De Novo Assembly and Discovery of Genes That Involved in Drought Tolerance in the Common Vetch

Abstract
:1. Introduction
2. Results and Discussion
2.1. Transcriptome Sequencing and De Novo Assembly
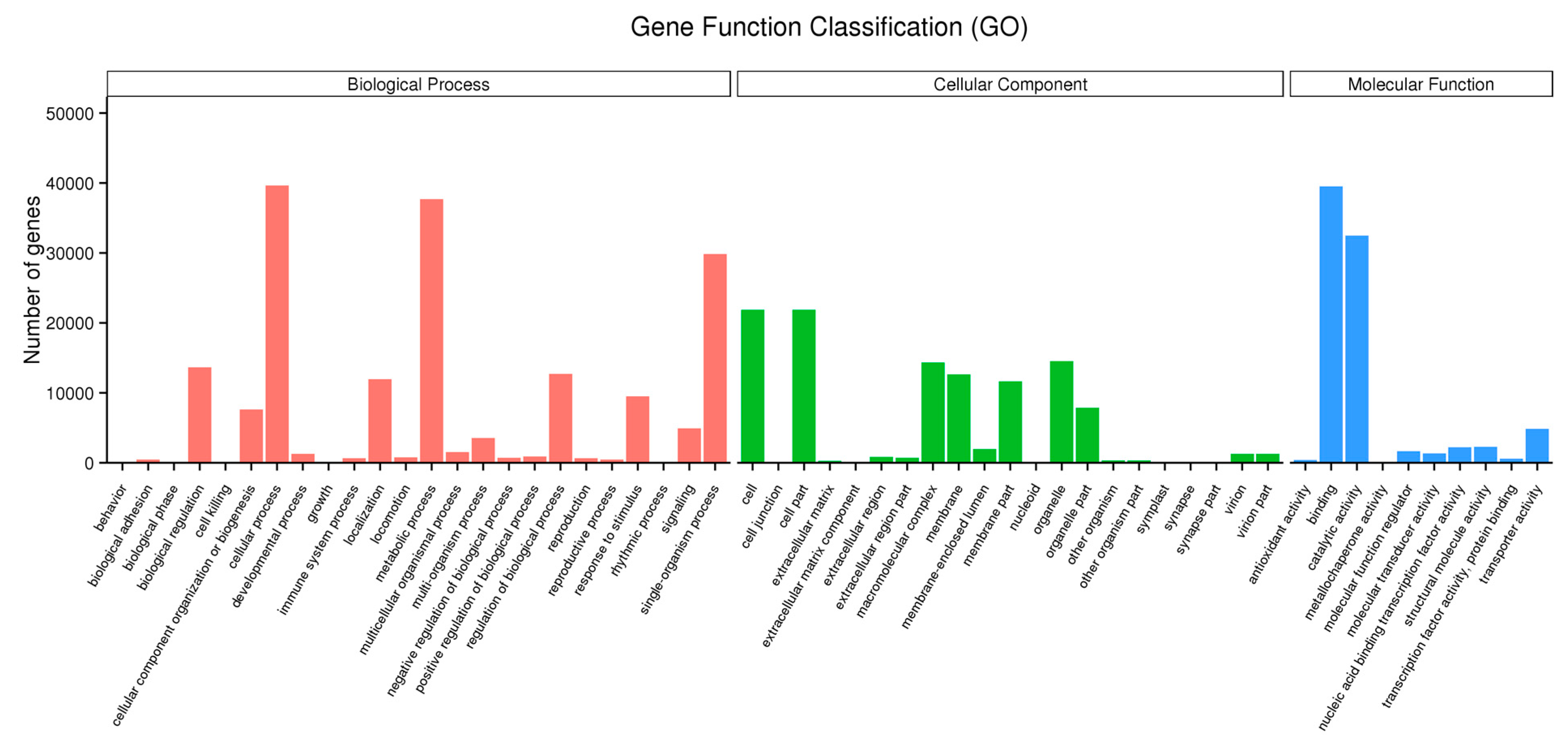
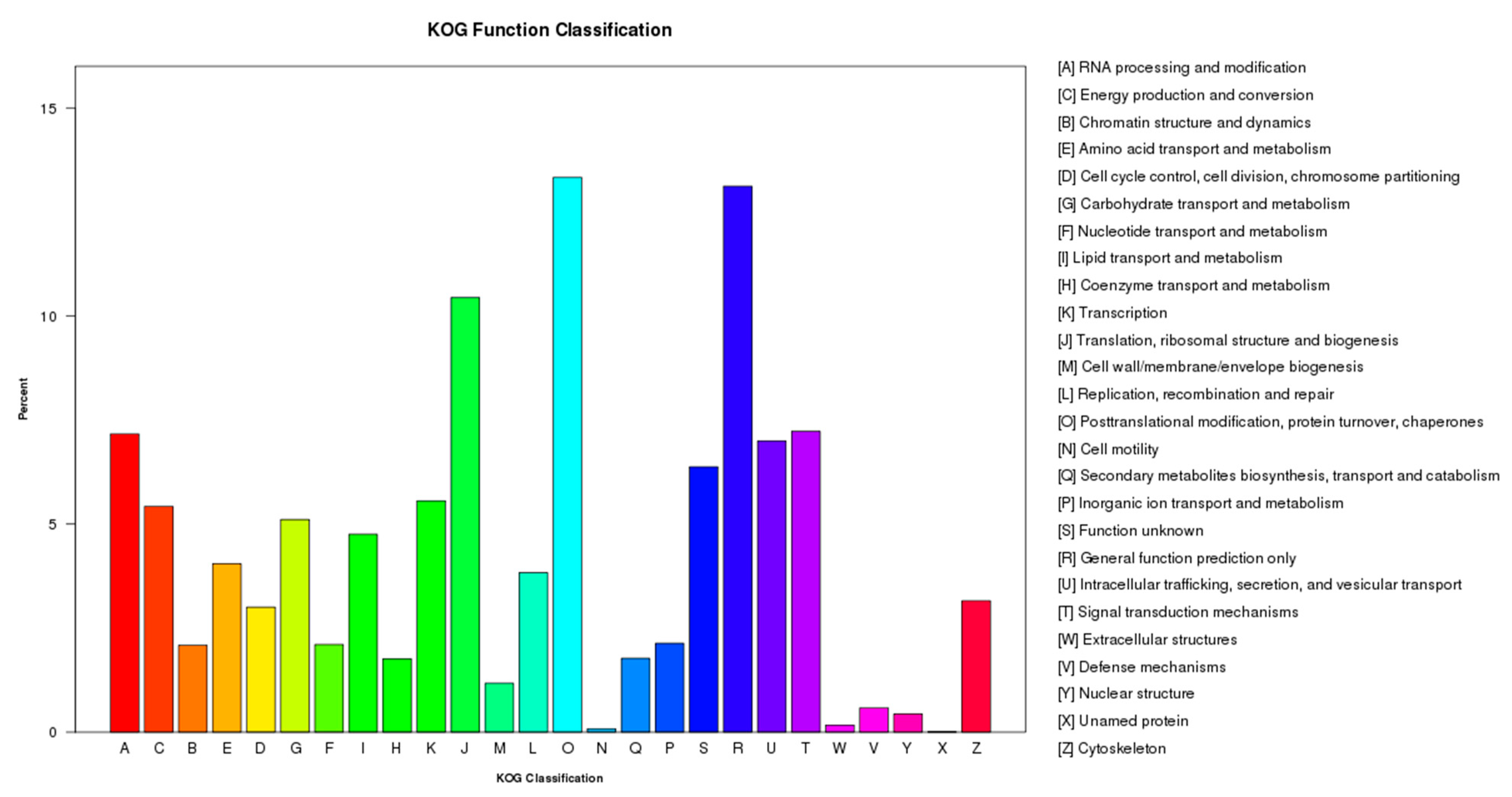
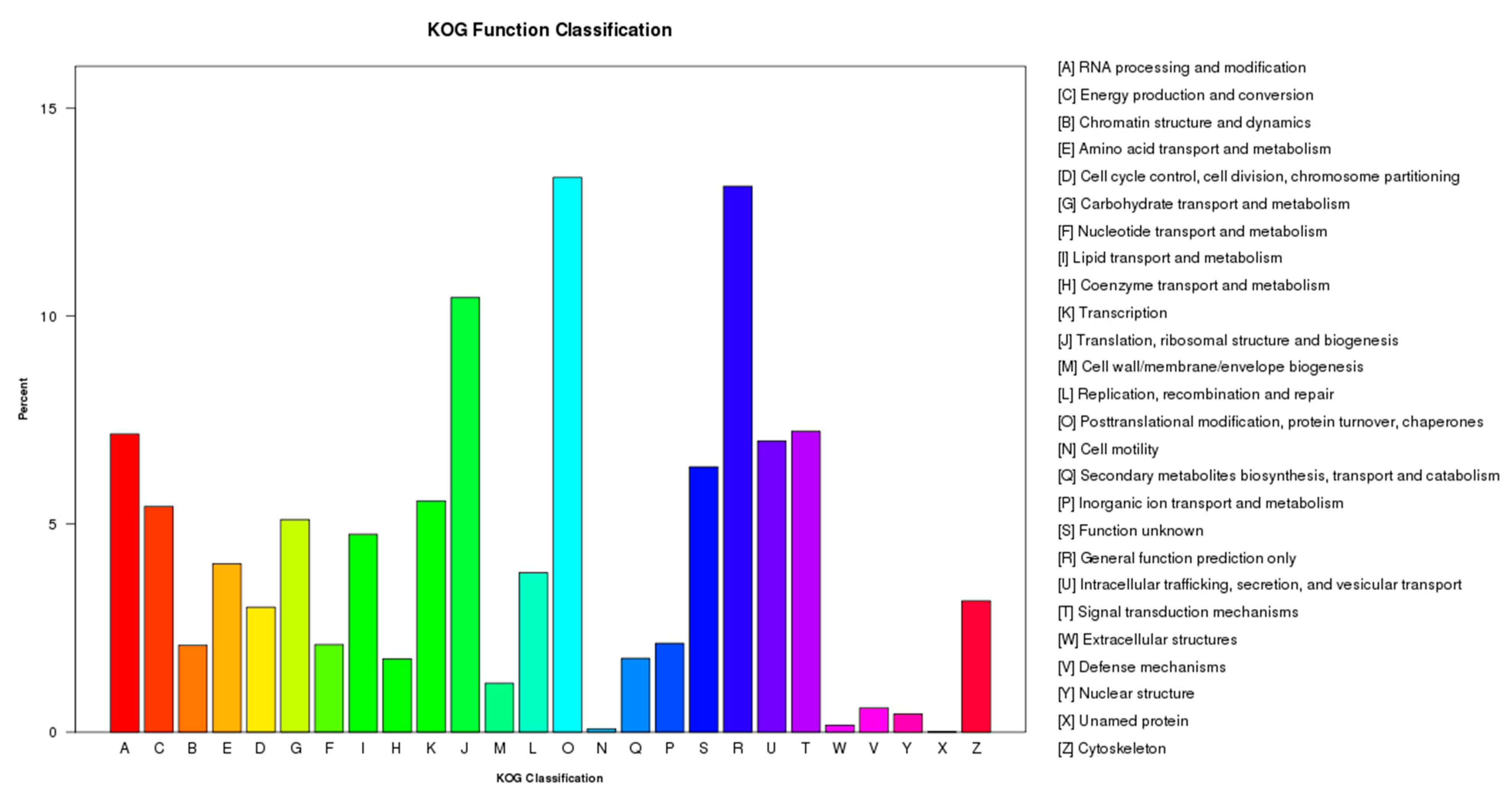
2.2. Functional Annotation and Pathway Assignment of Genes
2.3. Transcription Factors
2.4. SSR Identification
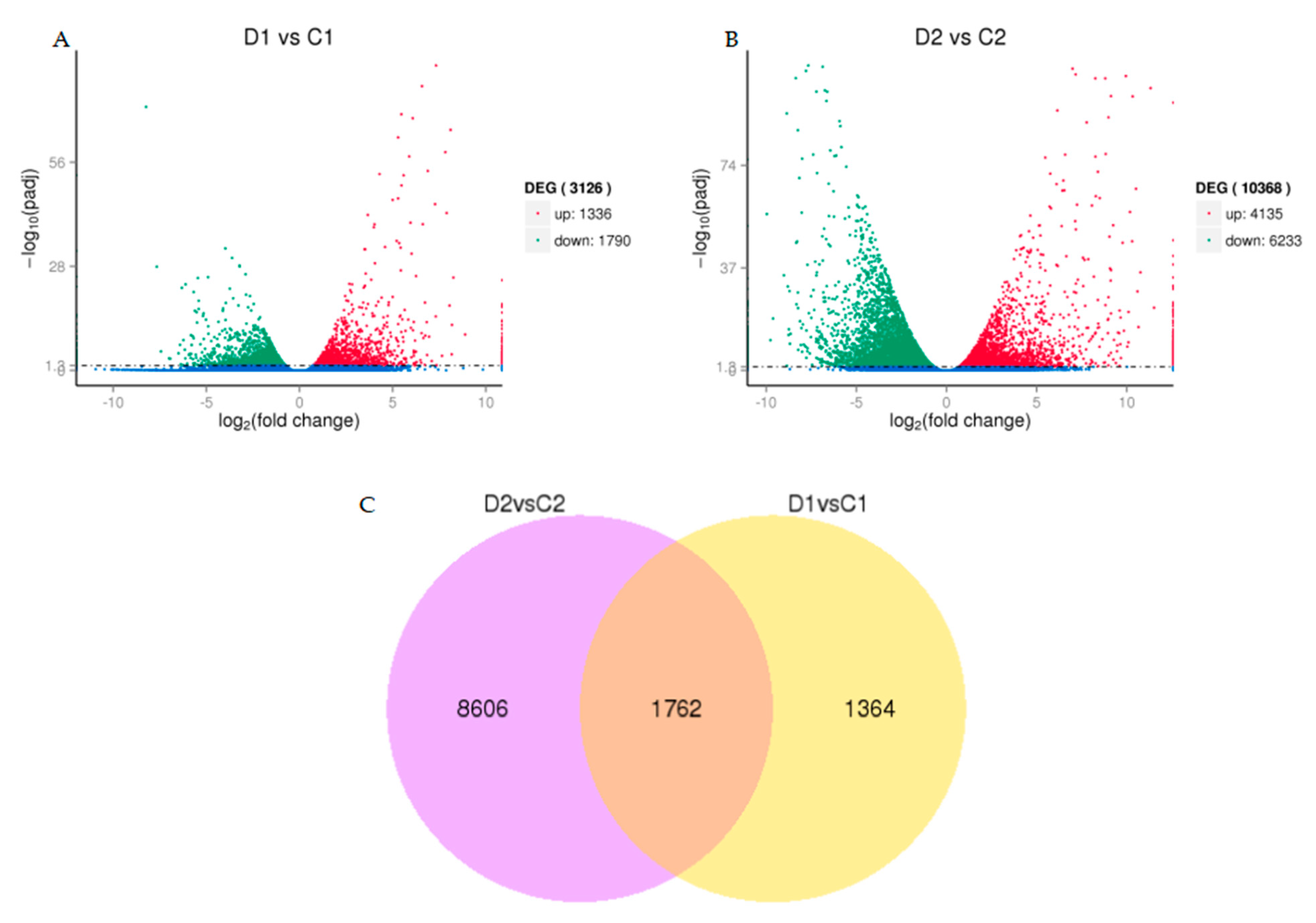
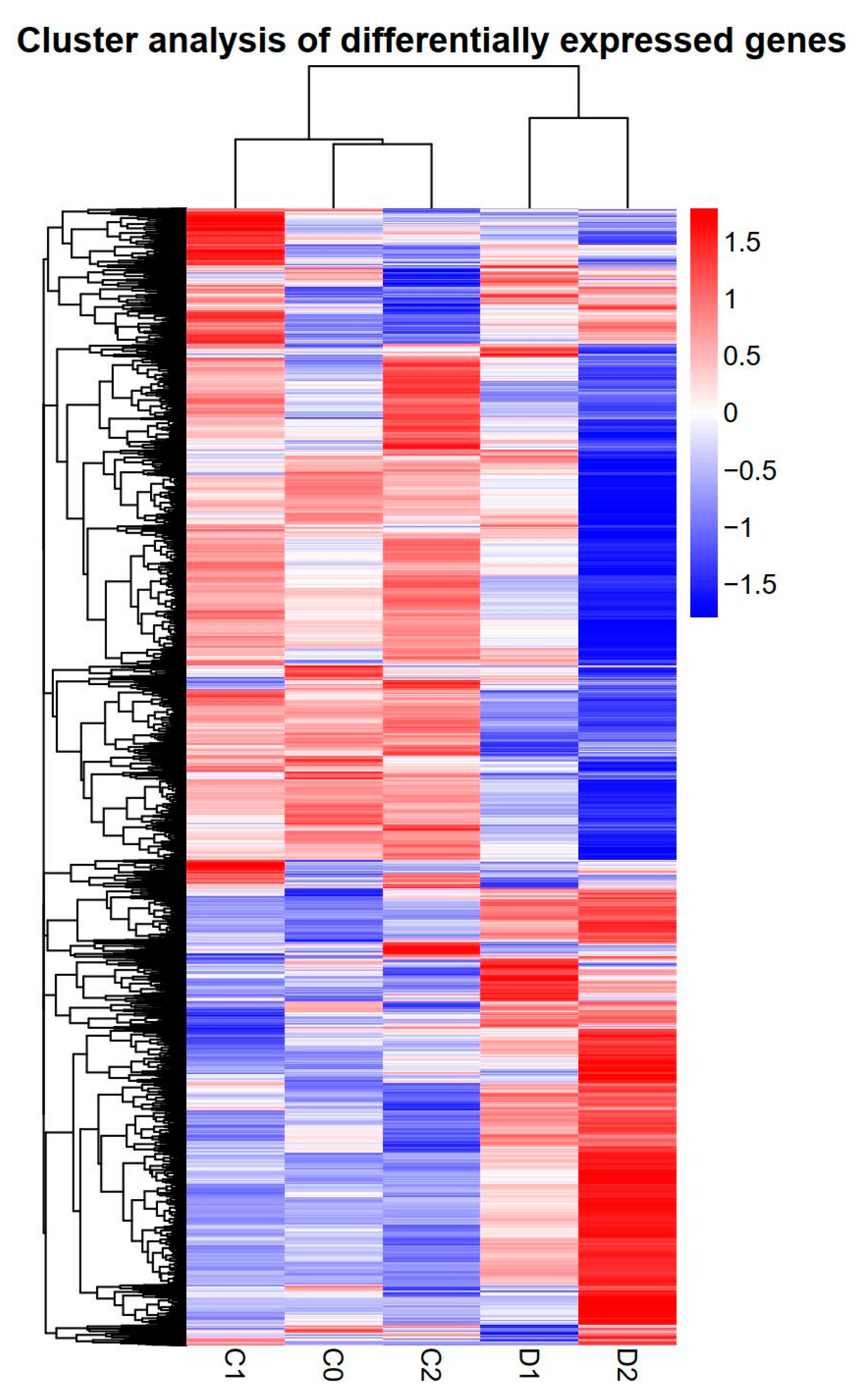
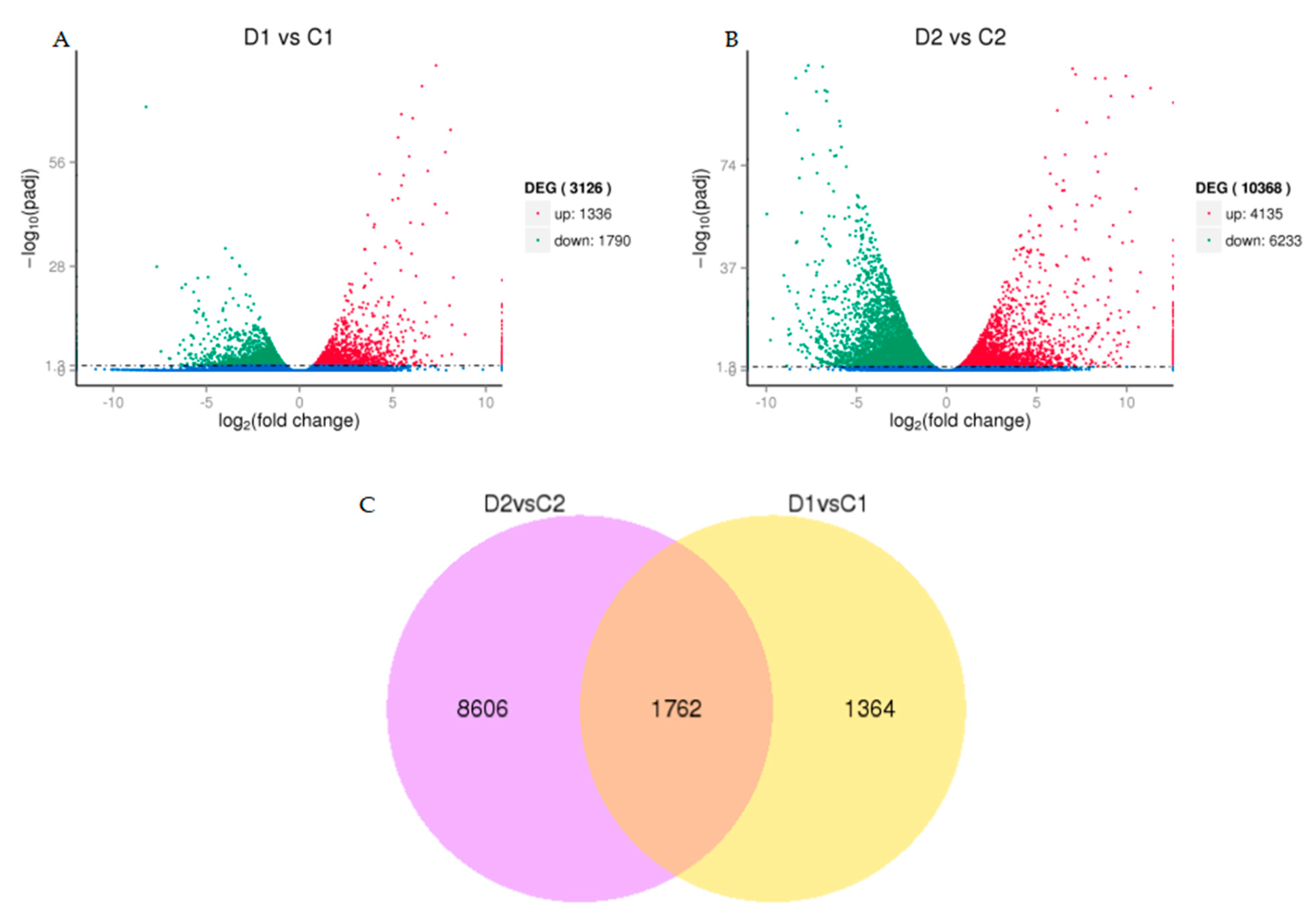
2.5. Differentially Expressed Genes under Drought Stress
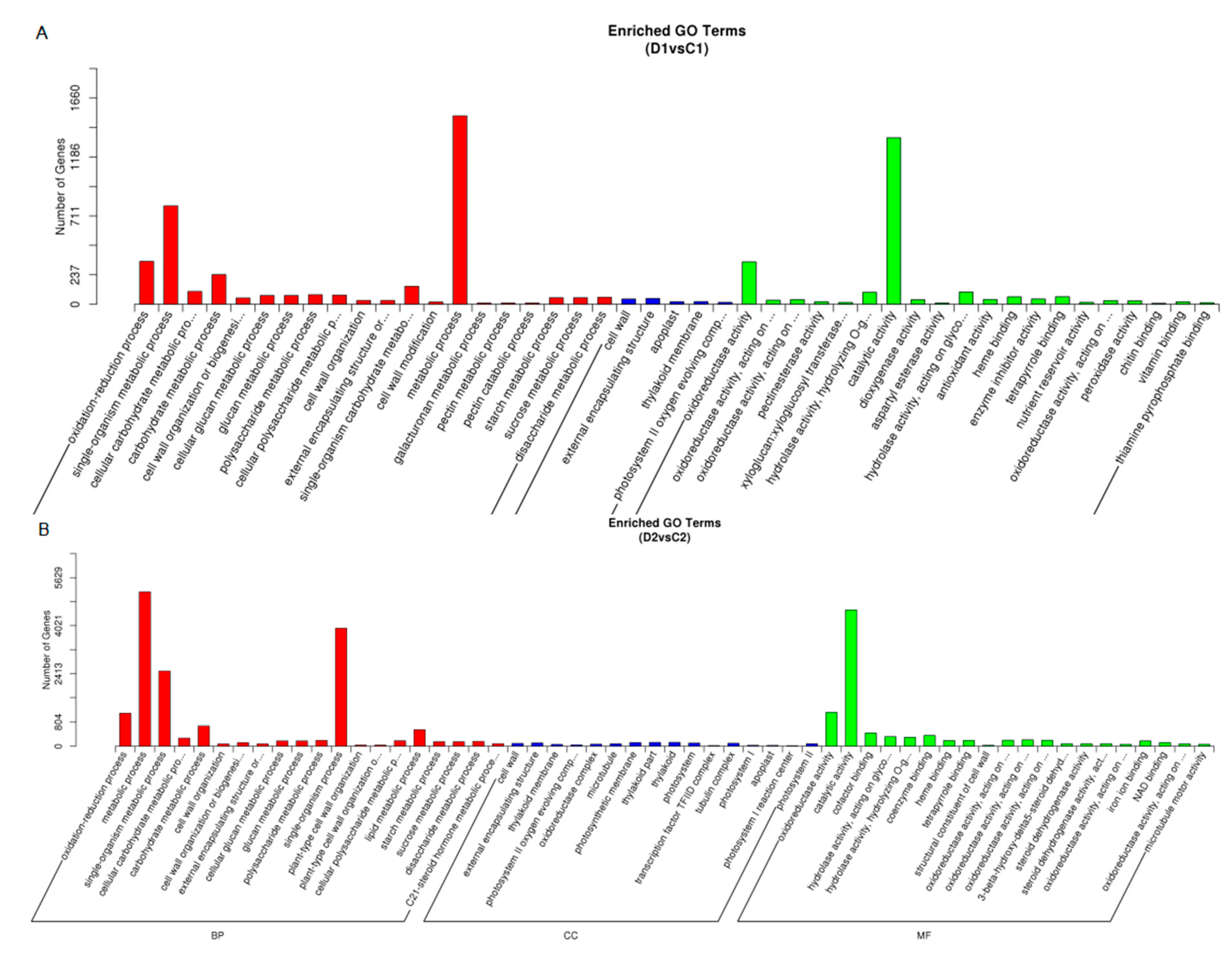
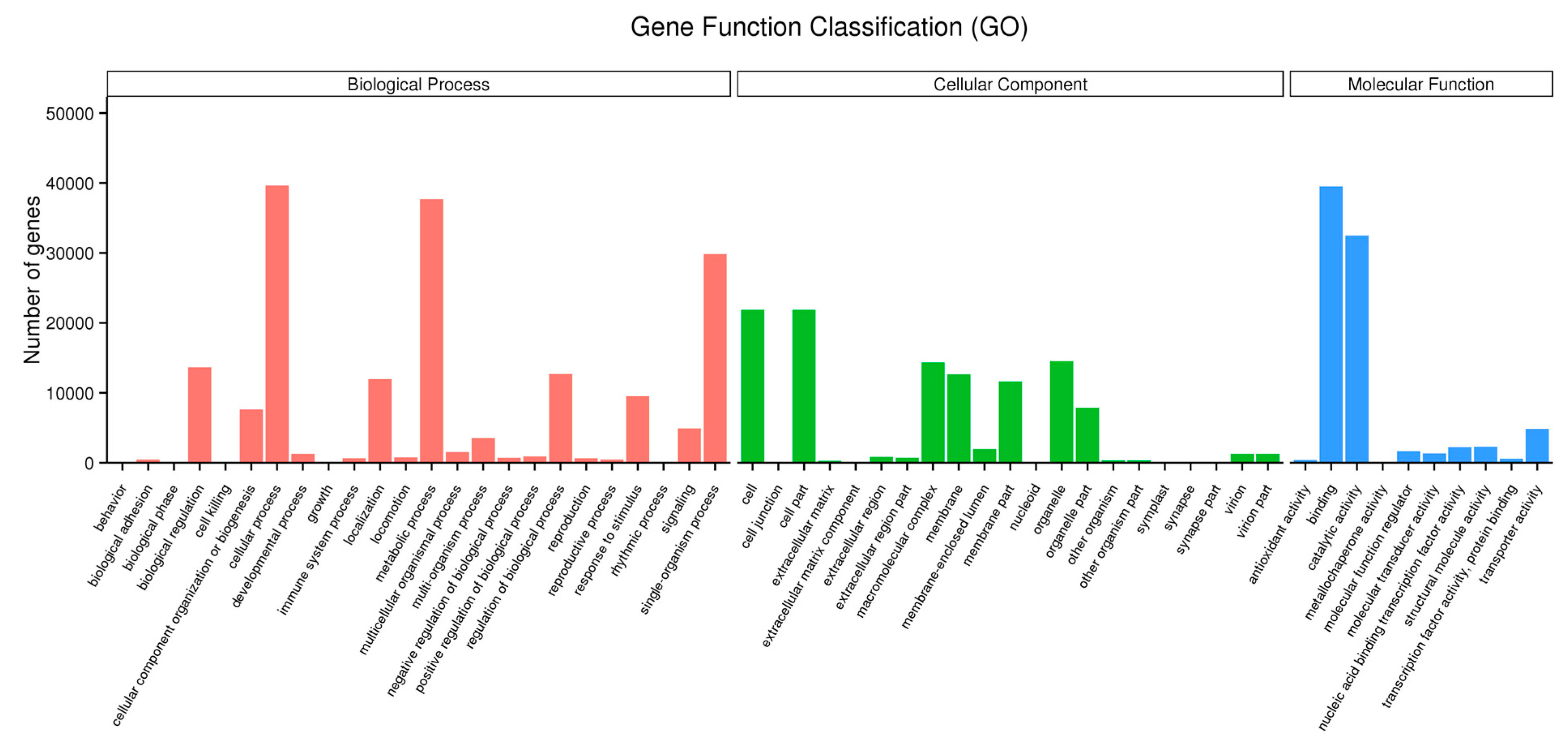
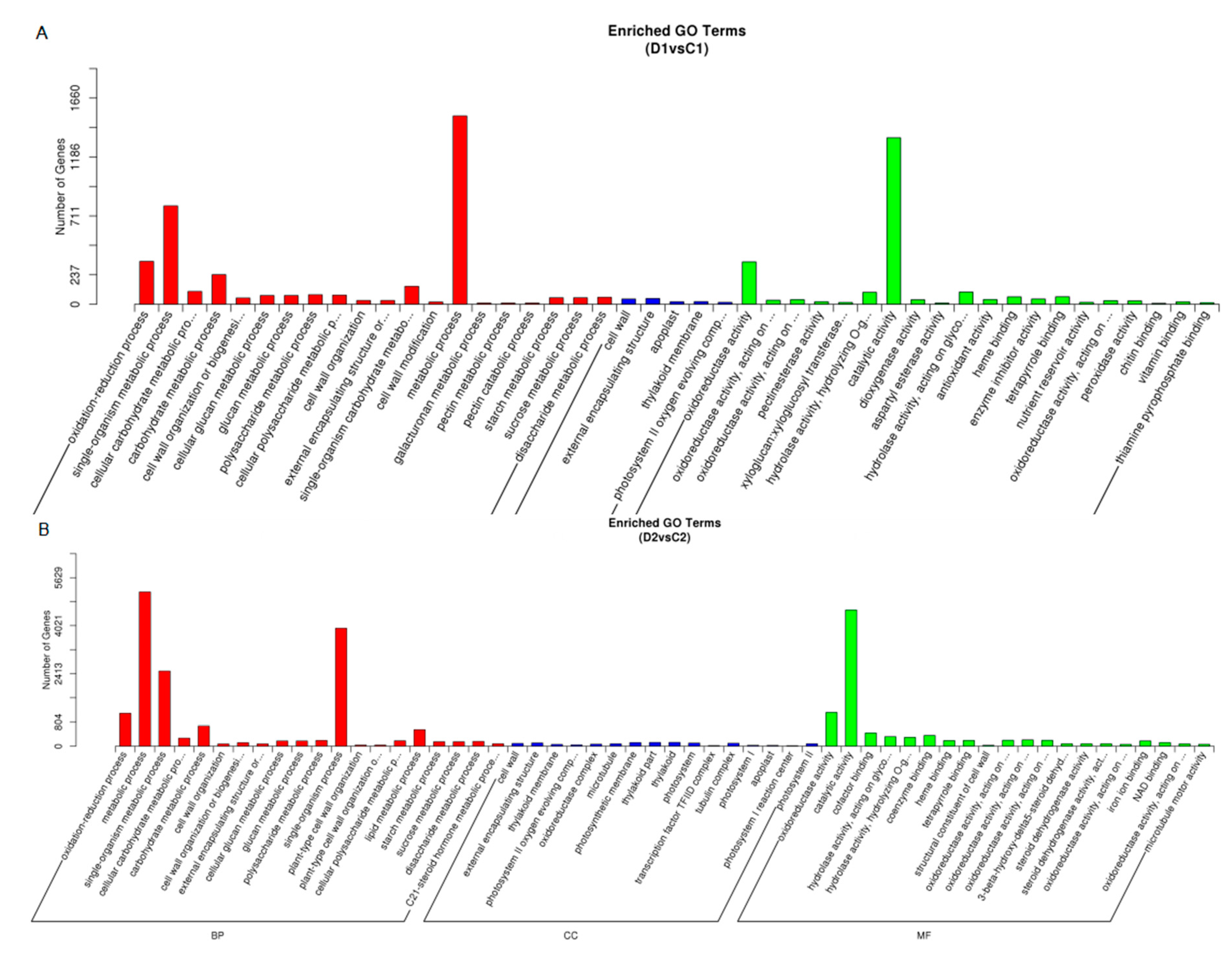
2.6. Functional Classification of the Drought-Responsive Stress Genes using Gene Ontology Analysis
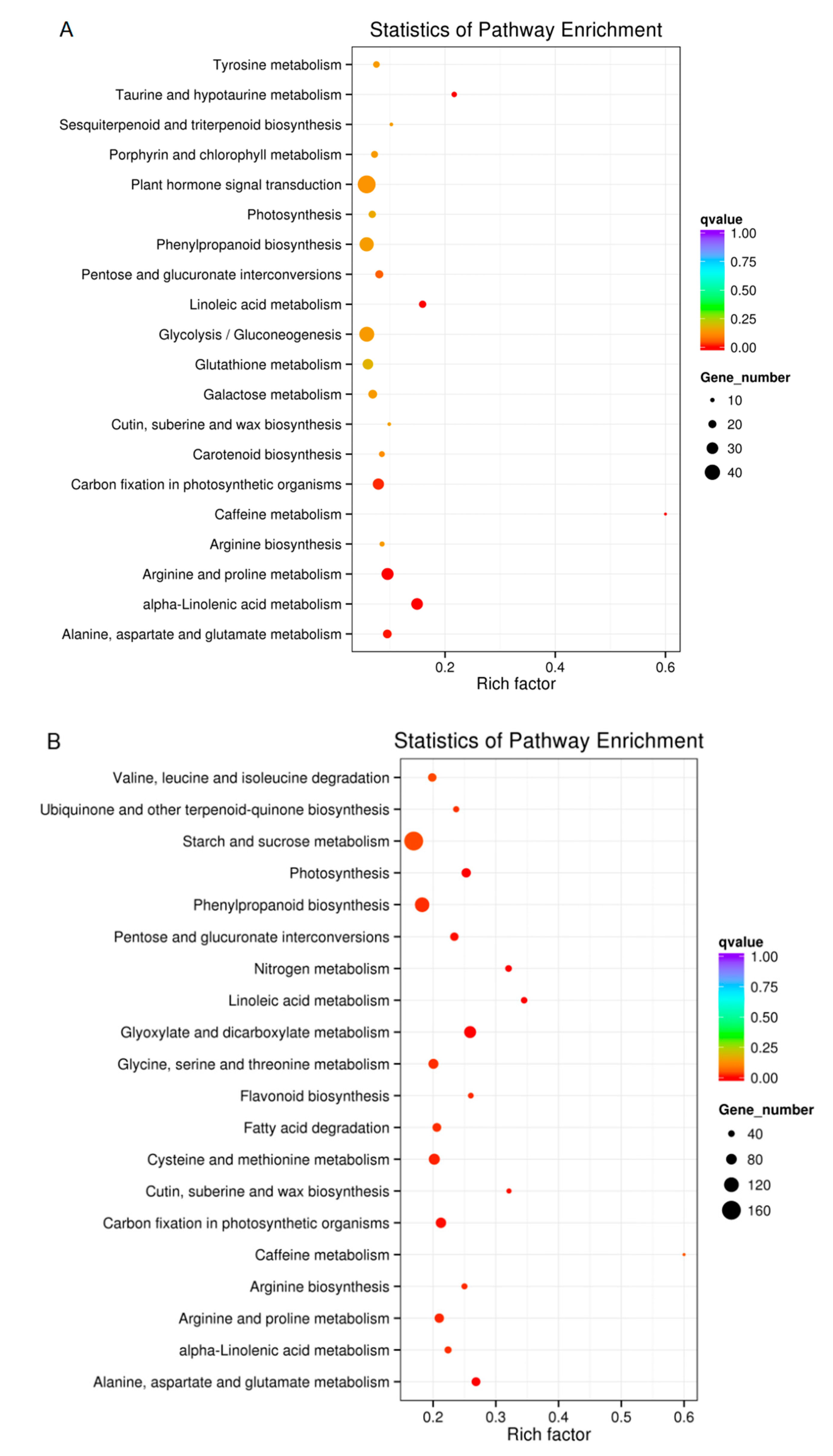
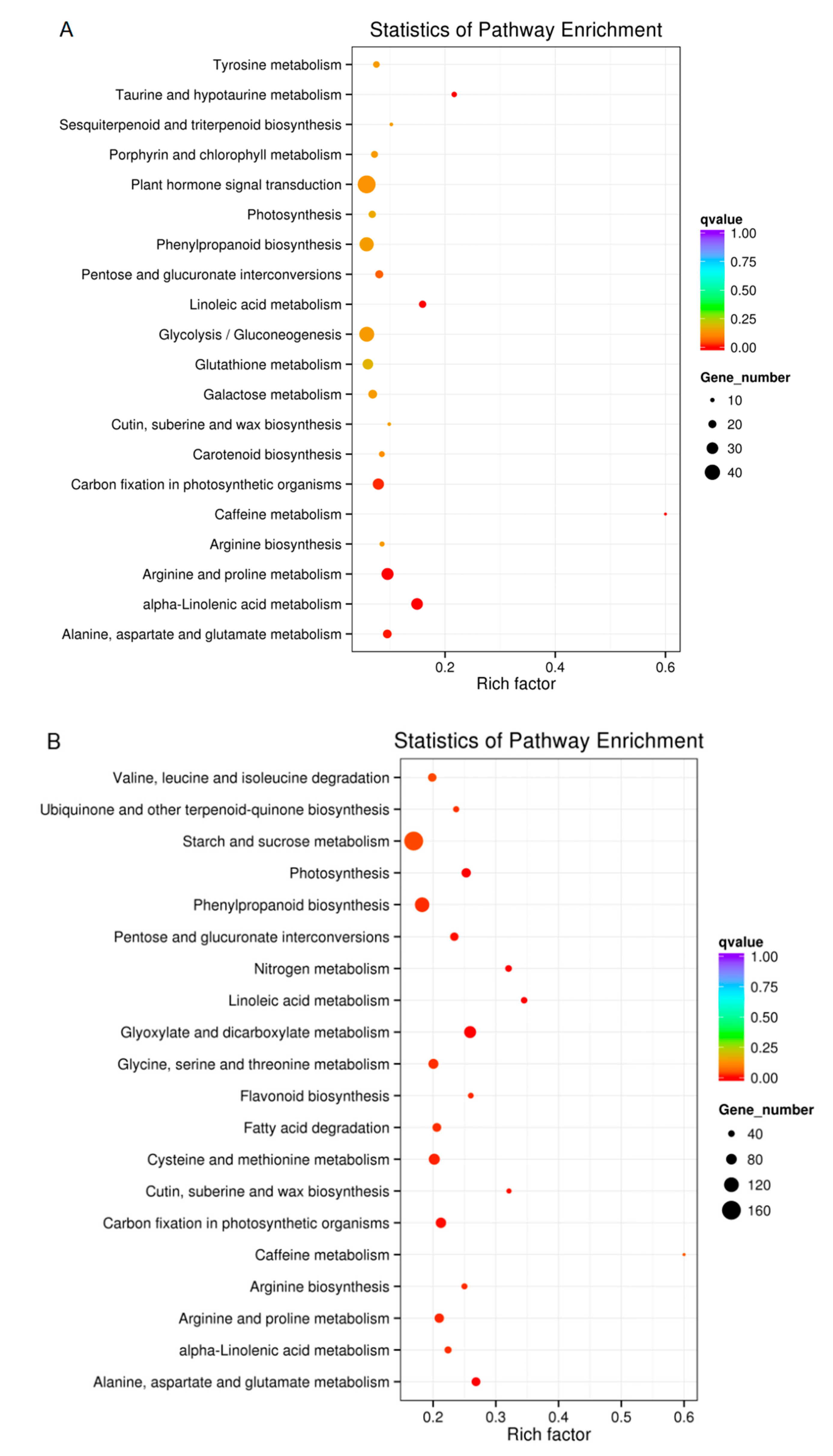
2.7. KEGG Pathway Analysis of DEGs in Plants Exposed to Drought Conditions
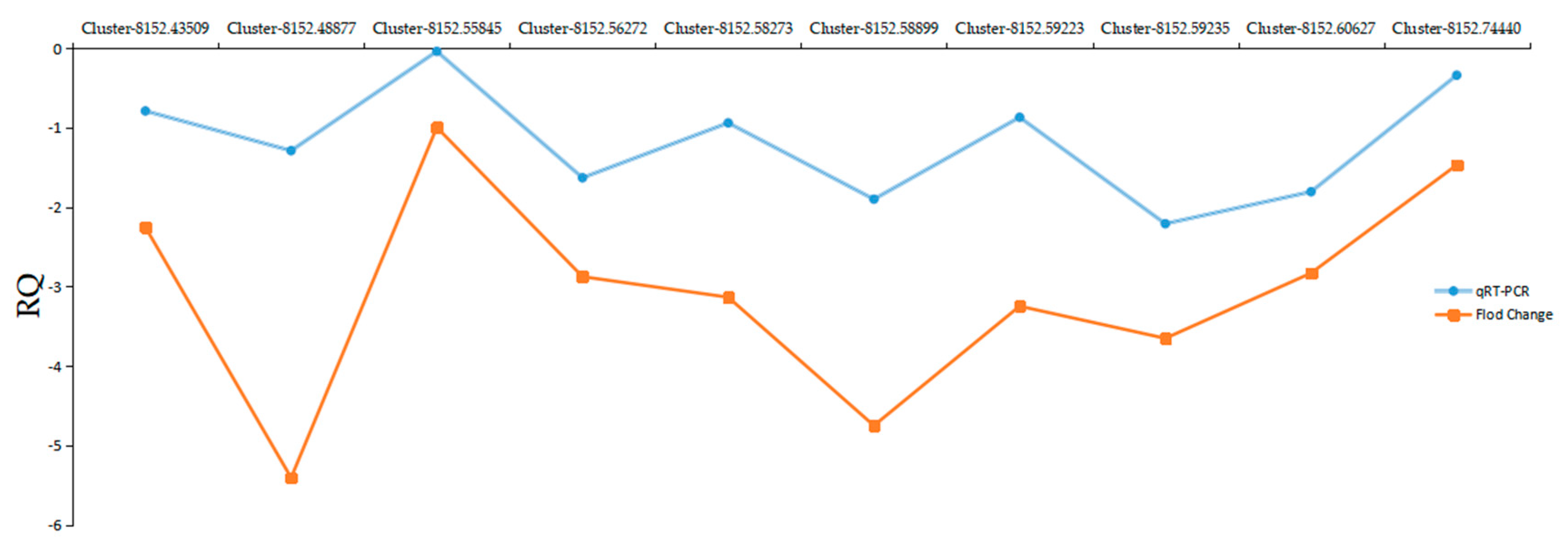
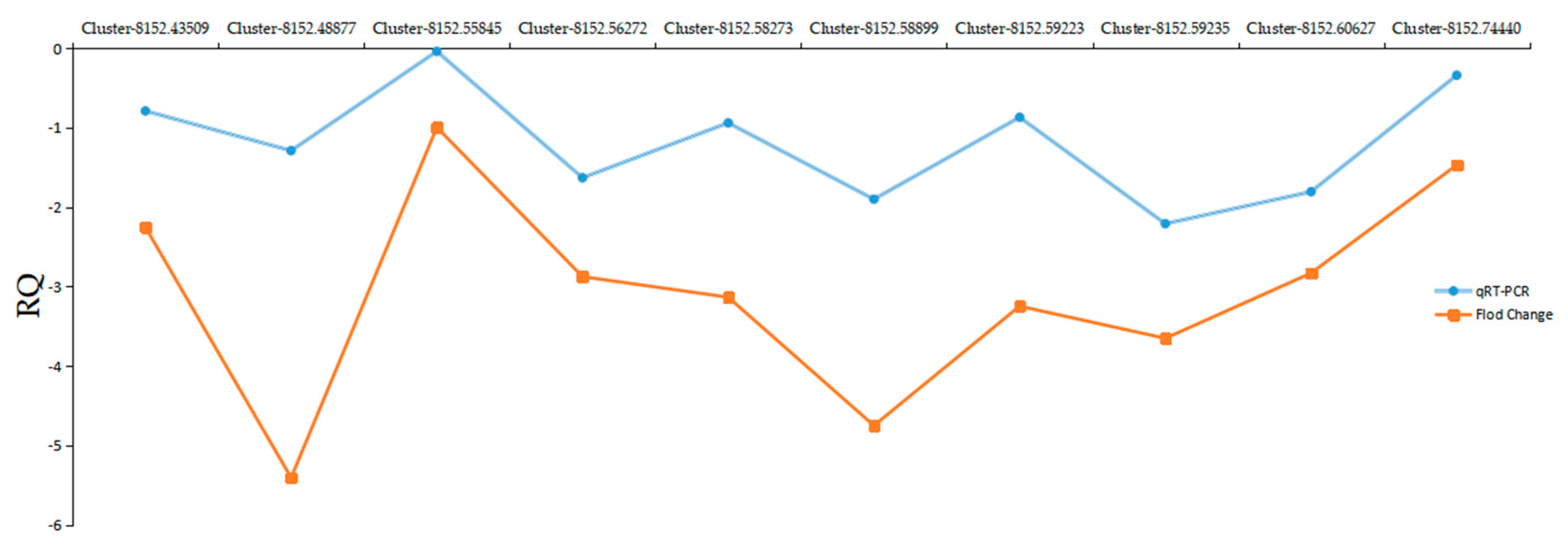
2.8. Quantitative Real-Time-PCR Validation of DEGs from RNA-Seq
3. Materials and Methods
3.1. Plant Material and Drought Treatment
3.2. RNA Extraction
3.3. Library Preparation for Transcriptome Sequencing
3.4. Sequence Read Mapping, Assembly and SSR Detection
3.5. Gene Expression Quantification and Differential Expression Analysis
3.6. Functional Annotation
3.7. Quantitative Real-Time-RCR Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| DGE | Digital Gene Expression |
| DEGs | Differentially Expressed Genes |
| TFs | Transcription factors |
| HPTs | Histidine-containing phosphotansfer proteins |
| PGAM | Putative Phosphoglycerate mutase |
| SCPL | Serine Carboxypeptidase-like |
References
- Mikić, A.; Mihailović, V.; Ćupina, B.; Vasiljević, S.; Milošević, B.; Katanski, S.; Matić, R.; Radojević, V.; Kraljevićbalalić, M. Agronomic characteristics related to grain yield and crude protein content in common vetch (Vicia sativa) accessions of diverse geographic origin. N. Zeal. J. Agric. Res. 2013, 56, 297–308. [Google Scholar] [CrossRef]
- Chung, J.W.; Kim, T.S.; Suresh, S.; Lee, S.Y.; Cho, G.T. Development of 65 novel polymorphic cDNA-SSR markers in common vetch (Vicia sativa subsp. sativa) using next generation sequencing. Molecules 2013, 18, 8376–8392. [Google Scholar] [CrossRef] [PubMed]
- Fırıncıoglu, H.K.; Erbektaş, E.; Dogruyol, L.; Mutlu, Z.; Ünal, S.; Karakurt, E. Phenotypic variation of autumn and spring-sown vetch (Vicia sativa ssp.) populations in central Turkey. Span. J. Agric. Res. 2009, 7, 596–606. [Google Scholar] [CrossRef]
- Chooi, W.Y. Variation in nuclear DNA content in the genus vicia. Genetics 1971, 68, 195–211. [Google Scholar] [PubMed]
- Cakmakci, S.; Aydinoglu, B.; Karaca, M.; Bilgen, M. Heritability of yield components in common vetch (Vicia sativa L.). Acta Agric. Scand. 2006, 56, 54–59. [Google Scholar]
- Dong, R.; Jahufer, M.; Dong, D.K.; Wang, Y.R.; Liu, Z.P. Characterisation of the morphological variation for seed traits among 537 germplasm accessions of common vetch (Vicia sativa L.) using digital image analysis. N. Zeal. J. Agric. Res. 2016, 59, 422–435. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, P.; Luo, D.; Liu, W.; Wang, Y. Exploiting Illumina sequencing for the development of 95 novel polymorphic EST-SSR markers in common vetch (Vicia sativa subsp. sativa). Molecules 2014, 19, 5777–5789. [Google Scholar] [CrossRef]
- Wang, W.; Vinocur, B.; Altman, A. Plant responses to drought, salinity and extreme temperatures: Towards genetic engineering for stress tolerance. Planta 2003, 218, 1–14. [Google Scholar] [CrossRef]
- Shanker, A.K.; Maheswari, M.; Yadav, S.K.; Desai, S.; Bhanu, D.; Attal, N.B.; Venkateswarlu, B. Drought stress responses in crops. Funct. Integr. Genom. 2014, 14, 11–22. [Google Scholar] [CrossRef]
- Golldack, D.; Li, C.; Mohan, H.; Probst, N. Tolerance to drought and salt stress in plants: Unraveling the signaling networks. Front. Plant Sci. 2014, 5, 151. [Google Scholar] [CrossRef]
- Hegedus, Z.; Zakrzewska, A.; Agoston, V.C.; Ordas, A.; Rácz, P.; Mink, M.; Spaink, H.P.; Meijer, A.H. Deep sequencing of the zebrafish transcriptome response to mycobacterium infection. Mol. Immunol. 2009, 46, 2918–2930. [Google Scholar] [CrossRef]
- Tian, D.Q.; Pan, X.Y.; Yu, Y.M.; Wang, W.Y.; Zhang, F.; Ge, Y.Y.; Shen, X.L.; Shen, F.Q.; Liu, X.J. De novo characterization of the Anthurium transcriptome and analysis of its digital gene expression under cold stress. BMC Genom. 2013, 14, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yates, S.A.; Swain, M.T.; Hegarty, M.J.; Chernukin, I.; Lowe, M.; Allison, G.G.; Ruttink, T.; Abberton, M.T.; Jenkins, G.; Skøt, L. De novo assembly of red clover transcriptome based on RNA-Seq data provides insight into drought response, gene discovery and marker identification. BMC Genom. 2014, 15, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.J.; Wei, W.; Song, Q.X.; Chen, H.W.; Zhang, Y.Q.; Wang, F.; Zou, H.F.; Lei, G.; Tian, A.G.; Zhang, W.K. Soybean NAC transcription factors promote abiotic stress tolerance and lateral root formation in transgenic plants. Plant J. 2011, 68, 302–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Dai, M.; Yao, J.; Xiao, B.; Li, X.; Zhang, Q.; Xiong, L. Overexpressing a NAM, ATAF, and CUC (NAC) transcription factor enhances drought resistance and salt tolerance in rice. Proc. Natl. Acad. Sci. USA 2006, 103, 12987–12992. [Google Scholar] [CrossRef] [Green Version]
- Seki, M.; Narusaka, M.; Ishida, J.; Nanjo, T.; Fujita, M.; Oono, Y.; Kamiya, A.; Nakajima, M.; Enju, A.; Sakurai, T. Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and high-salinity stresses using a full-length cDNA microarray. Plant J. 2010, 31, 279–292. [Google Scholar] [CrossRef]
- Seo, J.J.; Youn Shic, K.; Kwang Hun, B.; Harin, J.; Sun-Hwa, H.; Yang, D.C.; Minkyun, K.; Christophe, R.; Ju-Kon, K. Root-specific expression of OsNAC10 improves drought tolerance and grain yield in rice under field drought conditions. Plant Physiol. 2010, 153, 185–197. [Google Scholar]
- Takasaki, H.; Maruyama, K.; Kidokoro, S.; Ito, Y.; Fujita, Y.; Shinozaki, K.; Yamaguchi-Shinozaki, K.; Nakashima, K. The abiotic stress-responsive NAC-type transcription factor OsNAC5 regulates stress-inducible genes and stress tolerance in rice. Mol. Genet. Genom. 2010, 284, 173–183. [Google Scholar] [CrossRef]
- Zhou, Q.Y.; Tian, A.G.; Zou, H.F.; Xie, Z.M.; Lei, G.; Huang, J.; Wang, C.M.; Wang, H.W.; Zhang, J.S.; Chen, S.Y. Soybean WRKY-type transcription factor genes, GmWRKY13, GmWRKY21, and GmWRKY54, confer differential tolerance to abiotic stresses in transgenic Arabidopsis plants. Plant Biotechnol. J. 2010, 6, 486–503. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Gao, S.; Yang, Y.; Huang, M.; Cheng, L.; Wei, Q.; Fei, Z.; Gao, J.; Hong, B. Transcriptome sequencing and whole genome expression profiling of chrysanthemum under dehydration stress. BMC Genom. 2013, 14, 662. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhu, S.; Tang, Q.; Yu, Y.; Tang, S. Identification of drought stress-responsive transcription factors in ramie (Boehmeria nivea L. Gaud). BMC Plant Biol. 2013, 13, 130. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Zhou, X.; Cao, Y.; Zhou, M.; Mcneil, D.; Liang, S.; Yang, C. RNA-seq Analysis of Cold and Drought Responsive Transcriptomes of Zea mays ssp. mexicana L. Front. Plant Sci. 2017, 8, 136. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wang, X.; Huang, L.; Lin, C.; Zhang, X.; Xu, W.; Peng, J.; Li, Z.; Yan, H.; Luo, F. Transcriptomic Identification of Drought-Related Genes and SSR Markers in Sudan Grass Based on RNA-Seq. Front. Plant Sci. 2017, 8, 687. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Jing, S.; Lu, C. Identification of stress-responsive genes in Ammopiptanthus mongolicus using ESTs generated from cold- and drought-stressed seedlings. BMC Plant Biol. 2013, 13, 88. [Google Scholar] [CrossRef] [PubMed]
- Huie, L.; Weijie, Y.; Yaru, F.; Shaoke, L.; Qiqiang, G. De novo assembly and discovery of genes that are involved in drought tolerance in Tibetan Sophora moorcroftiana. PLoS ONE 2015, 10, e111054. [Google Scholar]
- Xiong, L.; Schumaker, K.S.; Zhu, J.K. Cell Signaling during Cold, Drought, and Salt Stress. Plant Cell 2002, 14 (Suppl. 1), S165. [Google Scholar] [CrossRef]
- Hwang, I.; Chen, H.C.; Sheen, J. Two-component signal transduction pathways in Arabidopsis. Plant Physiol. 2002, 129, 500. [Google Scholar] [CrossRef]
- Fran Oise, C.; Hélène, B.; Christiane, D.; Francis, D.; Domenico, M.; Sabine, C. Osmotic stress sensing in Populus: Components identification of a phosphorelay system. FEBS Lett. 2006, 580, 77–81. [Google Scholar]
- Broeckling, C.D.; Huhman, D.V.; Farag, M.A.; Smith, J.T.; May, G.D.; Mendes, P.; Dixon, R.A.; Sumner, L.W. Metabolic profiling of Medicago truncatula cell cultures reveals the effects of biotic and abiotic elicitors on metabolism. J. Exp. Bot. 2005, 56, 323–336. [Google Scholar] [CrossRef]
- Kovács, G.; Sorvari, S.; Scott, P.; Toldi, O. Pyrophosphate: Fructose 6-phosphate 1-phosphotransferase operates in net gluconeogenic direction in taproots of cold and drought stressed carrot plants. Acta Biol. Szeged 2006, 50, 25–30. [Google Scholar]
- Shaar-Moshe, L.; Hübner, S.; Peleg, Z. Identification of conserved drought-adaptive genes using a cross-species meta-analysis approach. BMC Plant Biol. 2015, 15, 111. [Google Scholar] [CrossRef] [PubMed]
- Guzman, F.; Almerão, M.P.; Körbes, A.P.; Loss-Morais, G.; Margis, R. Identification of MicroRNAs from Eugenia uniflora by High-Throughput Sequencing and Bioinformatics Analysis. PLoS ONE 2012, 7, e49811. [Google Scholar] [CrossRef] [PubMed]
- Cramer, G.R.; Sluyter, S.C.V.; Hopper, D.W.; Pascovici, D.; Keighley, T.; Haynes, P.A. Proteomic analysis indicates massive changes in metabolism prior to the inhibition of growth and photosynthesis of grapevine (Vitis vinifera L.) in response to water deficit. BMC Plant Biol. 2013, 13, 49. [Google Scholar] [CrossRef] [PubMed]
- Liebo, S.; Qiaojun, L.; Chenfei, M.; Wei, D.; Jia, Z.; Jinhong, W.; Fangjun, F.; Xin, L.; Lijun, L.; Guowang, X. Genetic, proteomic and metabolic analysis of the regulation of energy storage in rice seedlings in response to drought. Proteomics 2011, 11, 4122–4138. [Google Scholar] [Green Version]
- Tsuneaki, A.; Guillaume, T.; Joulia, P.; Willmann, M.R.; Wan-Ling, C.; Lourdes, G.G.; Thomas, B.; Ausubel, F.M.; Jen, S. MAP kinase signalling cascade in Arabidopsis innate immunity. Nature 2002, 415, 977–983. [Google Scholar]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11. [Google Scholar] [CrossRef] [PubMed]
- Marco, S.; Donaldson, J.G. Constitutive internalization of G protein-coupled receptors and G proteins via clathrin-independent endocytosis. J. Biol. Chem. 2009, 284, 3577–3585. [Google Scholar]
- Long, R.M.; Rodney, C. Preliminary assessment of the C13-side chain 2′-hydroxylase involved in taxol biosynthesis. Biochem. Biophys. Res. Commun. 2005, 338, 410–417. [Google Scholar] [CrossRef]
- Kevin, W.; Robert, L.; Rodney, C. The final acylation step in taxol biosynthesis: Cloning of the taxoid C13-side-chain N-benzoyltransferase from Taxus. Proc. Natl. Acad. Sci. USA 2002, 99, 9166–9171. [Google Scholar]
- Davies, P.J. The Plant Hormones: Their Nature, Occurrence, and Functions. In Plant Hormones; Springer: Dordrecht, The Netherlands, 2010; pp. 1–15. [Google Scholar]
- Aaron, S.; Mark, E. Recent advances and emerging trends in plant hormone signalling. Nature 2009, 459, 1071–1078. [Google Scholar]
- Nemhauser, J.L.; Hong, F.; Chory, J. Different Plant Hormones Regulate Similar Processes through Largely Nonoverlapping Transcriptional Responses. Cell 2006, 126, 467–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimoto, H.; Kondo, S.; Tanaka, T.; Imamura, C.; Muramoto, N.; Hattori, E.; Ogawa, K.; Mitsukawa, N.; Ohto, C. Overexpression of a novel Arabidopsis PP2C isoform, AtPP2CF1, enhances plant biomass production by increasing inflorescence stem growth. J. Exp. Bot. 2014, 65, 5385–5400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahlbrock, K.; Scheel, D. Physiology and molecular biology of phenylpropanoid metabolism. Ann. Rev. Plant Biol. 1989, 40, 347–369. [Google Scholar] [CrossRef]
- Bamforth, C.W.; Martin, H.L.; Wainwright, T. A role for carboxypeptidase in solubilization of barley β-glucan. J. Inst. Brew. 2013, 85, 334–338. [Google Scholar] [CrossRef]
- Bradley, D. Isolation and Characterization of a Gene Encoding a Carboxypeptidase Y-Like Protein from Arabidopsis thaliana. Plant Physiol. 1992, 98, 1526–1529. [Google Scholar] [CrossRef]
- Walker-Simmons, M.; Ryan, C.A. Isolation and properties of carboxypeptidase from leaves of wounded tomato plants. Phytochemistry 1980, 19, 43–47. [Google Scholar] [CrossRef]
- Washio, K.; Ishikawa, K. Organ-Specific and Hormone-Dependent Expression of Genes for Serine Carboxypeptidases during Development and Following Germination of Rice Grains. Plant Physiol. 1994, 105, 1275–1280. [Google Scholar] [CrossRef] [PubMed]
- Potts, J.R.M.; Weklych, R.; Conn, E.E. The 4-hydroxylation of cinnamic acid by sorghum microsomes and the requirement for cytochrome P-450. J. Biol. Chem. 1974, 249, 5019–5026. [Google Scholar]
- Russell, D.W. The metabolism of aromatic compounds in higher plants X. Properties of the cinnamic acid 4-hydroxylase of pea seedlings and some aspects of its metabolic and developmental control. J. Biol. Chem. 1971, 246, 3870–3878. [Google Scholar]
- Yannick, B.; Schalk, M.; Marie-Agnès, P.; Alfred, Z.; Durst, F.; Werck-Reichhart, D. Regulation of the Cinnamate 4-Hydroxylase (CYP73A1) in Jerusalem Artichoke Tubers in Response to Wounding and Chemical Treatments. Plant Physiol. 1997, 113, 951–959. [Google Scholar] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Moran, Y.; Levin, J.Z.; Thompson, D.A.; Ido, A.; Xian, A.; Lin, F.; Raktima, R.; Qiandong, Z. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Sch Ffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Minoru, K.; Michihiro, A.; Susumu, G.; Masahiro, H.; Mika, H.; Masumi, I.; Toshiaki, K.; Shuichi, K.; Shujiro, O.; Toshiaki, T. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, 480–484. [Google Scholar]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gategory | C0 | C1 | C2 | D1 | D2 | Total | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| C0a | C0b | C1a | C1b | C2a | C2b | D1a | D1b | D2a | D2b | ||
| Raw reads | 50,421,376 | 52,941,226 | 54,578,760 | 46,387,122 | 54,257,670 | 44,592,212 | 54,615,492 | 54,797,818 | 54,777,004 | 47,989,120 | 515,357,800 |
| Clean reads | 483,648,36 | 50,722,480 | 52,325,294 | 44,492,586 | 52,050,600 | 42,819,464 | 52,423,694 | 52,648,460 | 52,609,898 | 46,059,876 | 494,517,188 |
| Accounting | 95.92% | 95.81% | 95.87% | 95.92% | 95.93% | 96.02% | 95.99% | 96.08% | 96.04% | 95.98% | 95.96% |
| Clean bases (G) | 7.25 | 7.61 | 7.85 | 6.67 | 7.81 | 6.42 | 7.86 | 7.9 | 7.89 | 6.91 | 74.17 |
| Q20 (%) | 95.97 | 96.03 | 95.89 | 95.59 | 95.75 | 95.63 | 95.71 | 95.67 | 95.61 | 95.56 | - |
| Q30 (%) | 90 | 90.12 | 89.89 | 89.31 | 89.61 | 89.34 | 89.53 | 89.4 | 89.34 | 89.25 | - |
| GC (%) | 42.85 | 42.98 | 42.61 | 42.63 | 42.66 | 42.68 | 42.61 | 42.83 | 42.51 | 42.46 | - |
| Error (%) | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | - |
| Nucleotides Length (bp) | Transcripts | Unigenes |
|---|---|---|
| 200–500 | 75,068 | 23,874 |
| 500–1000 | 32,116 | 30,990 |
| 1000–2000 | 36,224 | 36,207 |
| >2000 | 31,228 | 31,228 |
| Total | 174,636 | 122,299 |
| Minimal length | 201 | 201 |
| Maximal length | 16,722 | 16,722 |
| Median length | 650 | 1142 |
| Average length | 1124 | 1483 |
| N50 | 1991 | 2127 |
| N90 | 442 | 722 |
| Public Database | Number of Unigenes | Percentage (%) |
|---|---|---|
| Annotated in Nr | 90,190 | 73.74 |
| Annotated in Nt | 90,947 | 74.36 |
| Annotated in KEGG | 37,056 | 30.29 |
| Annotated in Swiss-Prot | 71,241 | 58.25 |
| Annotated in Pfam | 65,975 | 53.94 |
| Annotated in GO | 67,889 | 55.51 |
| Annotated in KOG | 27,407 | 22.4 |
| Annotated in all Databases | 16,574 | 13.55 |
| Annotated in at least one Database | 102,106 | 83.48 |
| All assembled Unigenes | 122,299 | 100 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Y.; Liu, Q.; Xu, W.; Zhang, J.; Wang, X.; Nie, G.; Yao, L.; Wang, H.; Lin, C. De Novo Assembly and Discovery of Genes That Involved in Drought Tolerance in the Common Vetch. Int. J. Mol. Sci. 2019, 20, 328. https://doi.org/10.3390/ijms20020328
Zhu Y, Liu Q, Xu W, Zhang J, Wang X, Nie G, Yao L, Wang H, Lin C. De Novo Assembly and Discovery of Genes That Involved in Drought Tolerance in the Common Vetch. International Journal of Molecular Sciences. 2019; 20(2):328. https://doi.org/10.3390/ijms20020328
Chicago/Turabian StyleZhu, Yongqun, Qiuxu Liu, Wenzhi Xu, Jianhua Zhang, Xie Wang, Gang Nie, Li Yao, Hong Wang, and Chaowen Lin. 2019. "De Novo Assembly and Discovery of Genes That Involved in Drought Tolerance in the Common Vetch" International Journal of Molecular Sciences 20, no. 2: 328. https://doi.org/10.3390/ijms20020328