AZD1208, a Pan-Pim Kinase Inhibitor, Has Anti-Growth Effect on 93T449 Human Liposarcoma Cells via Control of the Expression and Phosphorylation of Pim-3, mTOR, 4EBP-1, S6, STAT-3 and AMPK


Abstract
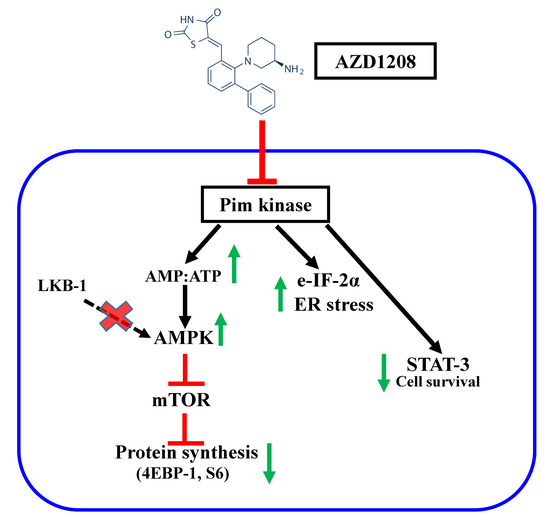
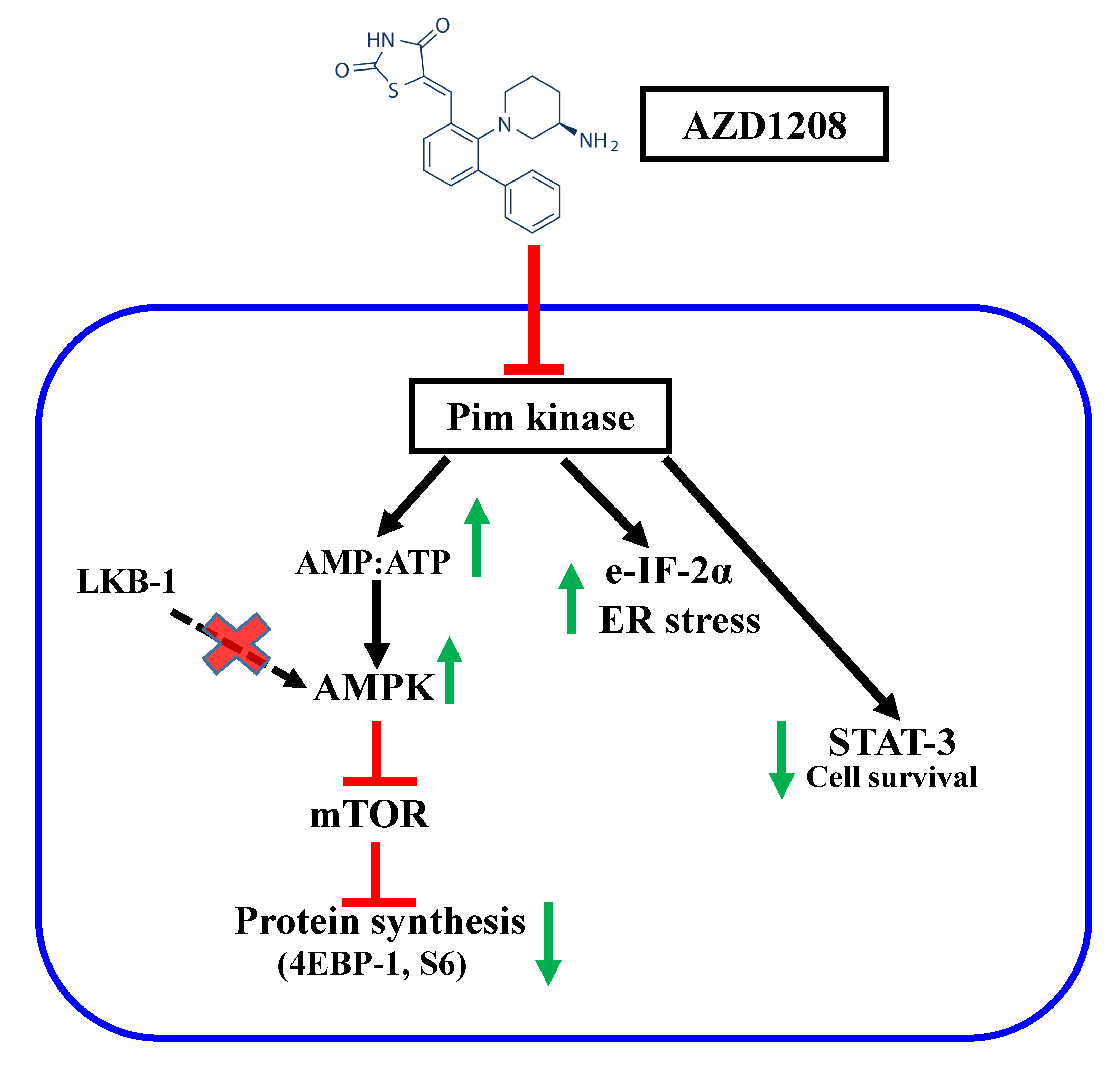{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. AZD1208 Strongly Inhibits Growth of 93T449 Human Liposarcoma Cells
2.2. AZD1208 Does Not Induce Apoptosis of 93T449 Human Liposarcoma Cells
2.3. AZD1208 Reduces Phosphorylation of STAT-3 in 93T449 Human Liposarcoma Cells and Pharmacological Inhibition of STAT-3 Leads to Reduction of the Cell Survival
2.4. AZD1208 Inhibits mTOR, S6 and eIF-2α in 93T449 Liposarcoma Cells
2.5. AZD1208 Induces Strong Phosphorylation of AMPK in 93T449 Cells and AMPK Knockdown Greatly Attenuates Reduction of the Cell Survival by AZD1208
2.6. AZD1208-Induced AMPK Phosphorylation in 93T449 Cells Appears to Be Not through LKB-1 But through Decrease in Cellular ATP Contents
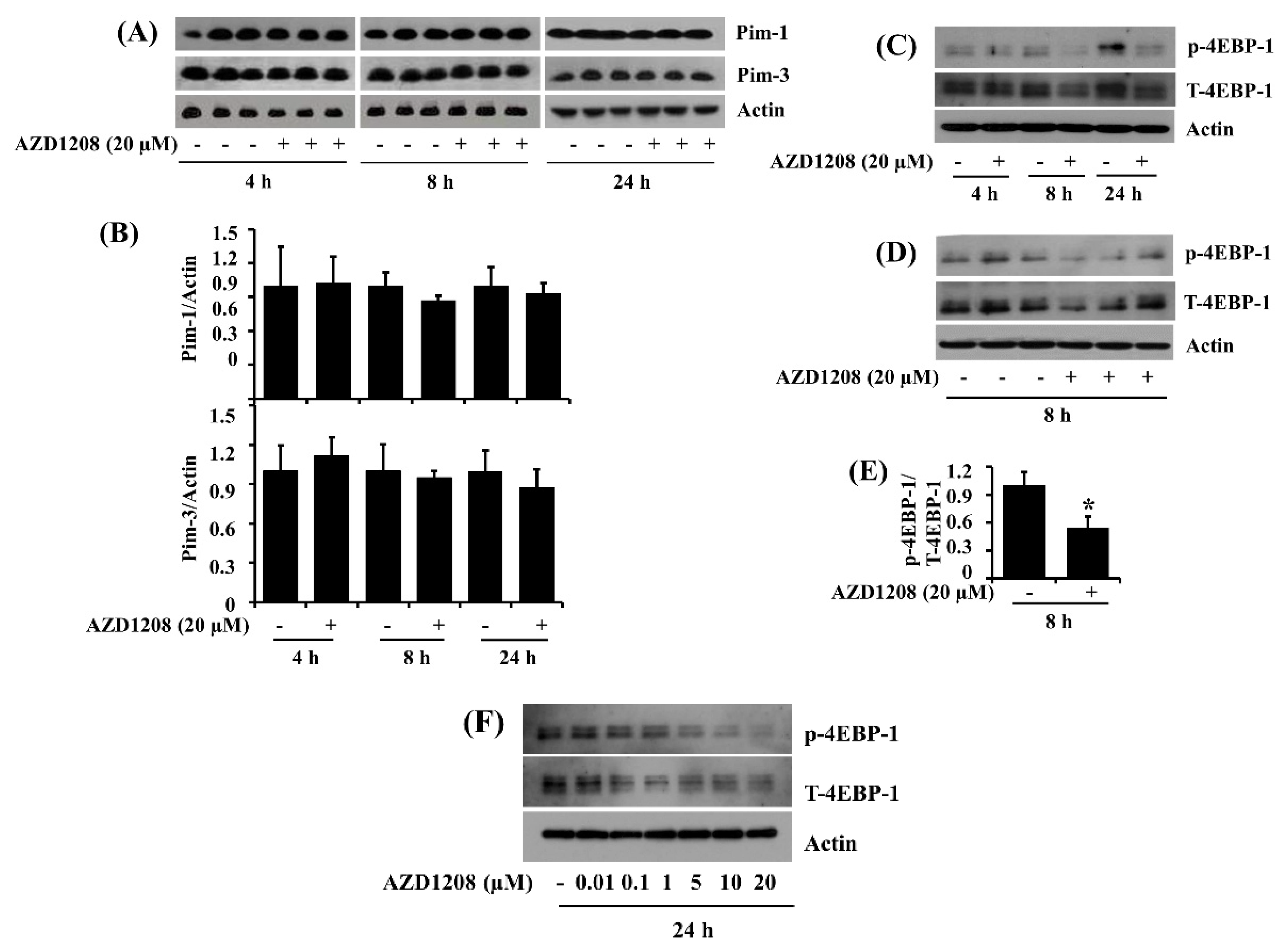
2.7. AZD1208 Does Not Affect Expression of Pim Kinases but It Reduces the Phosphorylation of 4EBP-1, a Downstream Effector of Pim Kinases, in 93T449 Human Liposarcoma Cells
2.8. Pim-3 Knock-Down Causes Not Only Growth Inhibition But Also Alteration of Phosphorylation of 4EBP-1, STAT-3, mTOR, AMPK and eIF-2a in 93T449 Human Liposarcoma Cells
3. Discussion
4. Materials and Methods
4.1. Chemicals and Antibodies
4.2. Cell Culture
4.3. Cell Count Assay and Cell Morphology Analysis
4.4. Measurement of DNA Fragmentation
4.5. Measurement of the Population of Sub G1 Phase by Flow Cytometry Analysis
4.6. Preparation of Whole Cell Lysates
4.7. Immunoblot Analysis
4.8. Small Interfering RNA (siRNA) Transfection
4.9. Measurement of Cellular ATP Contents
4.10. Statistical Analyses
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ECL | enhanced chemiluminescence |
| mTOR | mammalian target of rapamycin |
| FBS | fetal bovine serum |
| STAT-3 | signal transducer and activator of transcription-3 |
| e-IF-2α | eukaryotic initiation factor-2α |
| RT | room temperature |
References
- Cormier, J.N.; Pollock, R.E. Soft tissue sarcomas, CA. Cancer J. Clin. 2004, 54, 94–109. [Google Scholar] [CrossRef]
- Dalal, K.M.; Antonescu, C.R.; Singer, S. Diagnosis and Management of lipomatous tumors. J. Surg. Oncol. 2008, 97, 298–313. [Google Scholar] [CrossRef] [PubMed]
- N.C. Institute Site, General Information About Adult Soft Tissue Sarcoma. Available online: https://www.cancer.gov/types/soft-tissue-sarcoma/hp/adult-soft-tissue-treatment-pdq#cit/section_1.1 (accessed on 14 Januray 2018).
- Eilber, F.C.; Eilber, F.R.; Eckardt, J.; Rosen, G.; Riedel, E.; Maki, R.G.; Brennan, M.F.; Singer, S. The impact of chemotherapy on the survival of patients with high-grade primary extremity liposarcoma. Ann. Surg. 2004, 240, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Singer, S.; Antonescu, C.R.; Riedel, E.; Brennan, M.F. Histologic subtype and margin of resection predict pattern of recurrence and survival for retroperitoneal liposarcoma. Ann. Surg. 2003, 238, 358–370. [Google Scholar] [CrossRef]
- Steinmann, S.; Gali-Muhtasib, H.; Huebner, K.; Al-Halabi, R.; Abou Merhi, R.; Aman, P.; Agaimy, A.; Haller, F.; Schneider-Stock, R. Hsp90 inhibition by AUY922 as an effective treatment strategy against myxoid liposarcoma. Cancer Lett. 2015, 367, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Ries, L.; Young, J.; Keel, G.; Eisner, M.; Lin, Y.; Horner, M.-J. Cancer Survival among Adults-US SEER Program, 1988–2001, Patient and Tumor Characteristics. Available online: https://seer. cancer.gov/archive/publications/survival/index.html (accessed on 28 December 2017).
- Nawijn, M.C.; Alendar, A.; Berns, A. For better or for worse: The role of Pim oncogenes in tumorigenesis. Nat. Rev. Cancer 2011, 11, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.S.; Yang, J.Y.; Liang, H.; Cortes, J.E.; Gandhi, V. Protein profiling identifies mTOR pathway modulation and cytostatic effects of Pim kinase inhibitor, AZD1208, in acute myeloid leukemia. Leuk. Lymphoma 2016, 57, 2863–2873. [Google Scholar] [CrossRef]
- Beharry, Z.; Mahajan, S.; Zemskova, M.; Lin, Y.W.; Tholanikunnel, B.G.; Xia, Z.; Smith, C.D. The Pim protein kinases regulate energy metabolism and cell growth. Proc. Natl. Acad. Sci. USA 2011, 108, 528–533. [Google Scholar] [CrossRef]
- Chang, M.; Kanwar, N.; Feng, E.; Siu, A.; Liu, X.; Ma, D.; Jongstra, J. PIM kinase inhibitors downregulate STAT3(Tyr705) phosphorylation. Mol. Cancer Ther. 2010, 9, 2478–2487. [Google Scholar] [CrossRef]
- Decker, S.; Finter, J.; Forde, A.; Kissel, S.; Schwaller, J.; Mack, T.S.; Kuhn, A.; Gray, N.S.; Follo, M.; Jumaa, H.; et al. PIM kinases are essential for chronic lymphocytic leukemia cell survival (PIM2/3) and CXCR4 mediated microenvironmental interactions (PIM1). Mol. Cancer Ther. 2014, 13, 1231–1245. [Google Scholar] [CrossRef]
- Lu, J.; Zavorotinskaya, T.; Dai, Y.; Niu, X.H.; Castillo, J.; Sim, J.; Yu, J.; Wang, Y.; Langowski, J.L.; Holash, J.; et al. Pim2 is required for maintaining multiple myeloma cell growth through modulating TSC2 phosphorylation. Blood 2013, 122, 1610–1620. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Abad, C.; Pisonero, H.; Blanco-Aparicio, C.; Roncador, G.; González-Menchén, A.; Martinez-Climent, J.A.; Mata, E.; Rodríguez, M.E.; Muñoz-González, G.; Sánchez-Beato, M.; et al. PIM2 inhibition as a rational therapeutic approach in B-cell lymphoma. Blood 2011, 118, 5517–5527. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Mao, X.; Chen, J.; Huang, B.; Jin, C.; Xu, Z.; Qiu, S. Overexpression of Pim-1 in bladder cancer. J. Exp. Clin. Cancer Res. 2010, 29, 161. [Google Scholar] [CrossRef]
- Wang, J.; Anderson, P.D.; Luo, W.; Gius, D.; Roh, M.; Abdulkadir, S.A. Pim1 kinase is required to maintain tumorigenicity in MYC-expressing prostate cancer cells. Oncogene 2012, 31, 1794–1803. [Google Scholar] [CrossRef]
- Mikkers, H.; Nawijn, M.; Allen, J.; Brouwers, C.; Verhoeven, E.; Jonkers, J.; Berns, A. Mice deficient for all PIM kinases display reduced body size and impaired responses to hematopoietic growth factors. Mol. Cell. Biol. 2004, 24, 6104–6115. [Google Scholar] [CrossRef]
- Keeton, E.K.; McEachern, K.; Dillman, K.S.; Palakurthi, S.; Cao, Y.; Grondine, M.R.; Kaur, S.; Wang, S.; Chen, Y.; Wu, A.; et al. AZD1208, a potent and selective pan-Pim kinase inhibitor, demonstrates efficacy in preclinical models of acute myeloid leukemia. Blood 2014, 123, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Qian, K.C.; Wang, L.; Hickey, E.R.; Studts, J.; Barringer, K.; Peng, C.; Kronkaitis, A.; Li, J.; White, A.; Mische, S.; et al. Structural basis of constitutive activity and a unique nucleotide binding mode of human Pim-1 kinase. J. Biol. Chem. 2005, 280, 6130–6137. [Google Scholar] [CrossRef]
- Brasó-Maristany, F.; Filosto, S.; Catchpole, S.; Marlow, R.; Quist, J.; Francesch-Domenech, E.; Plumb, D.A.; Zakka, L.; Gazinska, P.; Liccardi, G.; et al. PIM1 kinase regulates cell death, Tumor growth and chemotherapy response in triple-negative breast cancer. Nat. Med. 2016, 22, 1303–1313. [Google Scholar] [CrossRef]
- Kirschner, A.N.; Wang, J.; van der Meer, R.; Anderson, P.D.; Franco-Coronel, O.E.; Kushner, M.H.; Everett, J.H.; Hameed, O.; Keeton, E.K.; Ahdesmaki, M.; et al. PIM kinase inhibitor AZD1208 for treatment of MYC-driven prostate cancer. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef]
- Kreuz, S.; Holmes, K.B.; Tooze, R.M.; Lefevre, P.F. Loss of PIM2 enhances the anti-proliferative effect of the pan-PIM kinase inhibitor AZD1208 in non-Hodgkin lymphomas. Mol. Cancer 2015, 14, 205. [Google Scholar] [CrossRef]
- Iqbal, A.; Eckerdt, F.; Bell, J.; Nakano, I.; Giles, F.J.; Cheng, S.Y.; Lulla, R.R.; Goldman, S.; Platanias, L.C. Targeting of glioblastoma cell lines and glioma stem cells by combined PIM kinase and PI3K-p110α inhibition. Oncotarget 2016, 7, 33192–33201. [Google Scholar] [CrossRef]
- Brunen, D.; de Vries, R.C.; Lieftink, C.; Beijersbergen, R.L.; Bernards, R. PIM Kinases Are a Potential Prognostic Biomarker and Therapeutic Target in Neuroblastoma. Mol. Cancer. Ther. 2018, 17, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.K.; Obiang-Obounou, B.W.; Lee, K.B.; Choi, J.S.; Jang, B.C. AZD1208, a pan-Pim kinase inhibitor, inhibits adipogenesis and induces lipolysis in 3T3-L1 adipocytes. J. Cell. Mol. Med. 2018, 22, 2488–2497. [Google Scholar] [CrossRef] [PubMed]
- Behan, J.W.; Yun, J.P.; Proektor, M.P.; Ehsanipour, E.A.; Arutyunyan, A.; Moses, A.S.; Avramis, V.I.; Louie, S.G.; Butturini, A.; Heisterkamp, N.; et al. Adipocytes impair leukemia treatment in mice. Cancer Res. 2009, 69, 7867–7874. [Google Scholar] [CrossRef] [PubMed]
- Nga, M.E.; Swe, N.N.; Chen, K.T.; Shen, L.; Lilly, M.B.; Chan, S.P.; Salto-Tellez, M.; Das, K. PIM-1 kinase expression in adipocytic neoplasms: Diagnostic and biological implications. Int. J. Exp. Pathol. 2010, 91, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Rajah, T.; Chow, S.C. The inhibition of human T cell proliferation by the caspase inhibitor z-VAD-FMK is mediated through oxidative stress. Toxicol. Appl. Pharmacol. 2014, 278, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Sahu, R.P.; Srivastava, S.K. The Role of STAT-3 in the Induction of Apoptosis in Pancreatic Cancer Cells by Benzyl Isothiocyanate. J. Natl. Cancer Inst. 2009, 101, 176–193. [Google Scholar]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.A.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef]
- Gowans, G.J.; Hardie, D.G. AMPK: A cellular energy sensor primarily regulated by AMP. Biochem. Soc. Trans. 2014, 42, 71–75. [Google Scholar] [CrossRef]
- Arczmar, G.S.; Arbeit, J.M.; Toy, B.J.; Speder, A.; Weiner, M.W. Selective depletion of tumor ATP by 2-deoxyglucose and insulin, detected by 31P magnetic resonance spectroscopy. Cancer Res. 1992, 52, 71–76. [Google Scholar]
- Yang, Q.; Chen, L.S.; Neelapu, S.S.; Miranda, R.N.; Medeiros, L.J.; Gandhi, V. Transcription and translation are primary targets of Pim kinase inhibitor SGI-1776 in mantle cell lymphoma. Blood 2012, 120, 3491–3500. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, B.A.; Altman, J.K.; Kaplan, J.B.; Ossenkoppele, G.; Swords, R.; Platanias, L.C.; Giles, F.J. Targeted therapy of acute myeloid leukemia. Expert Rev. Anticancer Ther. 2015, 15, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Cobb, M.G.; Gavilano, L.; Witherspoon, S.M.; Williams, D.; White, C.D.; Taverna, P.; Bednarski, B.K.; Kim, H.J.; Baldwin, A.S.; et al. Inhibition of oncogenic Pim-3 kinase modulates transformed growth and chemosensitizes pancreatic cancer cells to gemcitabine. Cancer Biol. Ther. 2013, 14, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.D.; Black, J.; Futer, O.; Swenson, L.; Hare, B.; Fleming, M.; Saxena, K. Pim-1 ligand-bound structures reveal the mechanism of serine/threonine kinase inhibition by LY294002. J. Biol. Chem. 2005, 280, 13728–13734. [Google Scholar] [CrossRef]
- More, K.N.; Jang, H.W.; Hong, V.S.; Lee, J. Pim kinase inhibitory and antiproliferative activity of a novel series of meridianin C derivatives. Bioorg. Med. Chem. Lett. 2014, 24, 2424–2428. [Google Scholar] [CrossRef] [PubMed]
- Ihle, J.N. STATs: Signal transducers and activators of transcription. Cell 1996, 84, 331–334. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr. STATs and gene regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Benekli, M.; Baer, M.R.; Baumann, H.; Wetzler, M. Signal transducer and activator of transcription proteins in leukemias. Blood 2003, 101, 2940–2954. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, C.; Tyagi, A.; Kaur, M.; Agarwal, R. Silibinin inhibits constitutive activation of Stat3, and causes caspase activation and apoptotic death of human prostate carcinoma DU145 cells. Carcinogenesis 2007, 28, 1463–1470. [Google Scholar] [CrossRef]
- Wang, X.; Goldstein, D.; Crowe, P.J.; Yang, J.L. S3I-201, a Novel STAT3 Inhibitor, Inhibits Growth of Human Soft Tissue Sarcoma Cell Lines. World J. Cancer Res. 2013, 1, 61–68. [Google Scholar] [CrossRef]
- Yue, P.; Turkson, J. Targeting STAT3 in cancer: How successful are we? Expert Opin. Investig. Drugs 2009, 18, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Amit-Vazina, M.; Shishodia, S.; Harris, D.; Van, Q.; Wang, M.; Weber, D.; Alexanian, R.; Talpaz, M.; Aggarwal, B.B.; Estrov, Z. Atiprimod blocks STAT3 phosphorylation and induces apoptosis in multiple myeloma cells. Br. J. Cancer. 2005, 93, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.F.; Kong, L.Y.; Jordan, J.; Conrad, C.; Madden, T.; Fokt, I.; Priebe, W.; Heimberger, A.B. A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res. 2007, 67, 9630–9636. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E., Jr. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer. 2002, 2, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Beharry, Z.M.; Harris, T.E.; Lilly, M.B.; Smith, C.D.; Mahajan, S.; Kraft, A.S. PIM1 protein kinase regulates PRAS40 phosphorylation and mTOR activity in FDCP1 cells. Cancer Biol. Ther. 2009, 8, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yue, P.; Chan, C.B.; Ye, K.; Ueda, T.; Watanabe-Fukunaga, R.; Fukunaga, R.; Fu, H.; Khuri, F.R.; Sun, S.Y. Inhibition of mammalian target of rapamycin induces phosphatidylinositol 3-kinase-dependent and Mnk-mediated eukaryotic translation initiation factor 4E phosphorylation. Mol. Cell. Biol. 2007, 27, 7405–7413. [Google Scholar] [CrossRef]
- Ip, C.K.; Wong, A.S. Exploiting p70 S6 kinase as a target for ovarian cancer. Expert. Opin. Ther. Targets 2012, 16, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Park, J.W.; Lee, J.S.; Lee, S.R.; Jang, B.C.; Suh, S.I. Silibinin inhibits expression of HIF-1alpha through suppression of protein translation in prostate cancer cells. Biochem. Biophys. Res. Commun. 2009, 390, 71–76. [Google Scholar] [CrossRef]
- Hardie, D.G. AMPK: A key regulator of energy balance in the single cell and the whole organism. Int. J. Obes. 2008, 32, S7–12. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef]
- Hou, X.; Xu, S.; Maitland-Toolan, K.A.; Sato, K.; Jiang, B.; Ido, Y.; Lan, F.; Walsh, K.; Wierzbicki, M.; Verbeuren, T.J.; et al. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J. Biol. Chem. 2008, 83, 20015–20026. [Google Scholar] [CrossRef]
- Huang, B.; Cheng, X.; Wang, D.; Peng, M.; Xue, Z.; Da, Y.; Zhang, N.; Yao, Z.; Li, M.; Xu, A.; et al. Adiponectin promotes pancreatic cancer progression by inhibiting apoptosis via the activation of AMPK/Sirt1/PGC-1α signaling. Oncotarget 2014, 5, 4732–4745. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, X.B.; Chen, L.Y.; Huang, L.; Dong, R.Z. Belinostat-induced apoptosis and growth inhibition in pancreatic cancer cells involve activation of TAK1-AMPK signaling axis. Biochem. Biophys. Res. Commun. 2013, 437, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Warfel, N.A.; Kraft, A.S. PIM kinase (and Akt) biology and signaling in tumors. Pharmacol. Ther. 2015, 151, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Park, N.S.; Park, Y.K.; Ramalingam, M.; Yadav, A.K.; Cho, H.R.; Hong, V.S.; More, K.N.; Bae, J.H.; Bishop-Bailey, D.; Kano, J.; et al. Meridianin C inhibits the growth of YD-10B human tongue cancer cells through macropinocytosis and the down-regulation of Dickkopf-related protein-3. J. Cell Mol. Med. 2018, 12, 5833–5846. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yadav, A.K.; Kumar, V.; Bailey, D.B.; Jang, B.-C. AZD1208, a Pan-Pim Kinase Inhibitor, Has Anti-Growth Effect on 93T449 Human Liposarcoma Cells via Control of the Expression and Phosphorylation of Pim-3, mTOR, 4EBP-1, S6, STAT-3 and AMPK. Int. J. Mol. Sci. 2019, 20, 363. https://doi.org/10.3390/ijms20020363
Yadav AK, Kumar V, Bailey DB, Jang B-C. AZD1208, a Pan-Pim Kinase Inhibitor, Has Anti-Growth Effect on 93T449 Human Liposarcoma Cells via Control of the Expression and Phosphorylation of Pim-3, mTOR, 4EBP-1, S6, STAT-3 and AMPK. International Journal of Molecular Sciences. 2019; 20(2):363. https://doi.org/10.3390/ijms20020363
Chicago/Turabian StyleYadav, Anil Kumar, Vinoth Kumar, David Bishop Bailey, and Byeong-Churl Jang. 2019. "AZD1208, a Pan-Pim Kinase Inhibitor, Has Anti-Growth Effect on 93T449 Human Liposarcoma Cells via Control of the Expression and Phosphorylation of Pim-3, mTOR, 4EBP-1, S6, STAT-3 and AMPK" International Journal of Molecular Sciences 20, no. 2: 363. https://doi.org/10.3390/ijms20020363
APA StyleYadav, A. K., Kumar, V., Bailey, D. B., & Jang, B.-C. (2019). AZD1208, a Pan-Pim Kinase Inhibitor, Has Anti-Growth Effect on 93T449 Human Liposarcoma Cells via Control of the Expression and Phosphorylation of Pim-3, mTOR, 4EBP-1, S6, STAT-3 and AMPK. International Journal of Molecular Sciences, 20(2), 363. https://doi.org/10.3390/ijms20020363