Endoplasmic Reticulum Stress Impaired Uncoupling Protein 1 Expression via the Suppression of Peroxisome Proliferator-Activated Receptor γ Binding Activity in Mice Beige Adipocytes

 ,
,
Abstract
1. Introduction
2. Results
2.1. ER Stress Decreases Ucp1 mRNA Level in Beige Adipocytes
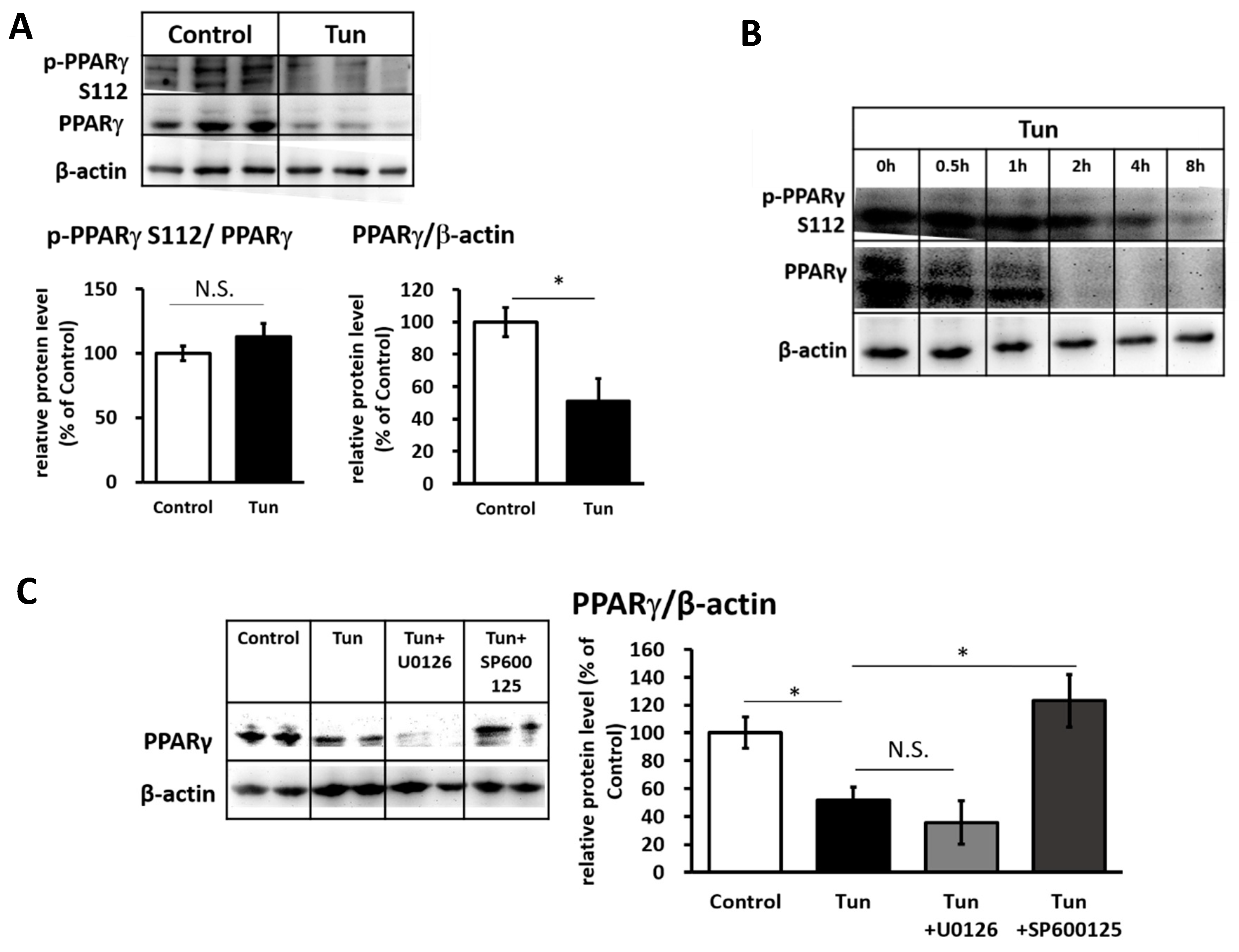
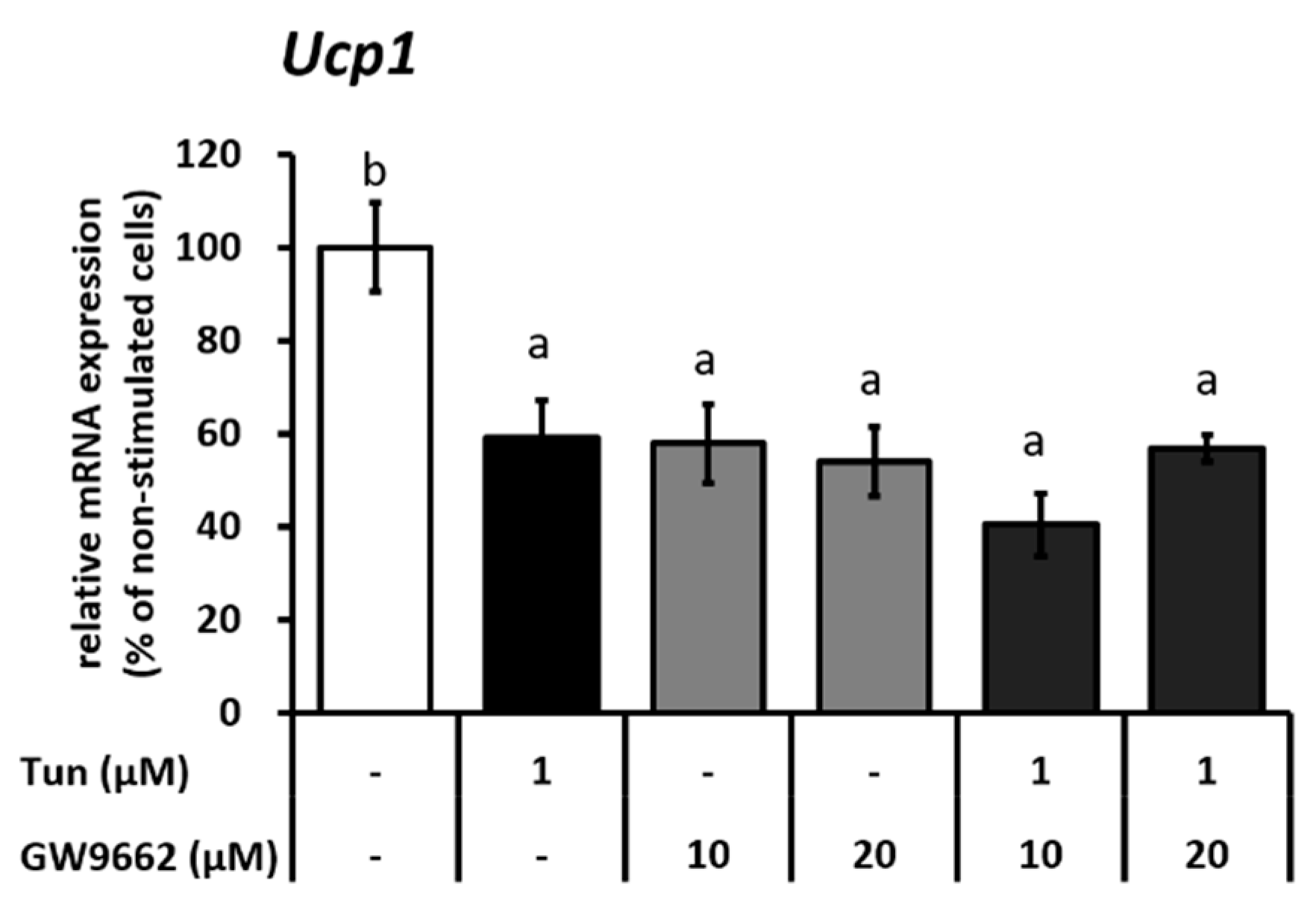
2.2. ER Stress Induces Downregulation of the Ucp1 Activator, Pparγ, Preferably via the JNK Pathway
2.3. ER Stress Stimulates Pparγ Degradation that Leads to the Reduced Binding Activity
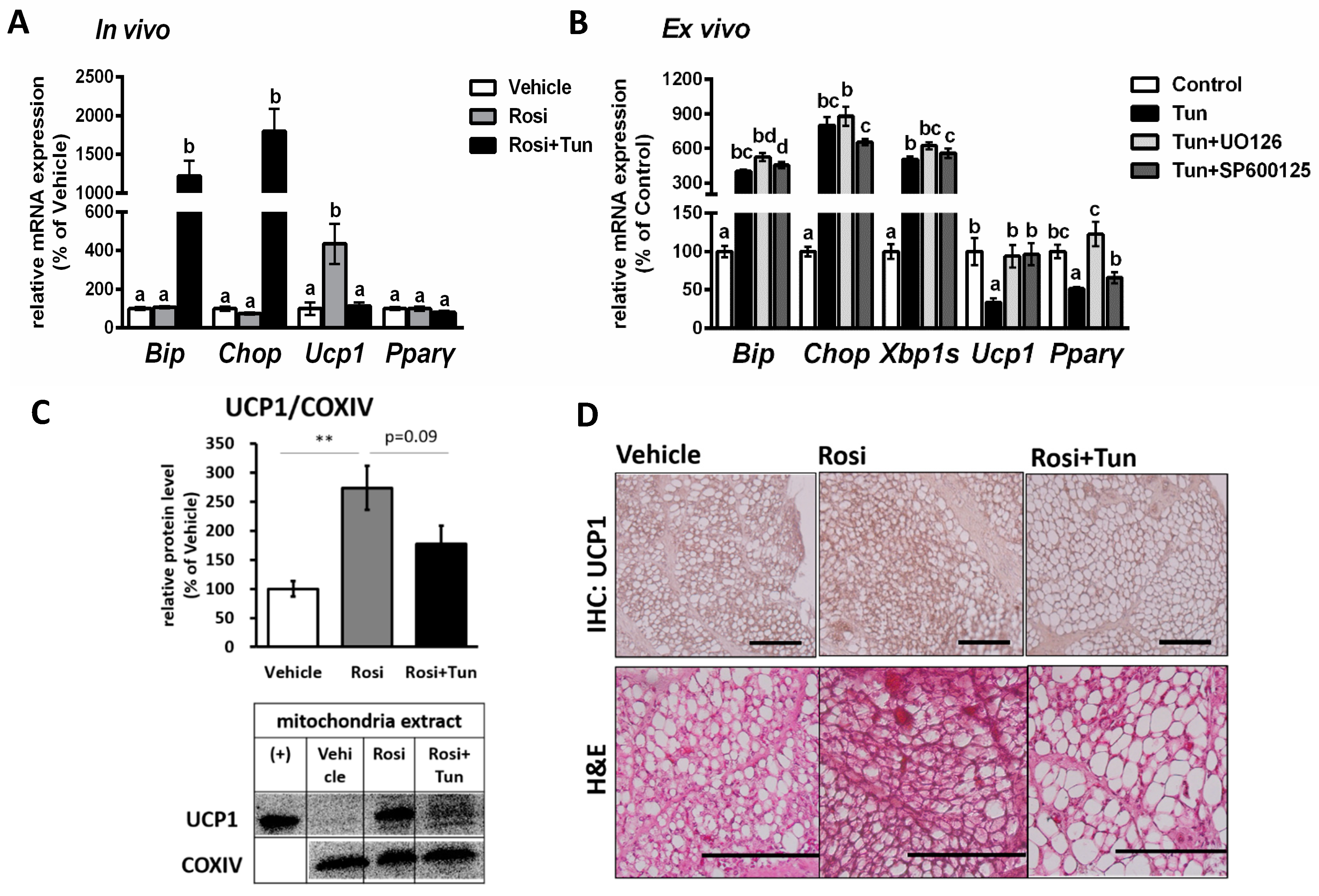
2.4. ER Stress Suppressed Both Ucp1 mRNA and Protein Expression in Adipose Tissue
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animal Experiment
4.3. Ex Vivo Experiment
4.4. Cell Culture
4.5. RNA Preparation and Quantification of Gene Expression
4.6. Protein Extraction and Western Blotting
4.7. Chromatin Immunoprecipitation (ChIP) Assay
4.8. Luciferase Assay
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| β-AR | beta-adrenergic receptor |
| BAT | brown adipose tissue |
| Bip | binding immunoglobulin protein |
| ChIP | chromatin immunoprecipitation |
| Chop | CCAAT-enhancer-binding protein homologous protein |
| COXIV | cytochrome c oxidase subunit IV |
| ER | endoplasmic reticulum |
| ERAD | ER-associated degradation |
| ERK | extracellular signal-regulated kinase |
| H&E | hematoxylin and eosin |
| IHC | immunohistochemical staining |
| IRE1α | inositol-requiring enzyme 1 alpha |
| IWAT | inguinal white adipose tissue |
| JNK | c-Jun N-terminal kinase |
| MAPK | mitogen-activated protein kinase |
| PBA | 4-phenylbutyrate |
| Pparγ | peroxisome proliferator-activated receptor gamma |
| PPRE | PPAR response element |
| Rosi | rosiglitazone |
| S112 | serine 112 |
| T3 | triiodo-L-thyronine |
| Ucp1 | uncoupling protein 1 |
| WAT | white adipose tissue |
| XBP1s | X-box binding protein 1 splicing |
References
- Villarroya, F.; Iglesias, R.; Giralt, M. PPARs in the control of uncoupling proteins gene expression. PPAR Res. 2007, 2007. [Google Scholar] [CrossRef] [PubMed]
- Harms, M.; Seale, P. Brown and beige fat: Development, function and therapeutic potential. Nat. Med. 2013, 19, 1252–1263. [Google Scholar] [CrossRef] [PubMed]
- Cannon, B.; Nedergaard, J. Brown adipose tissue: Function and physiological significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef] [PubMed]
- Vitali, A.; Murano, I.; Zingaretti, M.C.; Frontini, A.; Ricquier, D.; Cinti, S. The adipose organ of obesity-prone C57BL/6J mice is composed of mixed white and brown adipocytes. J. Lipid Res. 2012, 53, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Kalinovich, A.V.; de Jong, J.M.A.; Cannon, B.; Nedergaard, J. UCP1 in adipose tissues: Two steps to full browning. Biochimie 2017, 134, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Okla, M.; Wang, W.; Kang, I.; Pashaj, A.; Carr, T.; Chung, S. Activation of Toll-like receptor 4 (TLR4) attenuates adaptive thermogenesis via endoplasmic reticulum stress. J. Biol. Chem. 2015, 290, 26476–26490. [Google Scholar] [CrossRef]
- Lee, Y.K.; Cowan, C.A. White to brite adipocyte transition and back again. Nat. Cell Biol. 2013, 15, 568–569. [Google Scholar] [CrossRef]
- Rosenwald, M.; Perdikari, A.; Rülicke, T.; Wolfrum, C. Bi-directional interconversion of brite and white adipocytes. Nat. Cell Biol. 2013, 15, 659–667. [Google Scholar] [CrossRef]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.-H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hostamisligil, G.S. Endoplasmic Reticulum Stress Links Obesity, Insulin Action, and Type 2 Diabetes. Science 2003, 299, 1033–1036. [Google Scholar] [CrossRef]
- Lindholm, D.; Korhonen, L.; Eriksson, O.; Kõks, S. Recent Insights into the Role of Unfolded Protein Response in ER Stress in Health and Disease. Front. Cell Dev. Biol. 2017, 5, 1–16. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1085. [Google Scholar] [CrossRef]
- Hetz, C.; Chevet, E.; Harding, H.P. Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 2013, 12, 703–719. [Google Scholar] [CrossRef]
- Darling, N.J.; Cook, S.J. The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim. Biophys. Acta 2014, 1843, 2150–2163. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef]
- Kawasaki, N.; Asada, R.; Saito, A.; Kanemoto, S.; Imaizumi, K. Obesity-induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci. Rep. 2012, 2, 1–7. [Google Scholar] [CrossRef]
- Gregor, M.F.; Hotamisligil, G.S. Thematic review series: Adipocyte Biology. Adipocyte stress: The endoplasmic reticulum and metabolic disease. J. Lipid Res. 2007, 48, 1905–1914. [Google Scholar] [CrossRef]
- Han, J.; Kaufman, R.J. The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338. [Google Scholar] [CrossRef]
- Bartelt, A.; Widenmaier, S.B.; Schlein, C.; Johann, K.; Goncalves, R.L.S.; Eguchi, K.; Fischer, A.W.; Parlakgül, G.; Snyder, N.A.; Nguyen, T.B.; et al. Brown adipose tissue thermogenic adaptation requires Nrf1-mediated proteasomal activity. Nat. Med. 2018, 24, 292–303. [Google Scholar] [CrossRef]
- Salvadó, L.; Palomer, X.; Barroso, E.; Vázquez-Carrera, M. Targeting endoplasmic reticulum stress in insulin resistance. Trends Endocrinol. MeTable 2015, 26, 438–448. [Google Scholar] [CrossRef]
- Alcalá, M.; Calderon-Dominguez, M.; Bustos, E.; Ramos, P.; Casals, N.; Serra, D.; Viana, M.; Herrero, L. Increased inflammation, oxidative stress and mitochondrial respiration in brown adipose tissue from obese mice. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Deng, J.; Liu, S.; Zou, L.; Xu, C.; Geng, B.; Xu, G. Lipolysis response to endoplasmic reticulum stress in adipose cells. J. Biol. Chem. 2012, 287, 6240–6249. [Google Scholar] [CrossRef]
- Sakamoto, T.; Nitta, T.; Maruno, K.; Yeh, Y.-S.; Kuwata, H.; Tomita, K.; Goto, T.; Takahashi, N.; Kawada, T. Macrophage infiltration into obese adipose tissues suppresses the induction of UCP1 expression in mice. Am. J. Physiol. 2016. [Google Scholar] [CrossRef]
- Shan, B.; Wang, X.; Wu, Y.; Xu, C.; Xia, Z.; Dai, J.; Shao, M.; Zhao, F.; He, S.; Yang, L.; et al. The metabolic ER stress sensor IRE1α suppresses alternative activation of macrophages and impairs energy expenditure in obesity. Nat. Immunol. 2017, 18, 519–529. [Google Scholar] [CrossRef]
- Wagner, S.; Ribich, S.; Arrojo, R.; Castillo, M. The chemical chaperones tauroursodeoxycholic and 4-phenylbutyric acid accelerate thyroid hormone activation and energy expenditure. FEBS Lett. 2012, 585, 539–544. [Google Scholar] [CrossRef]
- Contreras, C.; González-García, I.; Seoane-Collazo, P.; Martínez-Sánchez, N.; Liñares-Pose, L.; Rial-Pensado, E.; Fernø, J.; Tena-Sempere, M.; Casals, N.; Diéguez, C.; et al. Reduction of hypothalamic endoplasmic reticulum stress activates browning of white fat and ameliorates obesity. Diabetes 2017, 66, 87–99. [Google Scholar] [CrossRef]
- Özcan, U.; Yilmaz, E.; Özcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Görgün, C.Z.; Hotamisligil, G.S. Chemical Chaperones Reduce ER stress and restore glucoes homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef]
- Gehart, H.; Kumpf, S.; Ittner, A.; Ricci, R. MAPK signalling in cellular metabolism: Stress or wellness? EMBO Rep. 2010, 11, 834–840. [Google Scholar] [CrossRef]
- Burgermeister, E.; Seger, R. MAPK kinases as nucleo-cytoplasmic shuttles for PPARγ. Cell Cycle 2007, 6, 1539–1548. [Google Scholar] [CrossRef]
- Sakamoto, T.; Takahashi, N.; Sawaragi, Y.; Naknukool, S.; Yu, R.; Goto, T.; Kawada, T. Inflammation induced by RAW macrophages suppresses UCP1 mRNA induction via ERK activation in 10T1/2 adipocytes. AJP Cell Physiol. 2013, 304, C729–C738. [Google Scholar] [CrossRef]
- Mukherjee, J.; Baranwal, A.N.; Schade, K. Classification of Therapeutic and Experimental Drugs for Brown Adipose Tissue Activation: Potential Treatment Strategies for Diabetes and Obesity. Curr. Diabetes Rev. 2016, 12, 414–428. [Google Scholar] [CrossRef]
- Salomone, S. Pleiotropic effects of glitazones: A double edge sword? Front. Pharmacol. 2011. [Google Scholar] [CrossRef]
- Digby, J.E.; Montague, C.T.; Sewter, C.P.; Sanders, L.; Wilkison, W.O.; O’Rahilly, S.; Prins, J.B. Thiazolidinedione exposure increases the expression of uncoupling protein 1 in cultured human preadipocytes. Diabetes 1998, 47, 138–141. [Google Scholar] [CrossRef]
- Asano, H.; Kanamori, Y.; Higurashi, S.; Nara, T.; Kato, K.; Matsui, T.; Funaba, M. Induction of Beige-Like Adipocytes in 3T3-L1 Cells. J. Vet. Med. Sci. 2014, 76, 57–64. [Google Scholar] [CrossRef]
- Miller, C.N.; Yang, J.Y.; England, E.; Yin, A.; Baile, C.A.; Rayalam, S. Isoproterenol increases uncoupling, glycolysis, and markers of beiging in mature 3T3-L1 adipocytes. PLoS ONE 2015, 10, 1–14. [Google Scholar] [CrossRef]
- Zhuo, X.Z.; Wu, Y.; Ni, Y.J.; Liu, J.H.; Gong, M.; Wang, X.H.; Wei, F.; Wang, T.Z.; Yuan, Z.; Ma, A.Q.; et al. Isoproterenol instigates cardiomyocyte apoptosis and heart failure via AMPK inactivation-mediated endoplasmic reticulum stress. Apoptosis 2013, 18, 800–810. [Google Scholar] [CrossRef]
- Soccio, R.E.; Chen, E.R.; Lazar, M.A. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell MeTable 2014, 20, 573–591. [Google Scholar] [CrossRef]
- Chen, H.Y.; Liu, Q.; Salter, A.M.; Lomax, M.A. Synergism between cAMP and PPAR γ signalling in the initiation of UCP1 gene expression in HIB1B brown adipocytes. PPAR Res. 2013, 2013. [Google Scholar] [CrossRef]
- Xue, B.; Coulter, A.; Rim, J.S.; Koza, R. a; Kozak, L.P. Transcriptional Synergy and the Regulation of Ucp1 during Brown Adipcytes Induction in White Fat Depots. Mol. Cell. Biol. 2005, 25, 8311–8322. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.-Z.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of Stress in the Endoplasmic Reticulum to Activation of JNK Protein Kinases by Transmembrane Protein Kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Hauser, S.; Adelmant, G.; Sarraf, P.; Wright, H.M.; Mueller, E.; Spiegelman, B.M. Degradation of the peroxisome proliferator-activated receptor γ is linked to ligand-dependent activation. J. Biol. Chem. 2000, 275, 18527–18533. [Google Scholar] [CrossRef]
- Camp, H.S.; Tafuri, S.R.; Leff, T.; Biology, C.; Biology, M. c-Jun N-Terminal Kinase Phosphorylates Peroxisome Proliferator-Activated Receptor- gamma 1 and Negatively Regulates Its Transcriptional Activity. Endocrinology 1999, 140, 392–397. [Google Scholar] [CrossRef]
- Molton, S.A.; Todd, D.E.; Cook, S.J. Selective activation of the c-Jun N-terminal kinase (JNK) pathway fails to elicit Bax activation or apoptosis unless the phosphoinositide 3′-kinase (PI3K) pathway is inhibited. Oncogene 2003, 22, 4690–4701. [Google Scholar] [CrossRef]
- Kim, B.J.; Ryu, S.W.; Song, B.J. JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J. Biol. Chem. 2006, 281, 21256–21265. [Google Scholar] [CrossRef]
- Zinszner, H.; Kuroda, M.; Wang, X.Z.; Batchvarova, N.; Lightfoot, R.T.; Remotti, H.; Stevens, J.L.; Ron, D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998, 12, 982–995. [Google Scholar] [CrossRef]
- Guilherme, A.; Tesz, G.J.; Guntur, K.V.P.; Czech, M.P. Tumor necrosis factor-α induces caspase-mediated cleavage of peroxisome proliferator-activated receptor γ in adipocytes. J. Biol. Chem. 2009, 284, 17082–17091. [Google Scholar] [CrossRef]
- Zha, B.S.; Zhou, H. ER stress and lipid metabolism in adipocytes. Biochem. Res. Int. 2012, 2012. [Google Scholar] [CrossRef]
- Lockhart, D.J.; Travers, K.J.; Walter, P.; Patil, C.K.; Wodicka, L.; Weissman, J.S.; Lockhart, D.J.; Weissman, J.S.; Walter, P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 2000, 101, 249–258. [Google Scholar] [CrossRef]
- Ohno, H.; Shinoda, K.; Spiegelman, B.M.; Kajimura, S. PPARγ agonists induce a white-to-brown fat conversion through stabilization of PRDM16 protein. Cell MeTable 2012, 15, 395–404. [Google Scholar] [CrossRef]
- Yuliana, A.; Jheng, H.-F.; Kawarasaki, S.; Nomura, W.; Takahashi, H.; Ara, T.; Kawada, T.; Goto, T. β-adrenergic Receptor Stimulation Revealed a Novel Regulatory Pathway via Suppressing Histone Deacetylase 3 to Induce Uncoupling Protein 1 Expression in Mice Beige Adipocyte. Int. J. Mol. Sci. 2018, 19, 2436. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| Ucp1 | 5′-CAAAGTCCGCCTTCAGATCC-3′ | 5′-AGCCGGCTGAGATCTTGTTT-3′ |
| Pparγ | 5′-GGAGATCTCCAGTGATATCGACCA-3′ | 5′-ACGGCTTCTACGGATCGAAAACT-3′ |
| 36b4 | 5′-TCCTTCTTCCAGGCTTTGGG-3′ | 5′-GACACCCTCCAGAAAGCGAG-3′ |
| Bip | 5′-GTTTGCTGAGGAAGACAAAAAGCTC-3′ | 5′-CACTTCCATAGAGTTTGCTGATAAT-3′ |
| Chop | 5′-GTCCAGCTGGGAGCTGGAAG-3′ | 5′-CTGACTGGAATCTGGAGAG-3′ |
| Gene | Forward | Reverse |
|---|---|---|
| Ucp1 enhancer | 5′-CTCCTCTACAGCGTCACAGAGG-3′ | 5-AGTCTGAGGAAAGGGTTGA-3′ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuliana, A.; Daijo, A.; Jheng, H.-F.; Kwon, J.; Nomura, W.; Takahashi, H.; Ara, T.; Kawada, T.; Goto, T. Endoplasmic Reticulum Stress Impaired Uncoupling Protein 1 Expression via the Suppression of Peroxisome Proliferator-Activated Receptor γ Binding Activity in Mice Beige Adipocytes. Int. J. Mol. Sci. 2019, 20, 274. https://doi.org/10.3390/ijms20020274
Yuliana A, Daijo A, Jheng H-F, Kwon J, Nomura W, Takahashi H, Ara T, Kawada T, Goto T. Endoplasmic Reticulum Stress Impaired Uncoupling Protein 1 Expression via the Suppression of Peroxisome Proliferator-Activated Receptor γ Binding Activity in Mice Beige Adipocytes. International Journal of Molecular Sciences. 2019; 20(2):274. https://doi.org/10.3390/ijms20020274
Chicago/Turabian StyleYuliana, Ana, Asumi Daijo, Huei-Fen Jheng, Jungin Kwon, Wataru Nomura, Haruya Takahashi, Takeshi Ara, Teruo Kawada, and Tsuyoshi Goto. 2019. "Endoplasmic Reticulum Stress Impaired Uncoupling Protein 1 Expression via the Suppression of Peroxisome Proliferator-Activated Receptor γ Binding Activity in Mice Beige Adipocytes" International Journal of Molecular Sciences 20, no. 2: 274. https://doi.org/10.3390/ijms20020274
APA StyleYuliana, A., Daijo, A., Jheng, H.-F., Kwon, J., Nomura, W., Takahashi, H., Ara, T., Kawada, T., & Goto, T. (2019). Endoplasmic Reticulum Stress Impaired Uncoupling Protein 1 Expression via the Suppression of Peroxisome Proliferator-Activated Receptor γ Binding Activity in Mice Beige Adipocytes. International Journal of Molecular Sciences, 20(2), 274. https://doi.org/10.3390/ijms20020274