Recent Advances on Microbiota Involvement in the Pathogenesis of Autoimmunity



Abstract

1. Introduction
2. Microbiome Composition and Autoimmune Conditions
2.1. Type 1 Diabetes
2.2. Rheumatoid Arthritis
2.3. Systemic Lupus Erythematosus
2.4. Behcet’s Disease
2.5. Inflammatory Bowel Disease
2.6. Autoimmune Skin Conditions
2.6.1. Vitiligo
2.6.2. Psoriasis Vulgaris
2.6.3. Atopic Dermatitis
2.7. Autoimmune Neurological Diseases
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACPA | anti–citrullinated protein antibody |
| AD | atopic dermatitis |
| AHR | aryl hydrocarbon receptor |
| BD | Behcet’s disease |
| BPB | butyrate-producing bacteria |
| CBMCs | cord-blood mononuclear cells |
| CCL20 | chemokine (C-C motif) ligand 20 |
| CD | Crohn’s disease |
| CIA | collagen-induced arthritis |
| CNS | central nervous system |
| CR | cellulose rich |
| CS | caesarian section |
| EAE | autoimmune encephalomyelitis |
| EVs | extracellular vesicles |
| FLNA | filamin A |
| FMT | fecal microbiota transplantation |
| GNS | N-acetylglucosamine-6-sulfatase |
| HbA1c | hemoglobin A1c |
| HCD | hydrolyzed casein diet |
| IBD | inflammatory bowel disease |
| IFN-I | type I interferon |
| IFN-γ | interferon gamma |
| IL | Interleukin |
| LPS | lipopolysaccharide |
| MOG | myelin oligodendrocyte glycoprotein |
| MS | multiple sclerosis |
| NF-κB | nuclear factor-κB |
| NOD | non-obese diabetic mice |
| OSE | opticospinal encephalomyelitis |
| PAD | peptidylarginine deiminase |
| PAMPs | pathogen associated molecular patterns |
| PSA | polysaccharide A |
| RA | rheumatoid arthritis |
| RRMS | relapsing remitting MS |
| SCFAs | short-chain fatty acids |
| SCFAs | short chain fatty acids |
| sEAE | spontaneous EAE |
| SFB | segmented filamentous bacteria |
| SLE | systemic lupus erythematosus |
| SPF | specific-pathogen-free |
| T1D | type I diabetes |
| Tc | cytotoxic T lymphocytes |
| TCRs | T cell receptors |
| Tfh | T follicular helper |
| Th | T lymphocytes. T-helper |
| Treg | regulatory T |
| Trp | tryptophan |
| UCs | ulcerative colitis |
References
- Ajayi, T.; Innes, C.L.; Grimm, S.A.; Rai, P.; Finethy, R.; Coers, J.; Wang, X.; Bell, D.A.; McGrath, J.A.; Schurman, S.H.; et al. Crohn’s disease IRGM risk alleles are associated with altered gene expression in human tissues. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G95–G105. [Google Scholar] [CrossRef] [PubMed]
- Batura, V.; Muise, A.M. Very early onset IBD: Novel genetic aetiologies. Curr. Opin. Allergy Clin. Immunol. 2018, 18, 470–480. [Google Scholar] [CrossRef]
- Zhang, J.; Meng, Y.; Wu, H.; Wu, Y.; Yang, B.; Wang, L. Association between PPP2CA polymorphisms and clinical features in southwest Chinese systemic lupus erythematosus patients. Medicine 2018, 97, e11451. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Ding, Y.P.; Gao, J.; Tanaka, Y.; Zhang, W. Risk factors and primary prevention trials for type 1 diabetes. Int. J. Biol. Sci. 2013, 9, 666–679. [Google Scholar] [CrossRef] [PubMed]
- Barbeau, W.E. What is the key environmental trigger in type 1 diabetes—Is it viruses, or wheat gluten, or both? Autoimmun. Rev. 2012, 12, 295–299. [Google Scholar] [CrossRef]
- Lindoso, L.; Mondal, K.; Venkateswaran, S.; Somineni, H.K.; Ballengee, C.; Walters, T.D.; Griffiths, A.; Noe, J.D.; Crandall, W.; Snapper, S.; et al. The Effect of Early-Life Environmental Exposures on Disease Phenotype and Clinical Course of Crohn’s Disease in Children. Am. J. Gastroenterol. 2018, 113, 1524–1529. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, E.; Marin, M.L.; Martin, R.; Odriozola, J.M.; Olivares, M.; Xaus, J.; Fernández, L.; Rodríguez, J.M. Is meconium from healthy newborns actually sterile? Res. Microbiol. 2008, 159, 187–193. [Google Scholar] [CrossRef]
- Vereecke, L.; Beyaert, R.; van Loo, G. Enterocyte death and intestinal barrier maintenance in homeostasis and disease. Trends Mol. Med. 2011, 17, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, L.A.; Jones, B.V. The human gut virome: A multifaceted majority. Front. Microbiol. 2015, 6, 918. [Google Scholar] [CrossRef]
- Gough, E.K.; Prendergast, A.J.; Mutasa, K.E.; Stoltzfus, R.J.; Manges, A.R. Sanitation Hygiene Infant Nutrition Efficacy (SHINE) Trial Team. Assessing the Intestinal Microbiota in the SHINE Trial. Clin. Infect. Dis. 2015, 61, S738–S744. [Google Scholar] [CrossRef]
- Kaiko, G.E.; Stappenbeck, T.S. Host-microbe interactions shaping the gastrointestinal environment. Trends Immunol. 2014, 35, 538–548. [Google Scholar] [CrossRef]
- Consortium, H.M.P. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar]
- DiGiulio, D.B.; Callahan, B.J.; McMurdie, P.J.; Costello, E.K.; Lyell, D.J.; Robaczewska, A.; Sun, C.L.; Goltsman, D.S.; Wong, R.J.; Shaw, G.; et al. Temporal and spatial variation of the human microbiota during pregnancy. Proc. Natl. Acad. Sci. USA 2015, 112, 11060–11065. [Google Scholar] [CrossRef] [PubMed]
- Gomez de Agüero, M.; Ganal-Vonarburg, S.C.; Fuhrer, T.; Rupp, S.; Uchimura, Y.; Li, H.; Steinert, A.; Heikenwalder, M.; Hapfelmeier, S.; Sauer, U.; et al. The maternal microbiota drives early postnatal innate immune development. Science 2016, 351, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Clausen, T.D.; Bergholt, T.; Eriksson, F.; Rasmussen, S.; Keiding, N.; Løkkegaard, E.C. Prelabor Cesarean Section and Risk of Childhood Type 1 Diabetes: A Nationwide Register-based Cohort Study. Epidemiology 2016, 27, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Gianchecchi, E.; Fierabracci, A. On the pathogenesis of insulin-dependent diabetes mellitus: The role of microbiota. Immunol. Res. 2017, 65, 242–256. [Google Scholar] [CrossRef] [PubMed]
- Cardwell, C.R.; Stene, L.C.; Joner, G.; Cinek, O.; Svensson, J.; Goldacre, M.J.; Parslow, R.C.; Pozzilli, P.; Brigis, G.; Stoyanov, D.; et al. Caesarean section is associated with an increased risk of childhood-onset type 1 diabetes mellitus: A meta-analysis of observational studies. Diabetologia 2008, 51, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Salminen, S.; Gibson, G.R.; McCartney, A.L.; Isolauri, E. Influence of mode of delivery on gut microbiota composition in seven year old children. Gut 2004, 53, 1388–1389. [Google Scholar] [CrossRef] [PubMed]
- Magne, F.; Puchi Silva, A.; Carvajal, B.; Gotteland, M. The Elevated Rate of Cesarean Section and Its Contribution to Non-Communicable Chronic Diseases in Latin America: The Growing Involvement of the Microbiota. Front. Pediatr. 2017, 5, 192. [Google Scholar] [CrossRef]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef] [PubMed]
- Clarke, G.; Stilling, R.M.; Kennedy, P.J.; Stanton, C.; Cryan, J.F.; Dinan, T.G. Minireview: Gut microbiota: The neglected endocrine organ. Mol. Endocrinol. 2014, 28, 1221–1238. [Google Scholar] [CrossRef] [PubMed]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef]
- Chen, J.; Rao, J.N.; Zou, T.; Liu, L.; Marasa, B.S.; Xiao, L.; Zeng, X.; Turner, D.J.; Wang, J.Y. Polyamines are required for expression of Toll-like receptor 2 modulating intestinal epithelial barrier integrity. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G568–G576. [Google Scholar] [CrossRef]
- Willemsen, L.E.M.; Koetsier, M.A.; van Deventer, S.J.H.; van Tol, E.A.F. Short chain fatty acids stimulate epithelial mucin 2 expression through differential effects on prostaglandin E1 and E2 production by intestinal myofibroblasts. Gut 2003, 52, 1442–1447. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; de Zoeten, E.F.; Ozkaynak, E.; Chen, C.; Wang, L.; Porrett, P.M.; Li, B.; Turka, L.A.; Olson, E.N.; Greene, M.I.; et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat. Med. 2007, 13, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Renelli, M.; Matias, V.; Lo, R.Y.; Beveridge, T.J. DNA-containing membrane vesicles of Pseudomonas aeruginosa PAO1 and their genetic transformation potential. Microbiology 2004, 50, 2161–2169. [Google Scholar] [CrossRef]
- Muraca, M.; Putignani, L.; Fierabracci, A.; Teti, A.; Perilongo, G. Gut microbiota-derived outer membrane vesicles: Under-recognized major players in health and disease? Discov. Med. 2015, 19, 343–348. [Google Scholar]
- Avila-Calderón, E.D.; Araiza-Villanueva, M.G.; Cancino-Diaz, J.C.; López-Villegas, E.O.; Sriranganathan, N.; Boyle, S.M.; et al. Roles of bacterial membrane vesicles. Arch. Microbiol. 2015, 197, 1–10. [Google Scholar] [CrossRef]
- Graham, S.; Courtois, P.; Malaisse, W.J.; Rozing, J.; Scott, F.W.; Mowat, A.M. Enteropathy precedes type 1 diabetes in the BB rat. Gut 2004, 53, 1437–1444. [Google Scholar] [CrossRef]
- Neu, J.; Reverte, C.M.; Mackey, A.D.; Liboni, K.; Tuhacek-Tenace, L.M.; Hatch, M.; Li, N.; Caicedo, R.A.; Schatz, D.A.; Atkinson, M. Changes in intestinal morphology and permeability in the biobreeding rat before the onset of type 1 diabetes. J. Pediatr. Gastroenterol. Nutr. 2005, 40, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Brugman, S.; Klatter, F.A.; Visser, J.T.; Wildeboer-Veloo, A.C.; Harmsen, H.J.; Rozing, J.; Bos, N.A. Antibiotic treatment partially protects against type 1 diabetes in the Bio-Breeding diabetes-prone rat. Is the gut flora involved in the development of type 1 diabetes? Diabetologia 2006, 49, 2105–2108. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Ley, R.E.; Volchkov, P.Y.; Stranges, P.B.; Avanesyan, L.; Stonebraker, A.C.; Hu, C.; Wong, F.S.; Szot, G.L.; Bluestone, J.A.; et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 2008, 455, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Valladares, R.; Sankar, D.; Li, N.; Williams, E.; Lai, K.K.; Abdelgeliel, A.S.; Gonzalez, C.F.; Wasserfall, C.H.; Larkin, J.; Schatz, D.; et al. Lactobacillus johnsonii N6.2 mitigates the development of type 1 diabetes in BB-DP rats. PLoS ONE 2010, 5, e10507. [Google Scholar] [CrossRef]
- Giongo, A.; Gano, K.A.; Crabb, D.B.; Mukherjee, N.; Novelo, L.L.; Casella, G.; Drew, J.C.; Ilonen, J.; Knip, M.; Hyöty, H.; et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. 2011, 5, 82–91. [Google Scholar] [CrossRef] [PubMed]
- de Goffau, M.C.; Luopajärvi, K.; Knip, M.; Ilonen, J.; Ruohtula, T.; Härkönen, T.; Orivuori, L.; Hakala, S.; Welling, G.W.; Harmsen, H.J.; et al. Fecal microbiota composition differs between children with β-cell autoimmunity and those without. Diabetes 2013, 62, 1238–1244. [Google Scholar] [CrossRef]
- Murri, M.; Leiva, I.; Gomez-Zumaquero, J.M.; Tinahones, F.J.; Cardona, F.; Soriguer, F.; Queipo-Ortuño, M.I. Gut microbiota in children with type 1 diabetes differs from that in healthy children: A case-control study. BMC Med. 2013, 11, 46. [Google Scholar] [CrossRef]
- Patrick, C.; Wang, G.S.; Lefebvre, D.E.; Crookshank, J.A.; Sonier, B.; Eberhard, C.; Mojibian, M.; Kennedy, C.R.; Brooks, S.P.; Kalmokoff, M.L.; et al. Promotion of autoimmune diabetes by cereal diet in the presence or absence of microbes associated with gut immune activation.; regulatory imbalance; and altered cathelicidin antimicrobial Peptide. Diabetes 2013, 62, 2036–2047. [Google Scholar] [CrossRef]
- Davis-Richardson, A.G.; Ardissone, A.N.; Dias, R.; Simell, V.; Leonard, M.T.; Kemppainen, K.M.; Drew, J.C.; Schatz, D.; Atkinson, M.A.; Kolaczkowski, B.; et al. Bacteroides dorei dominates gut microbiome prior to autoimmunity in Finnish children at high risk for type 1 diabetes. Front. Microbiol. 2014, 5, 678. [Google Scholar] [CrossRef]
- Mejía-León, M.E.; Petrosino, J.F.; Ajami, N.J.; Domínguez-Bello, M.G.; de la Barca, A.M. Fecal microbiota imbalance in Mexican children with type 1 diabetes. Sci. Rep. 2014, 4, 3814. [Google Scholar] [CrossRef]
- Soyucen, E.; Gulcan, A.; Aktuglu-Zeybek, A.C.; Onal, H.; Kiykim, E.; Aydin, A. Differences in the gut microbiota of healthy children and those with type 1 diabetes. Pediatr. Int. 2014, 56, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Alkanani, A.K.; Hara, N.; Gottlieb, P.A.; Ir, D.; Robertson, C.E.; Wagner, B.D.; Frank, D.N.; Zipris, D. Alterations in intestinal microbiota correlate with susceptibility to type 1 diabetes. Diabetes 2015, 64, 3510–3520. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Gevers, D.; Siljander, H.; Vatanen, T.; Hyötyläinen, T.; Hämäläinen, A.M.; Peet, A.; Tillmann, V.; Pöhö, P.; Mattila, I.; et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe 2015, 17, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Mejia-Leon, M.E.; Barca, A.M. Diet, microbiota and immune system in type 1 diabetes development and evolution. Nutrients 2015, 7, 9171–9184. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Furio, L.; Mecheri, R.; van der Does, A.M.; Lundeberg, E.; Saveanu, L.; Chen, Y.; van Endert, P.; Agerberth, B.; Diana, J. Pancreatic β-Cells Limit Autoimmune Diabetes via an Immunoregulatory Antimicrobial Peptide Expressed under the Influence of the Gut Microbiota. Immunity 2015, 43, 304–317. [Google Scholar] [CrossRef]
- Uusitalo, U.; Liu, X.; Yang, J.; Aronsson, C.A.; Hummel, S.; Butterworth, M.; Lernmark, Å.; Rewers, M.; Hagopian, W.; She, J.X.; et al. Association of Early Exposure of Probiotics and Islet Autoimmunity in the TEDDY Study. JAMA Pediatr. 2016, 170, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Pinto, E.; Anselmo, M.; Calha, M.; Bottrill, A.; Duarte, I.; Andrew, P.W.; Faleiro, M.L. The intestinal proteome of diabetic and control children is enriched with different microbial and host proteins. Microbiology 2017, 163, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Gavin, P.G.; Mullaney, J.A.; Loo, D.; Cao, K.L.; Gottlieb, P.A.; Hill, M.M.; Zipris, D.; Hamilton-Williams, E.E. Intestinal Metaproteomics Reveals Host-Microbiota Interactions in Subjects at Risk for Type 1 Diabetes. Diabetes Care 2018, 41, 2178–2186. [Google Scholar] [CrossRef]
- Gürsoy, S.; Koçkar, T.; Atik, S.U.; Önal, Z.; Önal, H.; Adal, E. Autoimmunity and intestinal colonization by Candida albicans in patients with type 1 diabetes at the time of the diagnosis. Korean J. Pediatr. 2018, 61, 217–220. [Google Scholar] [CrossRef]
- Henschel, A.M.; Cabrera, S.M.; Kaldunski, M.L.; Jia, S.; Geoffrey, R.; Roethle, M.F.; Lam, V.; Chen, Y.G.; Wang, X.; Salzman, N.H.; et al. Modulation of the diet and gastrointestinal microbiota normalizes systemic inflammation and β-cell chemokine expression associated with autoimmune diabetes susceptibility. PLoS ONE 2018, 13, e0190351. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, S.C.; Hu, J.; Ruan, H.B.; Guo, H.M.; Zhang, H.H.; Wang, X.; Pei, Y.F.; Pan, Y.; Fang, C. Gut microbiota profiling in Han Chinese with type 1 diabetes. Diabetes Res. Clin. Pract. 2018, 141, 256–263. [Google Scholar] [CrossRef]
- Mullaney, J.A.; Stephens, J.E.; Costello, M.E.; Fong, C.; Geeling, B.E.; Gavin, P.G.; Wright, C.M.; Spector, T.D.; Brown, M.A.; Hamilton-Williams, E.E. Correction to: Type 1 diabetes susceptibility alleles are associated with distinct alterations in the gut microbiota. Microbiome 2018, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.; Li, Z.; Wang, Y.; Shi, B.; Gong, Z.; Cheng, X. Porphyromonas gingivalis may play an important role in the pathogenesis of periodontitis-associated rheumatoid arthritis. Med. Hypotheses 2009, 72, 732–735. [Google Scholar] [CrossRef] [PubMed]
- de Aquino, S.G.; Abdollahi-Roodsaz, S.; Koenders, M.I.; van de Loo, F.A.; Pruijn, G.J.; Marijnissen, R.J.; Walgreen, B.; Helsen, M.M.; van den Bersselaar, L.A.; de Molon, R.S.; et al. Periodontal pathogens directly promote autoimmune experimental arthritis by inducing a TLR2- and IL-1-driven Th17 response. J. Immunol. 2014, 192, 4103–4111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, D.; Jia, H.; Feng, Q.; Wang, D.; Liang, D.; Wu, X.; Li, J.; Tang, L.; Li, Y.; et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 2015, 21, 895–905. [Google Scholar] [CrossRef]
- Chen, J.; Wright, K.; Davis, J.M.; Jeraldo, P.; Marietta, E.V.; Murray, J.; Nelson, H.; Matteson, E.L.; Taneja, V. An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Med. 2016, 8, 43. [Google Scholar] [CrossRef]
- Pianta, A.; Arvikar, S.L.; Strle, K.; Drouin, E.E.; Wang, Q.; Costello, C.E.; Steere, A.C. Two rheumatoid arthritis-specific autoantigens correlate microbial immunity with autoimmune responses in joints. J. Clin. Investig. 2017, 127, 2946–2956. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Takahashi, N.; Kato, T.; Matsuda, Y.; Yokoji, M.; Yamada, M.; Nakajima, T.; Kondo, N.; Endo, N.; Yamamoto, R.; et al. Aggravation of collagen-induced arthritis by orally administered Porphyromonas gingivalis through modulation of the gut microbiota and gut immune system. Sci. Rep. 2017, 7, 6955. [Google Scholar] [CrossRef]
- Teng, F.; Felix, K.M.; Bradley, C.P.; Naskar, D.; Ma, H.; Raslan, W.A.; Wu, H.J. The impact of age and gut microbiota on Th17 and Tfh cells in K/BxN autoimmune arthritis. Arthritis Res. Ther. 2017, 19, 188. [Google Scholar] [CrossRef]
- Jubair, W.K.; Hendrickson, J.D.; Severs, E.L.; Schulz, H.M.; Adhikari, S.; Ir, D.; Pagan, J.D.; Anthony, R.M.; Robertson, C.E.; Frank, D.N.; et al. Modulation of Inflammatory Arthritis in Mice by Gut Microbiota Through Mucosal Inflammation and Autoantibody Generation. Arthritis Rheumatol. 2018, 70, 1220–1233. [Google Scholar] [CrossRef]
- Picchianti-Diamanti, A.; Panebianco, C.; Salemi, S.; Sorgi, M.L.; Di Rosa, R.; Tropea, A.; Sgrulletti, M.; Salerno, G.; Terracciano, F.; D’Amelio, R.; et al. Analysis of Gut Microbiota in Rheumatoid Arthritis Patients: Disease-Related Dysbiosis and Modifications Induced by Etanercept. Int. J. Mol. Sci. 2018, 19, 2938. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, S.; Richards, A.; Maddison, P.; Scully, C. Gingival and periodontal health in systemic lupus erythematosus. Community Dent. Oral Epidemiol. 1993, 21, 158–161. [Google Scholar] [CrossRef] [PubMed]
- de Araújo Navas, E.A.F.; Sato, E.I.; Pereira, D.F.A.; Back-Brito, G.N.; Ishikawa, J.A.; Jorge, A.O.C.; Brighenti, F.L.; Koga-Ito, C.Y. Oral microbial colonization in patients with systemic lupus erythematous: Correlation with treatment and disease activity. Lupus 2012, 21, 969–977. [Google Scholar] [CrossRef]
- Hevia, A.; Milani, C.; López, P.; Cuervo, A.; Arboleya, S.; Duranti, S.; Turroni, F.; González, S.; Suárez, A.; Gueimonde, M.; et al. Intestinal dysbiosis associated with systemic lupus erythematosus. mBio 2014, 5, 1–10. [Google Scholar] [CrossRef]
- Calderaro, D.C.; Ferreira, G.A.; de Mendonça, S.M.S.; Corrêa, J.D.; Santos, F.X.; Sanção, J.G.C.; da Silva, T.A.; Teixeira, A.L. Há associação entre o lúpus eritematoso sistêmico e a doença periodontal? Rev. Bras. Reumatol. 2016, 56, 280–284. [Google Scholar] [CrossRef]
- Corrêa, J.D.; Calderaro, D.C.; Ferreira, G.A.; Mendonça, S.M.; Fernandes, G.R.; Xiao, E.; Teixeira, A.L.; Leys, E.J.; Graves, D.T.; Silva, T.A. Subgingival microbiota dysbiosis in systemic lupus erythematosus: Association with periodontal status. Microbiome 2017, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Ott, S.J.; Musfeldt, M.; Wenderoth, D.F.; Hampe, J.; Brant, O.; Fölsch, U.R.; Timmis, K.N.; Schreiber, S. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut 2004, 53, 685–693. [Google Scholar] [CrossRef]
- Gophna, U.; Sommerfeld, K.; Gophna, S.; Doolittle, W.F.; Veldhuyzen van Zanten, S.J. Differences between tissue-associated intestinal microfloras of patients with Crohn’s disease and ulcerative colitis. J. Clin. Microbiol. 2006, 44, 4136–4141. [Google Scholar] [CrossRef]
- Manichanh, C.; Rigottier-Gois, L.; Bonnaud, E.; Gloux, K.; Pelletier, E.; Frangeul, L.; Nalin, R.; Jarrin, C.; Chardon, P.; Marteau, P.; et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 2006, 55, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humarán, L.G.; Gratadoux, J.J.; Blugeon, S.; Bridonneau, C.; Furet, J.P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.W.; Sanderson, J.D.; Churcher, C.; Parkes, G.C.; Hudspith, B.N.; Rayment, N.; Brostoff, J.; Parkhill, J.; Dougan, G.; Petrovska, L. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 2011, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, L.; Zhou, R.; Wang, X.; Song, L.; Huang, S.; Wang, G.; Xia, B. Increased proportions of Bifidobacterium and the Lactobacillus group and loss of butyrate-producing bacteria in inflammatory bowel disease. J. Clin. Microbiol. 2014, 52, 398–406. [Google Scholar] [CrossRef]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease specific alterations in the enteric virome in inflammatory bowel disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Nishida, A.; Fujimoto, T.; Fujii, M.; Shioya, M.; Imaeda, H.; Inatomi, O.; Bamba, S.; Sugimoto, M.; Andoh, A. Reduced Abundance of Butyrate-Producing Bacteria Species in the Fecal Microbial Community in Crohn’s Disease. Digestion 2016, 93, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Wu, G.D.; Albenberg, L.; Tomov, V.T. Gut microbiota and IBD: Causation or correlation? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 573–584. [Google Scholar] [CrossRef]
- Cao, S.S. Cellular Stress Responses and Gut Microbiota in Inflammatory Bowel Disease. Gastroenterol. Res. Pract. 2018, 2018, 7192646. [Google Scholar] [CrossRef]
- Consolandi, C.; Turroni, S.; Emmi, G.; Severgnini, M.; Fiori, J.; Peano, C.; Biagi, E.; Grassi, A.; Rampelli, S.; Silvestri, E.; et al. Behcet’s syndrome patients exhibit specific microbiome signature. Autoimmun. Rev. 2015, 14, 269–276. [Google Scholar] [CrossRef]
- Seoudi, N.; Bergmeier, L.A.; Drobniewski, F.; Paster, B.; Fortune, F. The oral mucosal and salivary microbial community of Behcet’s syndrome and recurrent aphthous stomatitis. J. Oral Microbiol. 2015, 7, 27150. [Google Scholar] [CrossRef]
- Shimizu, J.; Kubota, T.; Takada, E.; Takai, K.; Fujiwara, N.; Arimitsu, N.; Ueda, Y.; Wakisaka, S.; Suzuki, T.; Suzuki, N. Bifidobacteria abundance-featured gut microbiota compositional change in patients with Behcet’s disease. PLoS ONE 2016, 11, e0153746. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Zhang, N.; Wu, C.; Zhang, X.; Wang, Q.; Huang, X.; Du, L.; Cao, Q.; Tang, J.; Zhou, C.; et al. A metagenomic study of the gut microbiome in Behcet’s disease. Microbiome 2018, 6, 135. [Google Scholar] [CrossRef] [PubMed]
- Ganju, P.; Nagpal, S.; Mohammed, M.H.; Nishal Kumar, P.; Pandey, R.; Natarajan, V.T.; Mande, S.S.; Gokhale, R.S. Microbial community profiling shows dysbiosis in the lesional skin of Vitiligo subjects. Sci. Rep. 2016, 6, 18761. [Google Scholar] [CrossRef]
- Gao, Z.; Tseng, C.; Strober, B.E.; Pei, Z.; Blaser, M.J. Substantial alterations of the cutaneous bacterial biota in psoriatic lesions. PLoS ONE 2008, 3, e2719. [Google Scholar] [CrossRef] [PubMed]
- Fahlen, A.; Engstrand, L.; Baker, B.S.; Powles, A.; Fry, L. Comparison of bacterial microbiota in skin biopsies from normal and psoriatic skin. Arch. Dermatol. Res. 2012, 304, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Alekseyenko, A.V.; Perez-Perez, G.I.; De Souza, A.; Strober, B.; Gao, Z.; Bihan, M.; Li, K.; Methé, B.A.; Blaser, M.J. Community differentiation of the cutaneous microbiota in psoriasis. Microbiome 2013, 1, 31. [Google Scholar] [CrossRef] [PubMed]
- Tett, A.; Pasolli, E.; Farina, S.; Truong, D.T.; Asnicar, F.; Zolfo, M.; Beghini, F.; Armanini, F.; Jousson, O.; De Sanctis, V.; et al. Unexplored diversity and strain-level structure of the skin microbiome associated with psoriasis. NPJ Biofilms Microbiomes 2017, 3, 14. [Google Scholar] [CrossRef]
- Chang, H.W.; Yan, D.; Singh, R.; Liu, J.; Lu, X.; Ucmak, D.; Lee, K.; Afifi, L.; Fadrosh, D.; Leech, J.; et al. Alteration of the cutaneous microbiome in psoriasis and potential role in Th17 polarization. Microbiome 2018, 6, 154. [Google Scholar] [CrossRef]
- Hong, S.W.; Kim, M.R.; Lee, E.Y.; Kim, J.H.; Kim, Y.S.; Jeon, S.G.; Yang, J.M.; Lee, B.J.; Pyun, B.Y.; Gho, Y.S. Extracellular vesicles derived from Staphylococcus aureus induce atopic dermatitis-like skin inflammation. Allergy 2011, 66, 351–359. [Google Scholar] [CrossRef]
- Kong, H.H.; Oh, J.; Deming, C.; Conlan, S.; Grice, E.A.; Beatson, M.A.; Nomicos, E.; Polley, E.C.; Komarow, H.D.; NISC Comparative Sequence Program; et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 2012, 22, 850–859. [Google Scholar] [CrossRef]
- Laborel-Préneron, E.; Bianchi, P.; Boralevi, F.; Lehours, P.; Fraysse, F.; Morice-Picard, F.; Sugai, M.; Sato’o, Y.; Badiou, C.; Lina, G.; et al. Effects of the Staphylococcus aureus and Staphylococcus epidermidis Secretomes Isolated from the Skin Microbiota of Atopic Children on CD4+ T Cell Activation. PLoS ONE 2015, 10, e0141067. [Google Scholar] [CrossRef]
- Williams, M.R.; Gallo, R.L. The role of the skin microbiome in atopic dermatitis. Curr. Allergy Asthma Rep. 2015, 15, 65. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Yoo, Y.; Hwang, J.; Na, Y.C.; Kim, H.S. Faecalibacterium prausnitzii subspecies-level dysbiosis in the human gut microbiome underlying atopic dermatitis. J. Allergy Clin. Immunol. 2016, 137, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, K.; Moriwaki, M.; Niitsu, Y.; Saino, M.; Takahagi, S.; Hisatsune, J.; Sugai, M.; Hide, M. Staphylococcus aureus from atopic dermatitis skin alters cytokine production triggered by monocyte-derived Langerhans cell. J. Dermatol. Sci. 2017, 88, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Francuzik, W.; Franke, K.; Schumann, R.R.; Heine, G.; Worm, M. Propionibacterium acnes Abundance Correlates Inversely with Staphylococcus aureus: Data from Atopic Dermatitis Skin Microbiome. Acta Derm. Venereol. 2018, 98, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Choi, S.J.; Choi, H.I.; Choi, J.P.; Park, H.K.; Kim, E.K.; Kim, M.J.; Moon, B.S.; Min, T.K.; Rho, M.; et al. Lactobacillus plantarum-derived Extracellular Vesicles Protect Atopic Dermatitis Induced by Staphylococcus aureus-derived Extracellular Vesicles. Allergy Asthma Immunol. Res. 2018, 10, 516–532. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Campos-Alberto, E.; Morita, Y.; Yamaguchi, M.; Toshimitsu, T.; Kimura, K.; Ikegami, S.; Katsuki, T.; Kohno, Y.; Shimojo, N. Low Interleukin 10 Production at Birth Is a Risk Factor for Atopic Dermatitis in Neonates with Bifidobacterium Colonization. Int. Arch. Allergy Immunol. 2018, 11, 1–8. [Google Scholar] [CrossRef]
- Berer, K.; Mues, M.; Koutrolos, M.; Rasbi, Z.A.; Boziki, M.; Johner, C.; Wekerle, H.; Krishnamoorthy, G. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature 2011, 479, 538–541. [Google Scholar] [CrossRef]
- Wang, Y.; Begum-Haque, S.; Telesford, K.M.; Ochoa-Reparaz, J.; Christy, M.; Kasper, E.J.; Robson, S.C.; Kasper, L.H. A commensal bacterial product elicits and modulates migratory capacity of CD39(+) CD4 T regulatory subsets in the suppression of neuroinflammation. Gut Microbes 2014, 5, 552–561. [Google Scholar] [CrossRef]
- Wang, Y.; Telesford, K.M.; Ochoa-Reparaz, J.; Haque-Begum, S.; Christy, M.; Kasper, E.J.; Wang, L.; Wu, Y.; Robson, S.C.; Kasper, D.L.; et al. An intestinal commensal symbiosis factor controls neuroinflammation via TLR2-mediated CD39 signalling. Nat. Commun. 2014, 5, 4432. [Google Scholar] [CrossRef]
- Miyake, S.; Kim, S.; Suda, W.; Oshima, K.; Nakamura, M.; Matsuoka, T.; Chihara, N.; Tomita, A.; Sato, W.; Kim, S.W.; et al. Dysbiosis in the Gut Microbiota of Patients with Multiple Sclerosis, with a Striking Depletion of Species Belonging to Clostridia XIVa and IV Clusters. PLoS ONE 2015, 10, e0137429. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, J.; Chia, N.; Kalari, K.R.; Yao, J.Z.; Novotna, M.; Paz Soldan, M.M.; Luckey, D.H.; Marietta, E.V.; Jeraldo, P.R.; et al. Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci. Rep. 2016, 6, 28484. [Google Scholar] [CrossRef] [PubMed]
- Jangi, S.; Gandhi, R.; Cox, L.M.; Li, N.; von Glehn, F.; Yan, R.; Patel, B.; Mazzola, M.A.; Liu, S.; Glanz, B.L.; et al. Alterations of the human gut microbiome in multiple sclerosis. Nat. Commun. 2016, 7, 12015. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.C.; Patel, B.; Yan, R.; Blain, M.; et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 2016, 22, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Berer, K.; Gerdes, L.A.; Cekanaviciute, E.; Jia, X.; Xiao, L.; Xia, Z.; Liu, C.; Klotz, L.; Stauffer, U.; Baranzini, S.E.; et al. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 10719–10724. [Google Scholar] [CrossRef] [PubMed]
- Cekanaviciute, E.; Yoo, B.B.; Runia, T.F.; Debelius, J.W.; Singh, S.; Nelson, C.A.; Kanner, R.; Bencosme, Y.; Lee, Y.K.; Hauser, S.L.; et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc. Natl. Acad. Sci. USA 2017, 114, 10713–10718. [Google Scholar] [CrossRef] [PubMed]
- Scher, J.U.; Sczesnak, A.; Longman, R.S.; Segata, N.; Ubeda, C.; Bielski, C.; Rostron, T.; Cerundolo, V.; Pamer, E.G.; Abramson, S.B.; et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife 2013, 2, e01202. [Google Scholar] [CrossRef] [PubMed]
- Terao, C.; Asai, K.; Hashimoto, M.; Yamazaki, T.; Ohmura, K.; Yamaguchi, A.; Takahashi, K.; Takei, N.; Ishii, T.; Kawaguchi, T.; et al. Significant association of periodontal disease with anticitrullinated peptide antibody in a Japanese healthy population—The Nagahama study. J. Autoimmun. 2015, 59, 85–90. [Google Scholar]
- Mendonça, S.M.S.; Corrêa, J.D.; Souza, A.F.; Travassos, D.V.; Calderaro, D.C.; Rocha, N.P.; Vieira, É.L.M.; Teixeira, A.L.; Ferreira, G.A.; Silva, T.A. Immunological signatures in saliva of systemic lupus erythematosus patients: Influence of periodontal condition. Clin. Exp. Rheumatol. 2018. [Google Scholar]
- Socransky, S.S.; Haffajee, A.D.; Cugini, M.A.; Smith, C.; Kent, R.L., Jr. Microbial complexes in subgingival plaque. J. Clin. Periodontol. 1998, 25, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Rehman, A.; Rausch, P.; Wang, J.; Skieceviciene, J.; Kiudelis, G.; Bhagalia, K.; Amarapurkar, D.; Kupcinskas, L.; Schreiber, S.; Rosenstiel, P.; et al. Geographical patterns of the standing and active human gut microbiome in health and IBD. Gut 2016, 65, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Assa, A.; Butcher, J.; Li, J.; Elkadri, A.; Sherman, P.M.; Muise, A.M.; Stintzi, A.; Mack, D. Mucosa-Associated Ileal Microbiota in New-Onset Pediatric Crohn’s Disease. Inflamm. Bowel Dis. 2016, 22, 1533–1539. [Google Scholar] [CrossRef]
- Wagner, J.; Maksimovic, J.; Farries, G.; Sim, W.H.; Bishop, R.F.; Cameron, D.J.; Catto-Smith, A.G.; Kirkwood, C.D. Bacteriophages in gut samples from pediatric Crohn’s disease patients: Metagenomic analysis using 454 pyrosequencing. Inflamm. Bowel Dis. 2013, 19, 1598–1608. [Google Scholar] [CrossRef] [PubMed]
- Grice, E.A.; Kong, H.H.; Conlan, S.; Deming, C.B.; Davis, J.; Young, A.C.; NISC Comparative Sequencing Program; Bouffard, G.G.; Blakesley, R.W.; Murray, P.R.; et al. Topographical and temporal diversity of the human skin microbiome. Science 2009, 324, 1190–1192. [Google Scholar] [CrossRef] [PubMed]
- Carratù, R.; Secondulfo, M.; de Magistris, L.; Iafusco, D.; Urio, A.; Carbone, M.G.; Pontoni, G.; Cartenì, M.; Prisco, F. Altered intestinal permeability to mannitol in diabetes mellitus type I. J. Pediatr. Gastroenterol. Nutr. 1999, 28, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Westerholm-Ormio, M.; Vaarala, O.; Pihkala, P.; Ilonen, J.; Savilahti, E. Immunologic activity in the small intestinal mucosa of pediatric patients with type 1 diabetes. Diabetes 2003, 52, 2287–2295. [Google Scholar] [CrossRef]
- Secondulfo, M.; Iafusco, D.; Carratù, R.; deMagistris, L.; Sapone, A.; Generoso, M.; Mezzogiomo, A.; Sasso, F.C.; Cartenì, M.; De Rosa, R.; et al. Ultrastructural mucosal alterations and increased intestinal permeability in non-celiac.; type I diabetic patients. Dig. Liver Dis. 2004, 36, 35–45. [Google Scholar] [CrossRef]
- Sapone, A.; de Magistris, L.; Pietzak, M.; Clemente, M.G.; Tripathi, A.; Cucca, F.; Lampis, R.; Kryszak, D.; Cartenì, M.; Generoso, M.; et al. Zonulin upregulation is associated with increased gut permeability in subjects with type 1 diabetes and their relatives. Diabetes 2006, 55, 1443–1449. [Google Scholar] [CrossRef]
- Bosi, E.; Molteni, L.; Radaelli, M.G.; Folini, L.; Fermo, I.; Bazzigaluppi, E.; Piemonti, L.; Pastore, M.R.; Paroni, R. Increased intestinal permeability precedes clinical onset of type 1 diabetes. Diabetologia 2006, 49, 2824–2827. [Google Scholar] [CrossRef]
- Brown, K.; Godovannyi, A.; Ma, C.; Zhang, Y.; Ahmadi-Vand, Z.; Dai, C.; Gorzelak, M.A.; Chan, Y.; Chan, J.M.; Lochner, A.; et al. Prolonged antibiotic treatment induces a diabetogenic intestinal microbiome that accelerates diabetes in NOD mice. ISME J. 2016, 10, 321–332. [Google Scholar] [CrossRef]
- Candon, S.; Perez-Arroyo, A.; Marquet, C.; Valette, F.; Foray, A.P.; Pelletier, B.; Milani, C.; Ventura, M.; Bach, J.F.; Chatenoud, L. Antibiotics in Early Life Alter the Gut Microbiome and Increase Disease Incidence in a Spontaneous Mouse Model of Autoimmune Insulin-Dependent Diabetes. PLoS ONE 2016, 11, e0147888. [Google Scholar] [CrossRef] [PubMed]
- Livanos, A.E.; Greiner, T.U.; Vangay, P.; Pathmasiri, W.; Stewart, D.; McRitchie, S.; Li, H.; Chung, J.; Sohn, J.; Kim, S.; et al. Antibiotic-mediated gut microbiome perturbation accelerates development of type 1 diabetes in mice. Nat. Microbiol. 2016, 1, 16140. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.J.; Ivanov, I.I.; Darce, J.; Hattori, K.; Shima, T.; Umesaki, Y.; Littman, D.R.; Benoist, C.; Mathis, D. Gut residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 2010, 32, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef]
- Teng, F.; Klinger, C.N.; Felix, K.M.; Bradley, C.P.; Wu, E.; Tran, N.L.; Umesaki, Y.; Wu, H.J. Gut microbiota drive autoimmune arthritis by promoting differentiation and migration of Peyer’s patch T follicular helper cells. Immunity 2016, 44, 875–888. [Google Scholar] [CrossRef] [PubMed]
- Bradley, C.P.; Teng, F.; Felix, K.M.; Sano, T.; Naskar, D.; Block, K.E.; Huang, H.; Knox, K.S.; Littman, D.R.; Wu, H.J. Segmented filamentous bacteria provoke lung autoimmunity by inducing gut–lung axis Th17 cells expressing dual TCRs. Cell Host Microbe 2017, 22, 697–704.e4. [Google Scholar] [CrossRef]
- Wegner, N.; Wait, R.; Sroka, A.; Eick, S.; Nguyen, K.A.; Lundberg, K.; Kinloch, A.; Culshaw, S.; Potempa, J.; Venables, P.J. Peptidylarginine deiminase from Porphyromonas gingivalis citrullinates human fibrinogen and alpha-enolase: Implications for autoimmunity in rheumatoid arthritis. Arthritis Rheum. 2010, 62, 2662–2672. [Google Scholar] [CrossRef]
- Montgomery, A.B.; Kopec, J.; Shrestha, L.; Thezenas, M.L.; Burgess-Brown, N.A.; Fischer, R.; Yue, W.W.; Venables, P.J. Crystal structure of Porphyromonas gingivalis peptidylarginine deiminase: Implications for autoimmunity in rheumatoid arthritis. Ann. Rheum. Dis. 2016, 75, 1255–1261. [Google Scholar] [CrossRef]
- Konig, M.F.; Abusleme, L.; Reinholdt, J.; Palmer, R.J.; Teles, R.P.; Sampson, K.; Rosen, A.; Nigrovic, P.A.; Sokolove, J.; Giles, J.T.; et al. Aggregatibacter actinomycetemcomitans-induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci. Transl. Med. 2016, 8, 369ra176. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef]
- Tanabe, S. The effect of probiotics and gut microbiota on Th17 cells. Int. Rev. Immunol. 2013, 32, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Zenewicz, L.A.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.J.; Stevens, S.; Flavell, R.A. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity 2008, 29, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Ezzedine, K.; Eleftheriadou, V.; Whitton, M.; van Geel, N. Vitiligo. Lancet 2015, 386, 74–84. [Google Scholar] [CrossRef]
- Lotti, T.; D’Erme, A.M. Vitiligo as a systemic disease. Clin. Dermatol. 2014, 32, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Lebwohl, M. Psoriasis. Lancet 2003, 361, 1197–1204. [Google Scholar] [CrossRef]
- Bach, J.F. The hygiene hypothesis in autoimmunity: The role of pathogens and commensals. Nat. Rev. Immunol. 2017, 18, 105–120. [Google Scholar] [CrossRef]
- Lynch, S.V.; Pedersen, O.P. The human intestinal microbiome in health and disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef]
- Ahern, P.P.; Faith, J.J.; Gordon, J.I. Mining the human gut microbiota for effector strains that shap the immune system. Immunity 2014, 40, 815–823. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 2009, 32, S62–S67. [Google Scholar] [CrossRef] [PubMed]
- Stechova, K.; Kolar, M.; Blatny, R.; Halbhuber, Z.; Vcelakova, J.; Hubackova, M.; Petruzelkova, L.; Sumnik, Z.; Obermannova, B.; Pithova, P.; et al. Healthy first degree relatives of patients with type 1 diabetes exhibit significant differences in basal gene expression pattern of immunocompetent cells compared to controls: Expression pattern as predeterminant of autoimmune diabetes. Scand. J. Immunol. 2011, 75, 210–219. [Google Scholar] [CrossRef] [PubMed]
- La Marca, V.; Gianchecchi, E.; Fierabracci, A. Type 1 Diabetes and Its Multi-Factorial Pathogenesis: The Putative Role of NK Cells. Int. J. Mol. Sci. 2018, 19, 794. [Google Scholar] [CrossRef] [PubMed]
- Silveira, P.A.; Grey, S.T. B cells in the spotlight: Innocent bystanders or major players in the pathogenesis of type 1 diabetes. Trends Endocrinol. Metab. 2006, 17, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Gianchecchi, E.; Palombi, M.; Fierabracci, A. The putative role of the C1858T polymorphismof protein tyrosine phosphatase PTPN22 gene in autoimmunity. Autoimmun. Rev. 2013, 12, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, F.; Nguyen, T.L.; Brinkman, B.; Yunta, R.G.; Cauwe, B.; Vandenabeele, P.; Liston, A.; Raes, J. Inflammation-associated enterotypes, host genotype, cage and inter-individual effects drive gut microbiota variation in common laboratory mice. Genome Biol. 2013, 14, R4. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, D.S.; Krych, Ł.; Buschard, K.; Hansen, C.H.; Hansen, A.K. Beyond genetics. Influence of dietary factors and gut microbiota on type 1 diabetes. FEBS Lett. 2014, 588, 4234–4243. [Google Scholar] [CrossRef] [PubMed]
- Roesch, L.F.; Lorca, G.L.; Casella, G.; Giongo, A.; Naranjo, A.; Pionzio, A.M.; Li, N.; Mai, V.; Wasserfall, C.H.; Schatz, D.; et al. Culture-independent identification of gut bacteria correlated with the onset of diabetes in a rat model. ISME J. 2009, 3, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Hornef, M.W.; Pabst, O. Real friends: Faecalibacterium prausnitzii supports mucosal immune homeostasis. Gut 2016, 65, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, L.R.; Garcia-Valladares, I. Of bugs and joints: The relationship between infection and joints. Reumatología Clínica 2013, 9, 229–238. [Google Scholar] [CrossRef]
- Taneja, V. Cytokines pre-determined by genetic factors are involved in pathogenesis of rheumatoid arthritis. Cytokine 2014, 75, 216–221. [Google Scholar] [CrossRef]
- Lehner, T. Immunopathogenesis of Behçet’s disease. Ann. Med. Interne 1999, 150, 483–487. [Google Scholar]
- Goodnow, C.C. Multistep pathogenesis of autoimmune disease. Cell 2007, 130, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Lee, W.W.; Kim, S.H.; Kang, Y.; Lee, N.; Shin, M.S.; Kang, S.W.; Kang, I. Age-associated alteration in naive and memory Th17 cell response in humans. Clin. Immunol. 2011, 140, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Zou, R.; Gan, H.; Liang, Z.; Li, F.; Lin, T.; Luo, Y.; Cai, X.; He, F.; Shen, E. The effect of aging on the frequency, phenotype and cytokine production of human blood CD4 + CXCR5 + T follicular helper cells: Comparison of aged and young subjects. Immun. Ageing 2014, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Malmstrom, V.; Catrina, A.I.; Klareskog, L. The immunopathogenesis of seropositive rheumatoid arthritis: From triggering to targeting. Nat. Rev. Immunol. 2017, 17, 60–75. [Google Scholar] [CrossRef] [PubMed]
- Bartold, P.M.; Marino, V.; Cantley, M.; Haynes, D.R. Effect of Porphyromonas gingivalis-induced inflammation on the development of rheumatoid arthritis. J. Clin. Periodontol. 2010, 37, 405–411. [Google Scholar] [CrossRef]
- Kotzin, B.L. Systemic lupus erythematosus. Cell 1996, 85, 303–306. [Google Scholar] [CrossRef]
- Pacheco, G.V.; Cruz, D.C.; González Herrera, L.J.; Pérez Mendoza, G.J.; Adrián Amaro, G.I.; Nakazawa Ueji, Y.E.; Angulo Ramírez, A.V. Copy number variation of TLR-7 gene and its association with the development of systemic lupus erythematosus in female patients from Yucatan Mexico. Genet. Epigenet. 2014, 6, 31–36. [Google Scholar] [CrossRef]
- Zhang, H.; Liao, X.; Sparks, J.B.; Luo, X.M. Dynamics of gut microbiota in autoimmune lupus. Appl. Environ. Microbiol. 2014, 80, 7551–7560. [Google Scholar] [CrossRef]
- Al-Mutairi, K.; Al-Zahrani, M.; Bahlas, S.; Kayal, R.; Zawawi, K. Periodontal findings in systemic lupus erythematosus patients and healthy controls. Saudi Med. J. 2015, 36, 463–468. [Google Scholar] [CrossRef]
- Sakane, T.; Takeno, M.; Suzuki, N.; Inaba, G. Behçet’s disease. N. Engl. J. Med. 1999, 341, 1284–1291. [Google Scholar] [CrossRef]
- Yurdakul, S.; Hamuryudan, V.; Yazici, H. Behçet syndrome. Curr. Opin. Rheumatol. 2004, 16, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Takada, Y.; Fujita, Y.; Igarashi, M.; Katsumata, T.; Okabe, H.; Saigenji, K.; Takahashi, T.; Atari, E. Intestinal Behçet’s disease-pathognomonic changes in intramucosal lymphoid tissues and effect of a “rest cure” on intestinal lesions. J. Gastroenterol. 1997, 32, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Karasneh, J.; Gül, A.; Ollier, W.E.; Silman, A.J. Worthington, J. Whole-genome screening for susceptibility genes in multicase families with Behçet’s disease. Arthritis Rheum. 2005, 52, 1836–1842. [Google Scholar] [CrossRef] [PubMed]
- Melikoglu, M.; Uysal, S.; Krueger, J.G.; Kaplan, G.; Gogus, F.; Yazici, H.; Oliver, S. Characterization of the divergent wound-healing responses occurring in the pathergy reaction and normal healthy volunteers. J. Immunol. 2006, 177, 6415–6421. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Kastner, D.L.; Remmers, E.F. The immunogenetics of Behcet’s disease: A comprehensive review. J. Autoimmun. 2015, 64, 137–148. [Google Scholar] [CrossRef]
- Zeidan, M.J.; Saadoun, D.; Garrido, M.; Klatzmann, D.; Six, A.; Cacoub, P. Behcet’s disease physiopathology: A contemporary review. Auto. Immun. Highlights 2016, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Geremia, A.; Biancheri, P.; Allan, P.; Corazza, G.R.; Di Sabatino, A. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun. Rev. 2014, 13, 3–10. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Bernstein, C.N.; Iliopoulos, D.; Macpherson, A.; Neurath, M.F.; Ali, R.A.R.; Vavricka, S.R.; Fiocchi, C. Environmental triggers in IBD: A review of progress and evidence. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 39–49. [Google Scholar] [CrossRef]
- Kellermayer, R.; Mir, S.A.; Nagy-Szakal, D.; Cox, S.B.; Dowd, S.E.; Kaplan, J.L.; Sun, Y.; Reddy, S.; Bronsky, J.; Winter, H.S. Microbiota separation and C-reactive protein elevation in treatment-naïve pediatric granulomatous Crohn disease. J. Pediatr. Gastroenterol. Nutr. 2012, 55, 243–250. [Google Scholar] [CrossRef]
- Shah, R.; Cope, J.L.; Nagy-Szakal, D.; Dowd, S.; Versalovic, J.; Hollister, E.B.; Kellermayer, R. Composition and function of the pediatric colonic mucosal microbiome in untreated patients with ulcerative colitis. Gut Microbes 2016, 7, 384–396. [Google Scholar] [CrossRef]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vázquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 2017, 21, 301–304. [Google Scholar] [CrossRef]
- Akazawa, Y.; Isomoto, H.; Matsushima, K.; Kanda, T.; Minami, H.; Yamaghchi, N.; Taura, N.; Shiozawa, K.; Ohnita, K.; Takeshima, F.; et al. Endoplasmic reticulum stress contributes to Helicobacter pylori VacA induced apoptosis. PLoS ONE 2013, 8, e82322. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Haynes, M.; Hanson, N.; Angly, F.E.; Heath, A.C.; Rohwer, F.; Gordon, J.I. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 2010, 466, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Minot, S.; Sinha, R.; Chen, J.; Li, H.; Keilbaugh, S.A.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. The human gut virome: Inter-individual variation and dynamic response to diet. Genome Res. 2011, 21, 1616–1625. [Google Scholar] [CrossRef] [PubMed]
- Van Belleghem, J.D.; Clement, F.; Merabishvili, M.; Lavigne, R.; Vaneechoutte, M. Pro- and anti-inflammatory responses of peripheral blood mononuclear cells induced by Staphylococcus aureus and Pseudomonas aeruginosa phages. Sci. Rep. 2017, 7, 8004. [Google Scholar] [CrossRef] [PubMed]
- Magin, W.S.; Van Kruiningen, H.J.; Colombel, J.F. Immunohistochemical search for viral and bacterial antigens in Crohn’s disease. J. Crohns Colitis. 2013, 7, 161–166. [Google Scholar] [CrossRef]
- Monteleone, I.; Rizzo, A.; Sarra, M.; Sica, G.; Sileri, P.; Biancone, L.; MacDonald, T.T.; Pallone, F.; Monteleone, G. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology. 2011, 141, 237–248.e1. [Google Scholar] [CrossRef]
- Sugimoto, K.; Ogawa, A.; Mizoguchi, E.; Shimomura, Y.; Andoh, A.; Bhan, A.K.; Blumberg, R.S.; Xavier, R.J.; Mizoguchi, A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Investig. 2008, 118, 534–544. [Google Scholar] [CrossRef]
- Guerra, L.; Dellambra, E.; Brescia, S.; Raskovic, D. Vitiligo: Pathogenetic hypotheses and targets for current therapies. Curr. Drug Metab. 2010, 11, 451–467. [Google Scholar] [CrossRef]
- Malhotra, N.; Dytoc, M. The pathogenesis of vitiligo. J. Cutan. Med. Surg. 2013, 17, 153–172. [Google Scholar] [CrossRef]
- Amerio, P.; Tracanna, M.; De Remigis, P.; Betterle, C.; Vianale, L.; Marra, M.E.; Di Rollo, D.; Capizzi, R.; Feliciani, C.; Tulli, A. Vitiligo associated with other autoimmune diseases: Polyglandular autoimmune syndrome types 3B+C and 4. Clin. Exp. Dermatol. 2006, 31, 746–749. [Google Scholar] [CrossRef] [PubMed]
- Lowes, M.A.; Suárez-Fariñas, M.; Krueger, J.G. Immunology of psoriasis. Annu. Rev. Immunol. 2014, 32, 227–255. [Google Scholar] [CrossRef] [PubMed]
- Nestle, F.O.; Kaplan, D.H.; Barker, J. Psoriasis. N. Engl. J. Med. 2009, 361, 496–509. [Google Scholar] [CrossRef] [PubMed]
- Perera, G.K.; Di Meglio, P.; Nestle, F.O. Psoriasis. Annu. Rev. Pathol. Mech. Dis. 2012, 7, 385–422. [Google Scholar] [CrossRef]
- Loesche, M.A.; Farahi, K.; Capone, K.; Fakharzadeh, S.; Blauvelt, A.; Duffin, K.C.; DePrimo, S.E.; Muñoz-Elías, E.J.; Brodmerkel, C.; Dasgupta, B.; et al. Longitudinal study of the psoriasis-associated skin microbiome during therapy with ustekinumab in a randomized phase 3b clinical trial. J. Investig. Dermatol. 2018, 138, 1973–1981. [Google Scholar] [CrossRef] [PubMed]
- Sabin, B.R.; Peters, N.; Peters, A.T. Chapter 20: Atopic dermatitis. Allergy Asthma Proc. 2012, 33, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.C.; Kim, S.R.; Yoon, Y.J.; Park, K.S.; Kim, J.H.; Lee, J.; Kim, O.Y.; Choi, E.J.; Kim, D.K.; Choi, D.S.; et al. In vivo kinetic biodistribution of nano-sized outer membrane vesicles derived from bacteria. Small 2015, 11, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Koch-Henriksen, N.; Sorensen, P.S. The changing demographic pattern of multiple sclerosis epidemiology. Lancet Neurol. 2010, 9, 520–532. [Google Scholar] [CrossRef]
- Matveeva, O.; Bogie, J.F.J.; Hendriks, J.J.A.; Linker, R.A.; Haghikia, A.; Kleinewietfeld, M. Western lifestyle and immunopathology of multiple sclerosis. Ann. N. Y. Acad. Sci. 2018, 1417, 71–86. [Google Scholar] [CrossRef]
- Maslowski, K.M.; Mackay, C.R. Diet, gut microbiota and immune responses. Nat. Immunol. 2011, 12, 5–9. [Google Scholar] [CrossRef]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Cox, L.M.; Cho, I.; Young, S.A.; Anderson, W.H.; Waters, B.J.; Hung, S.C.; Gao, Z.; Mahana, D.; Bihan, M.; Alekseyenko, A.V.; et al. The nonfermentable dietary fiber hydroxypropyl methylcellulose modulates intestinal microbiota. FASEB J. 2012, 27, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Bindels, L.B.; Segura Munoz, R.R.; Gomes-Neto, J.C.; Mutemberezi, V.; Martínez, I.; Salazar, N.; Cody, E.A.; Quintero-Villegas, M.I.; Kittana, H.; de Los Reyes-Gavilán, C.G.; et al. Resistant starch can improve insulin sensitivity independently of the gut microbiota. Microbiome 2017, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Farinotti, M.; Simi, S.; Di Pietrantonj, C.; McDowell, N.; Brait, L.; Lupo, D.; Filippini, G. Dietary interventions for multiple sclerosis. Cochrane Database Syst. Rev. 2012, 12, Cd004192. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, T.; Miyake, S. Diet, Gut Flora, and Multiple Sclerosis: Current Research and Future Perspectives. In Multiple Sclerosis Immunology: A Foundation for Current and Future Treatments; Springer: New York, NY, USA, 2013; Volume 6, pp. 115–126. [Google Scholar]
- Quintana, F.J.; Sherr, D.H. Aryl hydrocarbon receptor control of adaptive immunity. Pharmacol. Rev. 2013, 65, 1148–1161. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, G.; Lassmann, H.; Wekerle, H.; Holz, A. Spontaneous opticospinal encephalomyelitis in a double-transgenic mouse model of autoimmune T cell/B cell cooperation. J. Clin. Investig. 2006, 116, 2385–2392. [Google Scholar] [CrossRef] [PubMed]
- Berer, K.; Martínez, I.; Walker, A.; Kunkel, B.; Schmitt-Kopplin, P.; Walter, J.; Krishnamoorthy, G. Dietary non-fermentable fiber prevents autoimmune neurological disease by changing gut metabolic and immune status. Sci. Rep. 2018, 8, 10431. [Google Scholar] [CrossRef]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly-Y, M.; Glickman, J.N.; Garrett, W.S. The Microbial Metabolites, Short-Chain Fatty Acids, Regulate Colonic Treg Cell Homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef]
- Trompette, A.; Gollwitzer, E.S.; Yadava, K.; Sichelstiel, AK.; Sprenger, N.; Ngom-Bru, C.; Blanchard, C.; Junt, T.; Nicod, L.P.; Harris, N.L.; et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat. Med. 2014, 20, 159–166. [Google Scholar] [CrossRef]
- Haghikia, A.; Jörg, S.; Duscha, A.; Berg, J.; Manzel, A.; Waschbisch, A.; Hammer, A.; Lee, D.H.; May, C.; Wilck, N.; et al. Dietary fatty acids directly impact central nervous system autoimmunity via the small intestine. Immunity 2015, 43, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Mariño, E.; Richards, J.L.; McLeod, K.H.; Stanley, D.; Yap, Y.A.; Knight, J.; McKenzie, C.; Kranich, J.; Oliveira, A.C.; Rossello, F.J.; et al. Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nat. Immunol. 2017, 18, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Marinho, F.A.; Pacifico, L.G.; Miyoshi, A.; Azevedo, V.; Le Loir, Y.; Guimarães, V.D.; Langella, P.; Cassali, G.D.; Fonseca, C.T.; Oliveira, S.C. An intranasal administration of Lactococcus lactis strains expressing recombinat interleukin-10 modulates acute allergic airway inflammation in a murine model. Clin Exp Allergy 2010, 40, 1541–1551. [Google Scholar] [CrossRef]
- Braat, H.; Rottiers, P.; Hommes, D.W.; Huyghebaert, N.; Remaut, E.; Remon, J.P.; van Deventer, S.J.; Neirynck, S.; Peppelenbosch, M.P.; Steidler, L. A phase I trial with transgenic bacteria expressing interleukin-10 in Crohn’s disease. Clin. Gastroenterol. Hepatol. 2006, 4, 745–749. [Google Scholar] [CrossRef] [PubMed]
- Breton, J.; Tennoune, N.; Lucas, N.; Francois, M.; Legrand, R.; Jacquemot, J.; Goichon, A.; Guérin, C.; Peltier, J.; Pestel-Caron, M.; et al. Gut commensal E. Coli proteins activate host satiety pathways following nutrient-induced bacterial growth. Cell Metab. 2016, 23, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Duan, F.F.; Liu, J.H.; March, J.C. Engineered commensal bacteria reprogram intestinal cells into glucose-responsive insulin-secreting cells for the treatment of diabetes. Diabetes 2015, 64, 1794–1803. [Google Scholar] [CrossRef] [PubMed]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojärvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916.e7. [Google Scholar] [CrossRef] [PubMed]

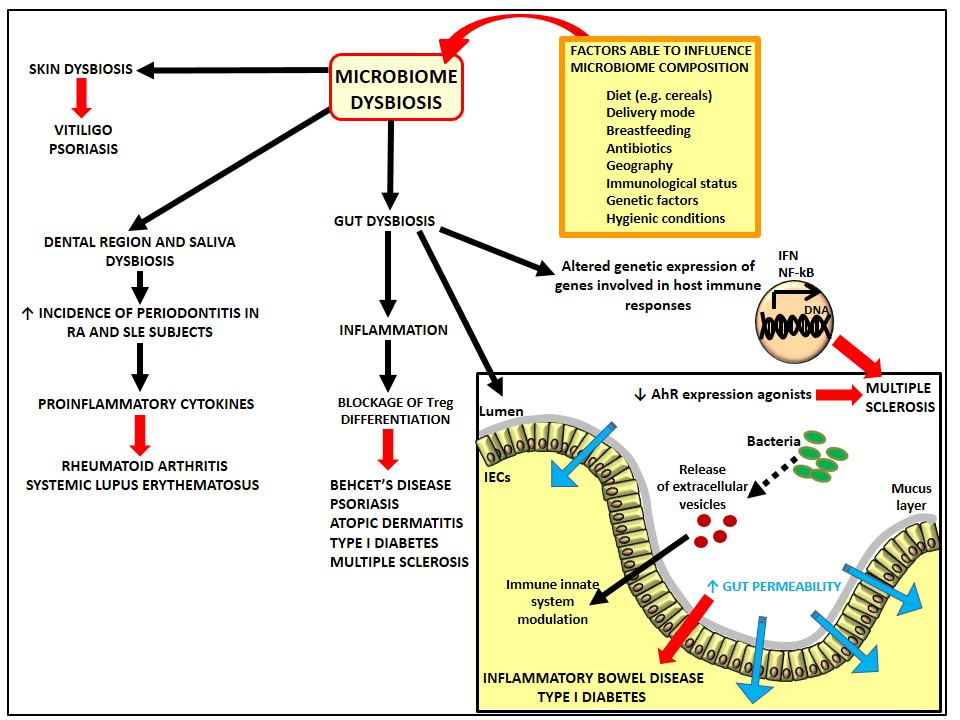{kind=link}
| Autoimmune Conditions | Microbiome Alterations in Autoimmunity Respect to Healthy Subjects | References |
|---|---|---|
| Type I diabetes | ↓ bacterial diversity in high-risk children | [35,42,43] |
| ↑ Bacteroidetes/Firmicutes ratio | [36,44,51] | |
| ↑ Bacteroidetes, Clostridium, and Veillonella; ↓ Bifidobacterium, Lactobacillus, Blautia coccoides/Eubacterium rectale group, and Prevotella | [37] | |
| ↑ Bacteroides dorei in high-risk children | [39] | |
| ↑ Bacteroides and ↓ Prevotella in newly diagnosed T1D patients | [40] | |
| ↓ Bifidobacterium | [36,41] | |
| ↑ Bacteroides and Clostridium cluster XVa and cluster IV; ↓ Bifidobacterium | [47] | |
| ↑ Candida albicans and Enterobacteriaceae | [41] | |
| Rheumatoid arthritis | ↑ of the pathobiont Prevotella (Prevotella copri) in new-onset RA subjects | [107] |
| Gut and oral microbiome dysbiosis; ↓ Haemophilus spp. and ↑ Lactobacillus salivarius | [56] | |
| ↓ gut bacterial diversity and expansion of rare lineage intestinal microbes | [57] | |
| Association between periodontal infection due to Porphyromonas gingivalis and RA | [54,108] | |
| ↑ Fretibacterium, Selenomonas and Prevotella nigrescens | [55] | |
| ↑ Bacilli and Lactobacillales; ↓ genus Faecalibacterium and the specie Faecalibacterium prausnitzii; Absence of the genus Flavobacterium and the species Blautia coccoides in RA patients present instead in controls | [62] | |
| ↑ Prevotella copri and ↓ Bacteroides in new-onset untreated RA patients | [107] | |
| Systemic lupus erythematosus | ↑ Bacteroidetes/Firmicutes ratio | [65] |
| Association between SLE and periodontal disease; Dysbiosis of the subgingival microbiota; ↑ subgingival bacterial load; ↓ subgingival microbial diversity at diseased sites | [67] | |
| ↑ Fretibacterium, Selenomonas, and Prevotella nigrescens; | [109] | |
| Association with periodontal pathogens (Treponema denticola, Porphyromonas gingivalis, Fretibacterium fastidiosum and Tannerella forsythia | [109,110] | |
| Behcet’s disease | ↓ Roseburia and Subdoligranulum genera | [79] |
| ↑ Bifidobacterium and Eggerthella genera and ↓ Megamonas and Prevotella genera | [81] | |
| ↑ Bilophila spp. and several opportunistic pathogens (e.g., Parabacteroides spp. and Paraprevotella spp.); ↓ butyrate-producing bacteria Clostridium spp. and methanogens (Methanoculleus spp. and Methanomethylophilus spp.). | [82] | |
| Inflammatory bowel disease | ↓ gut bacterial diversity | [68] |
| ↓ diversity in the bacterial phylum Firmicutes faecal; ↓ Clostridium leptum phylogenetic group | [70] | |
| ↑ Proteobacteria phylum including Escherichia coli; ↓ Firmicutes phylum was reduced | [69,70,73,111] | |
| ↓ Faecalibacterium prausnitzii is associated with an ↑ risk of postoperative recurrence of ileal CD | [72] | |
| ↓ in several butyrate-producing bacteria species | [74,76] | |
| ↓ of the genera Bacteroides, Eubacterium, Faecalibacterium and Ruminococcus; ↑ genera Actinomyces and Bifidobacterium; ↓ butyrate-producing bacterial species, as Blautia faecis, Roseburia inulinivorans, Ruminococcus torques, Clostridium lavalense, Bacteroides uniformis, and Faecalibacterium prausnitzii | [76] | |
| Dysbiosis | [112] | |
| ↑ Caudovirales bacteriophages and fungal composition | [113] | |
| Vitiligo | ↑ Actinobacteria, Proteobacteria, Firmicutes, and Bacteroidetes | [83,114] |
| ↓ bacterial diversity | [83] | |
| Psoriasis vulgaris | ↑ Proteobacteria; ↓ Staphylococci and Propionibacteria | [85] |
| ↓ bacterial diversity; ↑ Corynebacterium, Propionibacterium, Staphylococcus, and Streptococcus; ↓ Cupriavidus, Flavisolibacter, Methylobacterium, and Schlegelella genera. Association between lesion samples with Firmicutes-associated microbiota | [86] | |
| ↓ diversity and ↑ Staphylococcus in psoriatic ear sites | [87] | |
| ↑ diversity and ↑ heterogeneity ↑ Staphylococcus aureus; ↓ Staphylococcus epidermidis and Propionibacterium acnes | [88] | |
| Atopic dermatitis | ↑ Faecalibacterium prausnitzii subspecies | [93] |
| ↑ Staphylococcus aureus | [94] | |
| ↓ Propionibacterium acnes and Lawsonella clevelandensis; ↑ Staphylococcus aureus in non-lesional relative to lesional AD patients | [95] | |
| Autoimmune neurological diseases | ↓ species belonging to Clostridia XIVa and IV Clusters | [101] |
| ↑ Pseudomonas, Mycoplana, Haemophilus, Blautia, and Dorea in relapsing remitting MS patients; ↓ Parabacteroides, Adlercreutzia, and Prevotella genera | [102] | |
| ↑ Methanobrevibacter and Akkermansia; ↓ Butyricimonas | [103] | |
| ↑ Akkermansia muciniphila and Acinetobacter calcoaceticus; ↓ Parabacteroides distasonis | [106] |
| Autoimmune Disorders | Bacterial Associated Mechanisms Promoting Autoimmunity | References |
|---|---|---|
| Type I diabetes | Perturbation in the integrity of epithelial barrier | [49,115,116,117,118,119] |
| Changes in gut microbiota following antibiotic treatment | [120,121,122] | |
| Functional enrichment in core energy metabolism proteins, in particular on sugar transport and processing | [47] | |
| Perturbations in the integrity of epithelial tight junctions | [44] | |
| Blockage of Treg differentiation via products generated with the anaerobic respiration, such as acetate and succinate | [44] | |
| Increased intestinal inflammation and reduced barrier activity due to depletion in microbiota taxa related with host proteins implicated in the maintenance of mucous barrier functionality, microvilli adhesion, and exocrine pancreas | [48] | |
| Reduced presence of beneficial anaerobic gut bacteria Lactobacillus, Bifidobacterium, and Bacteroides species exerting an inhibitory function by synthetizing short-chain fatty acids and antimicrobial compounds | [36,41] | |
| Bacterial metabolites can affect host immune system leading to pro- or anti-inflammatory reactions | [53] | |
| Lung autoimmunity in rheumatoid arthritis | Modulation of host immune response by gut-residing segmented filamentous bacteria via increased Th17cells percentages induced by the strong Th17 chemoattractant CCL20 | [123,124,125] |
| Rheumatoid arthritis | Promotion of autoantibodies by gut residing segmented filamentous bacteria during the pre-arthritic phase | [126] |
| Molecular mimicry between several gut microbe epitopes and two autoantigens N-acetylglucosamine-6-sulfatase and filamin A greatly expressed in inflamed synovial tissue | [58] | |
| Citrullinated proteins via the peptidylarginine deiminase (PAD) enzyme derived from bacteria | [127,128,129] | |
| Systemic lupus erythematosus | Correlation between dysbiotic periodontal inflammation and more severe SLE scores | [67] |
| Behcet’s disease | Altered Th1, Th17, and Treg functions | [130,131] |
| Reduction in butyrate-producing bacteria and methanogens, with enhanced oxidation-reduction process, capsular polysaccharide transport system, and type III and IV secretion systems | [82] | |
| Strong intraocular inflammatory reaction | [82] | |
| Inflammatory bowel disease | Impaired epithelial barrier and increased intestinal permeability | [111] |
| Cellular stress responses interacting with microbiome in the gastrointestinal tract. Interaction between bacteria and endoplasmic reticulum | [78] | |
| Reduction of several butyrate-producing bacteria | [74,76] | |
| Reduced AHR agonists in the inflamed intestinal tissue samples that modulate T cell responses AHR agonists exert an anti-inflammatory effect inducing IL-22 | [132,133,134] | |
| Vitiligo | Aberrant intra-community network in the lesional skin areas respect to those of non-lesional sites. Dysbiosis of diverse microbial community in vitiligo lesional skin | [135] |
| Psoriasis vulgaris | Increased Th17 response which could have a role in IL-17-driven inflammation | [88] |
| Atopic dermatitis | Increase of IgE response, inflammatory and Th2/Th22 transcripts, promotion of Th2 activation, and suppression of resident Treg cells by secretomes of skin microbiota | [91] |
| Induction of an imbalanced Th1/Th2 skin immunity | [94] | |
| Decrease of butyrate and propionate producers (molecules with an anti-inflammatory activity) | [93] | |
| EVs derived from the microbiome and increase of inflammation | [89,136] | |
| Multiple sclerosis | Reduced levels of circulating AHR agonists and reduced AHR agonistic activity | [104] |
| Modulation by fecal microbiota abundance of expression of host immune genes involved in dendritic cell maturation, and interferon and NF-kB signaling pathways in circulating T lymphocytes and monocytes | [103] | |
| Reduced Treg compartment associated with increased percentages of effector CD4+ lymphocytes that differentiated into IFNγ-producing Th1 cells Reduced IL-10+ Treg subset in mice transplanted with microbiota from MS patients | [106] | |
| Diet skewed gut microbial and metabolic profiles | [105,137,138] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gianchecchi, E.; Fierabracci, A. Recent Advances on Microbiota Involvement in the Pathogenesis of Autoimmunity. Int. J. Mol. Sci. 2019, 20, 283. https://doi.org/10.3390/ijms20020283
Gianchecchi E, Fierabracci A. Recent Advances on Microbiota Involvement in the Pathogenesis of Autoimmunity. International Journal of Molecular Sciences. 2019; 20(2):283. https://doi.org/10.3390/ijms20020283
Chicago/Turabian StyleGianchecchi, Elena, and Alessandra Fierabracci. 2019. "Recent Advances on Microbiota Involvement in the Pathogenesis of Autoimmunity" International Journal of Molecular Sciences 20, no. 2: 283. https://doi.org/10.3390/ijms20020283
APA StyleGianchecchi, E., & Fierabracci, A. (2019). Recent Advances on Microbiota Involvement in the Pathogenesis of Autoimmunity. International Journal of Molecular Sciences, 20(2), 283. https://doi.org/10.3390/ijms20020283