Yu, Q.; Tu, P.; Wang, X. Schizandrin A Inhibits Microglia-Mediated Neuroninflammation through Inhibiting TRAF6-NF-kappaB and Jak2-Stat3 Signaling Pathways. PLoS ONE 2016, 11, e0149991. [Google Scholar]
- Gomez-Nicola, D.; Fransen, N.L.; Suzzi, S.; Perry, V.H. Regulation of microglial proliferation during chronic neurodegeneration. J. Neurosci. 2013, 33, 2481–2493. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef]
- Penn, J.S.; Madan, A.; Caldwell, R.B.; Bartoli, M.; Caldwell, R.W.; Hartnett, M.E. Vascular endothelial growth factor in eye disease. Prog. Retin. Eye Res. 2008, 27, 331–371. [Google Scholar] [CrossRef]
- Naeimi, R.; Safarpour, F.; Hashemian, M.; Tashakorian, H.; Ahmadian, S.R.; Ashrafpour, M.; Ghasemi-Kasman, M. Curcumin-loaded nanoparticles ameliorate glial activation and improve myelin repair in lyolecithin-induced focal demyelination model of rat corpus callosum. Neurosci. Lett. 2018, 674, 1–10. [Google Scholar] [CrossRef]
- Nomura, T.; Bando, Y.; You, H.; Tanaka, T.; Yoshida, S. Yokukansan Reduces Cuprizone-Induced Demyelination in the Corpus Callosum Through Anti-inflammatory Effects on Microglia. Neurochem. Res. 2017, 42, 3525–3536. [Google Scholar] [CrossRef]
- Rosenberg, G.A. Matrix metalloproteinases in neuroinflammation. Glia 2002, 39, 279–291. [Google Scholar] [CrossRef]
- Wang, X.; Jung, J.; Asahi, M.; Chwang, W.; Russo, L.; Moskowitz, M.A.; Dixon, C.E.; Fini, M.E.; Lo, E.H. Effects of matrix metalloproteinase-9 gene knock-out on morphological and motor outcomes after traumatic brain injury. J. Neurosci. 2000, 20, 7037–7042. [Google Scholar] [CrossRef]
- Ding, Z.; Mathur, V.; Ho, P.P.; James, M.L.; Lucin, K.M.; Hoehne, A.; Alabsi, H.; Gambhir, S.S.; Steinman, L.; Luo, J.; et al. Antiviral drug ganciclovir is a potent inhibitor of microglial proliferation and neuroinflammation. J. Exp. Med. 2014, 211, 189–198. [Google Scholar] [CrossRef]
- Mrvova, N.; Skandik, M.; Kuniakova, M.; Rackova, L. Modulation of BV-2 microglia functions by novel quercetin pivaloyl ester. Neurochem. Int. 2015, 90, 246–254. [Google Scholar] [CrossRef]
- Suh, H.S.; Kim, M.O.; Lee, S.C. Inhibition of granulocyte-macrophage colony-stimulating factor signaling and microglial proliferation by anti-CD45RO: Role of Hck tyrosine kinase and phosphatidylinositol 3-kinase/Akt. J. Immunol. 2005, 174, 2712–2719. [Google Scholar] [CrossRef]
- Tufekci, K.U.; Genc, S.; Genc, K. The endotoxin-induced neuroinflammation model of Parkinson’s disease. Parkinsons Dis. 2011, 2011, 487450. [Google Scholar] [CrossRef]
- Aid, S.; Langenbach, R.; Bosetti, F. Neuroinflammatory response to lipopolysaccharide is exacerbated in mice genetically deficient in cyclooxygenase-2. J. Neuroinflamm. 2008, 5, 17. [Google Scholar] [CrossRef]
- Dello Russo, C.; Lisi, L.; Tringali, G.; Navarra, P. Involvement of mTOR kinase in cytokine-dependent microglial activation and cell proliferation. Biochem. Pharmacol. 2009, 78, 1242–1251. [Google Scholar] [CrossRef]
- Iravani, M.M.; Leung, C.C.; Sadeghian, M.; Haddon, C.O.; Rose, S.; Jenner, P. The acute and the long-term effects of nigral lipopolysaccharide administration on dopaminergic dysfunction and glial cell activation. Eur. J. Neurosci. 2005, 22, 317–330. [Google Scholar] [CrossRef]
- Perry, V.H.; Newman, T.A.; Cunningham, C. The impact of systemic infection on the progression of neurodegenerative disease. Nat. Rev. Neurosci. 2003, 4, 103–112. [Google Scholar] [CrossRef]
- Oliveira, L.S.; de Queiroz, N.M.; Veloso, L.V.; Moreira, T.G.; Oliveira, F.S.; Carneiro, M.B.; Faria, A.M.; Vieira, L.Q.; Oliveira, S.C.; Horta, M.F. A defective TLR4 signaling for IFN-beta expression is responsible for the innately lower ability of BALB/c macrophages to produce NO in response to LPS as compared to C57BL/6. PLoS ONE 2014, 9, e98913. [Google Scholar] [CrossRef]
- Przanowski, P.; Dabrowski, M.; Ellert-Miklaszewska, A.; Kloss, M.; Mieczkowski, J.; Kaza, B.; Ronowicz, A.; Hu, F.; Piotrowski, A.; Kettenmann, H.; et al. The signal transducers Stat1 and Stat3 and their novel target Jmjd3 drive the expression of inflammatory genes in microglia. J. Mol. Med. 2014, 92, 239–254. [Google Scholar] [CrossRef]
- Kim, H.S.; Ye, S.K.; Cho, I.H.; Jung, J.E.; Kim, D.H.; Choi, S.; Kim, Y.S.; Park, C.G.; Kim, T.Y.; Lee, J.W.; et al. 8-hydroxydeoxyguanosine suppresses NO production and COX-2 activity via Rac1/STATs signaling in LPS-induced brain microglia. Free Radic. Biol. Med. 2006, 41, 1392–1403. [Google Scholar] [CrossRef]
- Kim, H.J.; Rowe, M.; Ren, M.; Hong, J.S.; Chen, P.S.; Chuang, D.M. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: Multiple mechanisms of action. J. Pharmacol. Exp. Ther. 2007, 321, 892–901. [Google Scholar] [CrossRef]
- Harrison, I.F.; Dexter, D.T. Epigenetic targeting of histone deacetylase: Therapeutic potential in Parkinson’s disease? Pharmacol. Ther. 2013, 140, 34–52. [Google Scholar] [CrossRef]
- Durham, B.S.; Grigg, R.; Wood, I.C. Inhibition of histone deacetylase 1 or 2 reduces induced cytokine expression in microglia through a protein synthesis independent mechanism. J. Neurochem. 2017, 143, 214–224. [Google Scholar] [CrossRef]
- Yang, S.S.; Zhang, R.; Wang, G.; Zhang, Y.F. The development prospection of HDAC inhibitors as a potential therapeutic direction in Alzheimer’s disease. Transl. Neurodegener. 2017, 6, 19. [Google Scholar] [CrossRef]
- Lee, H.Y.; Fan, S.J.; Huang, F.I.; Chao, H.Y.; Hsu, K.C.; Lin, T.E.; Yeh, T.K.; Lai, M.J.; Li, Y.H.; Huang, H.L.; et al. 5-Aroylindoles Act as Selective Histone Deacetylase 6 Inhibitors Ameliorating Alzheimer’s Disease Phenotypes. J. Med. Chem. 2018, 61, 7087–7102. [Google Scholar] [CrossRef]
- Yang, Y.C.; Chen, C.N.; Wu, C.I.; Huang, W.J.; Kuo, T.Y.; Kuan, M.C.; Tsai, T.H.; Huang, J.S.; Huang, C.Y. NBM-T-L-BMX-OS01, Semisynthesized from Osthole, Is a Novel Inhibitor of Histone Deacetylase and Enhances Learning and Memory in Rats. Evid.-Based Complement. Alternat. Med. 2013, 2013, 514908. [Google Scholar] [CrossRef]
- Wang, G.; Shi, Y.; Jiang, X.; Leak, R.K.; Hu, X.; Wu, Y.; Pu, H.; Li, W.W.; Tang, B.; Wang, Y.; et al. HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3beta/PTEN/Akt axis. Proc. Natl. Acad. Sci. USA 2015, 112, 2853–2858. [Google Scholar] [CrossRef]
- Li, Y.; Yuan, Z.; Liu, B.; Sailhamer, E.A.; Shults, C.; Velmahos, G.C.; Demoya, M.; Alam, H.B. Prevention of hypoxia-induced neuronal apoptosis through histone deacetylase inhibition. J. Trauma 2008, 64, 863–870. [Google Scholar] [CrossRef]
- Kiernan, R.; Bres, V.; Ng, R.W.; Coudart, M.P.; El Messaoudi, S.; Sardet, C.; Jin, D.Y.; Emiliani, S.; Benkirane, M. Post-activation turn-off of NF-kappa B-dependent transcription is regulated by acetylation of p65. J. Biol. Chem. 2003, 278, 2758–2766. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.M.; Chen, C.Q.; Wang, L.Y.; Hong, L.L.; Wu, J.B.; Dong, P.H.; Yu, F.J. Histone deacetylases inhibitor sodium butyrate inhibits JAK2/STAT signaling through upregulation of SOCS1 and SOCS3 mediated by HDAC8 inhibition in myeloproliferative neoplasms. Exp. Hematol. 2013, 41, 261–270.e4. [Google Scholar] [CrossRef]
- Turnquist, C.; Wang, Y.; Severson, D.T.; Zhong, S.; Sun, B.; Ma, J.; Constaninescu, S.N.; Ansorge, O.; Stolp, H.B.; Molnar, Z.; et al. STAT1-induced ASPP2 transcription identifies a link between neuroinflammation, cell polarity, and tumor suppression. Proc. Natl. Acad. Sci. USA 2014, 111, 9834–9839. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, B.; Ek, C.J.; Sun, Y.; Zhu, C.; Sandberg, M.; Mallard, C. GSK3beta inhibition protects the immature brain from hypoxic-ischaemic insult via reduced STAT3 signalling. Neuropharmacology 2016, 101, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.J.; Wang, Y.C.; Chao, S.W.; Yang, C.Y.; Chen, L.C.; Lin, M.H.; Hou, W.C.; Chen, M.Y.; Lee, T.L.; Yang, P.; et al. Synthesis and biological evaluation of ortho-aryl N-hydroxycinnamides as potent histone deacetylase (HDAC) 8 isoform-selective inhibitors. Chem. Med. Chem. 2012, 7, 1815–1824. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.L.; Hsiao, C.J.; Lin, F.L.; Jan, J.S.; Chou, Y.C.; Lin, Y.Y.; Chen, C.K.; Lam, K.K.; Hsiao, G. Haloperidol Abrogates Matrix Metalloproteinase-9 Expression by Inhibition of NF-kappaB Activation in Stimulated Human Monocytic Cells. Mediat. Inflamm. 2018, 2018, 9541459. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.L.; Lin, C.H.; Ho, J.D.; Yen, J.L.; Chang, H.M.; Chiou, G.C.; Cheng, Y.W.; Hsiao, G. The natural retinoprotectant chrysophanol attenuated photoreceptor cell apoptosis in an N-methyl-N-nitrosourea-induced mouse model of retinal degenaration. Sci. Rep. 2017, 7, 41086. [Google Scholar] [CrossRef]
- Chou, Y.C.; Sheu, J.R.; Chung, C.L.; Chen, C.Y.; Lin, F.L.; Hsu, M.J.; Kuo, Y.H.; Hsiao, G. Nuclear-targeted inhibition of NF-kappaB on MMP-9 production by N-2-(4-bromophenyl) ethyl caffeamide in human monocytic cells. Chem. Biol. Interact. 2010, 184, 403–412. [Google Scholar] [CrossRef]
- Hsiao, C.J.; Hsiao, G.; Chen, W.L.; Wang, S.W.; Chiang, C.P.; Liu, L.Y.; Guh, J.H.; Lee, T.H.; Chung, C.L. Cephalochromin induces G0/G1 cell cycle arrest and apoptosis in A549 human non-small-cell lung cancer cells by inflicting mitochondrial disruption. J. Nat. Prod. 2014, 77, 758–765. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, F.-L.; Yen, J.-L.; Kuo, Y.-C.; Kang, J.-J.; Cheng, Y.-W.; Huang, W.-J.; Hsiao, G. HDAC8 Inhibitor WK2-16 Therapeutically Targets Lipopolysaccharide-Induced Mouse Model of Neuroinflammation and Microglial Activation. Int. J. Mol. Sci. 2019, 20, 410. https://doi.org/10.3390/ijms20020410
Lin F-L, Yen J-L, Kuo Y-C, Kang J-J, Cheng Y-W, Huang W-J, Hsiao G. HDAC8 Inhibitor WK2-16 Therapeutically Targets Lipopolysaccharide-Induced Mouse Model of Neuroinflammation and Microglial Activation. International Journal of Molecular Sciences. 2019; 20(2):410. https://doi.org/10.3390/ijms20020410
Chicago/Turabian StyleLin, Fan-Li, Jing-Lun Yen, Yu-Cheng Kuo, Jaw-Jou Kang, Yu-Wen Cheng, Wei-Jan Huang, and George Hsiao. 2019. "HDAC8 Inhibitor WK2-16 Therapeutically Targets Lipopolysaccharide-Induced Mouse Model of Neuroinflammation and Microglial Activation" International Journal of Molecular Sciences 20, no. 2: 410. https://doi.org/10.3390/ijms20020410
APA StyleLin, F.-L., Yen, J.-L., Kuo, Y.-C., Kang, J.-J., Cheng, Y.-W., Huang, W.-J., & Hsiao, G. (2019). HDAC8 Inhibitor WK2-16 Therapeutically Targets Lipopolysaccharide-Induced Mouse Model of Neuroinflammation and Microglial Activation. International Journal of Molecular Sciences, 20(2), 410. https://doi.org/10.3390/ijms20020410