Linking Endoplasmic Reticular Stress and Alternative Splicing



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Model Systems for Studying ER Stress
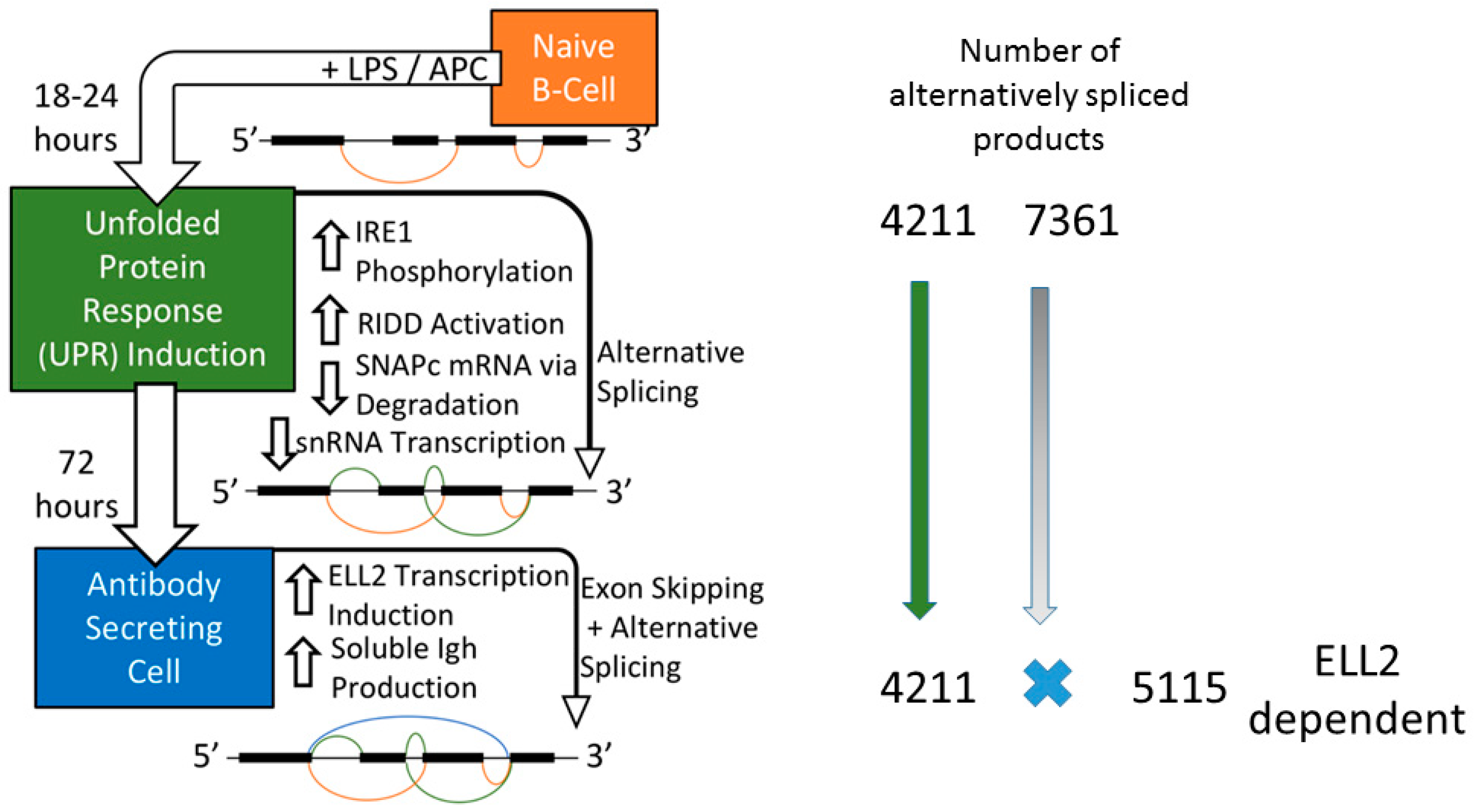
2.1. B cell Activation to Antibody Secreting Cells
2.2. Liver Secretion and the Effects of RIDD
2.3. Drosophila
2.4. Pancreatic Beta Cells
3. Ire1 Activation and ELL2 Induction with Alternative Splicing
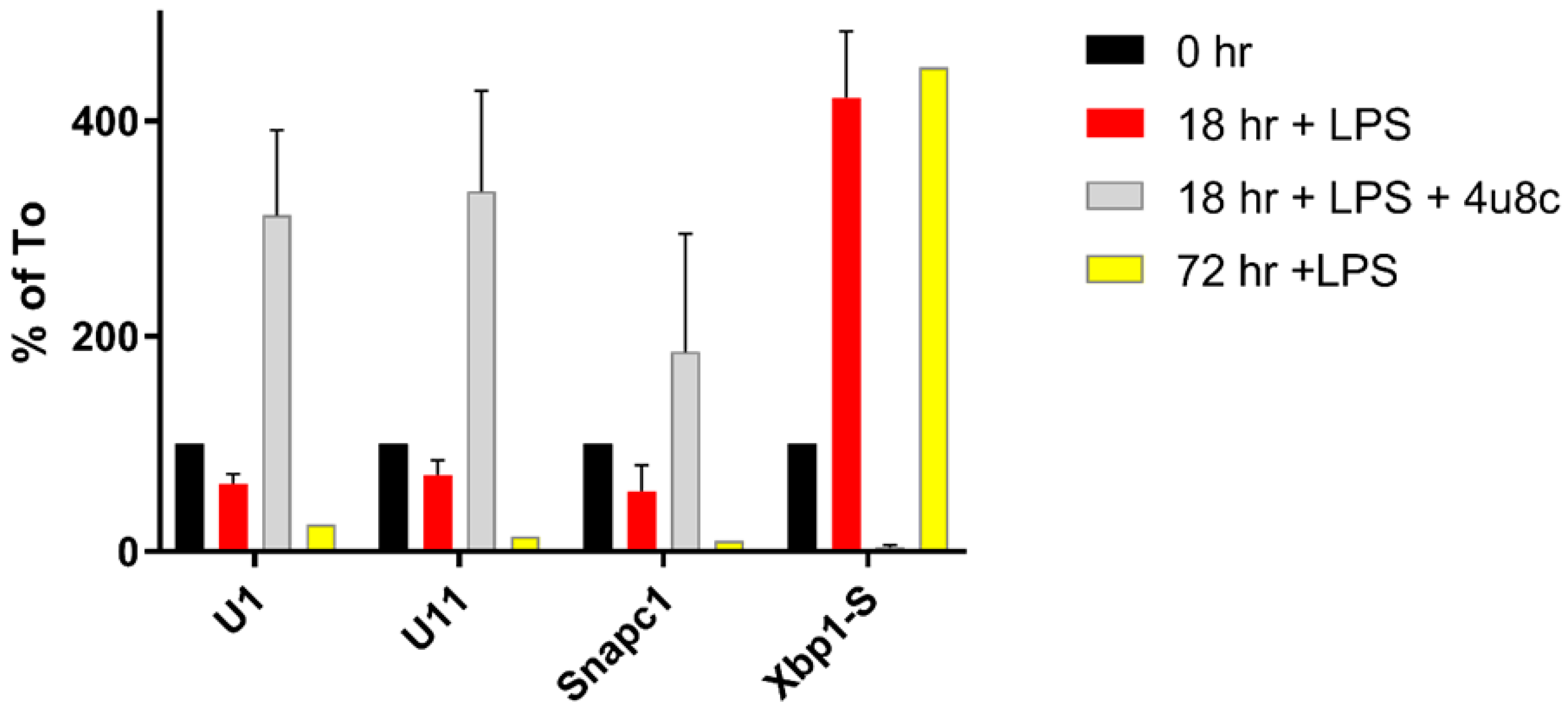
3.1. ER Stress Activates RIDD
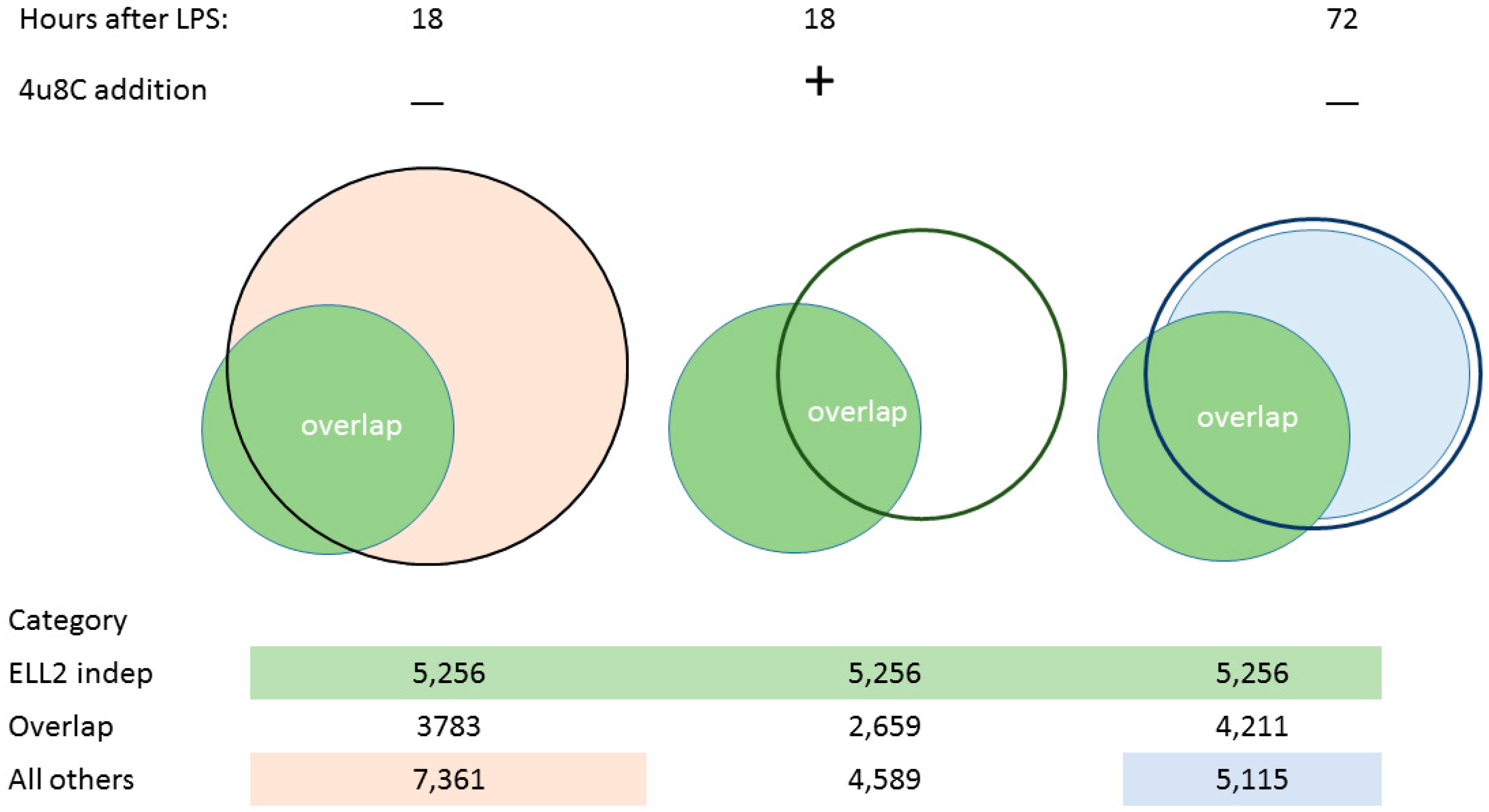
3.2. Splicing Patterns Are Changed When B Cells Are Activated
3.3. Changes in snRNA Are Correlated with the Timing of RIDD
3.4. snRNA Levels in Cell Growth
4. Changes in snRNA Levels Can Alter Gene Expression
5. Other Changes Associated with Stress That Could Change snRNAs and Splicing Patterns
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ASC | antibody-secreting cells |
| ELL2 | Eleven-nineteen lysine-rich leukemia gene 2 |
| ER | endoplasmic reticulum |
| Ig | immunoglobulin |
| IgH | immunoglobulin heavy chain |
| LPS | lipopolysaccharide, a polyclonal B cell activator |
| RIDD | regulated Ire1-dependent mRNA decay |
| UPR | unfolded protein response |
References
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef] [PubMed]
- Manz, R.A.; Radbruch, A. Plasma cells for a lifetime? Eur. J. Immunol. 2002, 32, 923–927. [Google Scholar] [CrossRef]
- Vincenz-Donnelly, L.; Hipp, M.S. The endoplasmic reticulum: A hub of protein quality control in health and disease. Free Radic. Biol. Med. 2017, 108, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Orthwein, A.; Patenaude, A.-M.; Affar, E.B.; Lamarre, A.; Young, J.C.; Di Noia, J.M. Regulation of activation-induced deaminase stability and antibody gene diversification by Hsp90. J. Exp. Med. 2010, 207, 2751–2765. [Google Scholar] [CrossRef] [PubMed]
- Hendershot, L.; Bole, D.; Kearney, J.F. The role of immunoglobulin heavy chain binding protein: In immunoglobulin transport. Immunol. Today 1987, 8, 111–114. [Google Scholar] [CrossRef]
- Penke, B.; Bogár, F.; Crul, T.; Sántha, M.; Tóth, M.; Vígh, L. Heat Shock Proteins and Autophagy Pathways in Neuroprotection: From Molecular Bases to Pharmacological Interventions. Int. J. Mol. Sci. 2018, 19, 325. [Google Scholar] [CrossRef] [PubMed]
- Back, S.H.; Schröder, M.; Lee, K.; Zhang, K.; Kaufman, R.J. ER stress signaling by regulated splicing: IRE1/HAC1/XBP1. Methods 2005, 35, 395–416. [Google Scholar] [CrossRef]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Bayles, I.; Milcarek, C. Plasma cell formation, secretion, and persistence: The short and the long of it. Crit. Rev. Immunol. 2014, 34, 481–499. [Google Scholar] [CrossRef]
- Li, G.; Mongillo, M.; Chin, K.-T.; Harding, H.; Ron, D.; Marks, A.R.; Tabas, I. Role of ERO1-α–mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J. Cell Biol. 2009, 186, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Shimizu, Y.; Mann, M.J.; Jin, Y.; Hendershot, L.M. Plasma cell differentiation initiates a limited ER stress response by specifically suppressing the PERK-dependent branch of the unfolded protein response. Cell Stress Chaperones 2010, 15, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Gass, J.N.; Jiang, H.-Y.; Wek, R.C.; Brewer, J.W. The unfolded protein response of B-lymphocytes: PERK-independent development of antibody-secreting cells. Mol. Immunol. 2008, 45, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Aragon, I.V.; Barrington, R.A.; Jackowski, S.; Mori, K.; Brewer, J.W. The Specialized Unfolded Protein Response of B Lymphocytes: ATF6α-Independent Development of Antibody-Secreting B cells. Mol. Immunol. 2012, 51, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Price, M.J.; Patterson, D.G.; Scharer, C.D.; Boss, J.M. Progressive Upregulation of Oxidative Metabolism Facilitates Plasmablast Differentiation to a T-Independent Antigen. Cell Rep. 2018, 23, 3152–3159. [Google Scholar] [CrossRef] [PubMed]
- Kumazaki, K.; Tirosh, B.; Maehr, R.; Boes, M.; Honjo, T.; Ploegh, H.L. AID−/−μs−/− mice are agammaglobulinemic and fail to maintain B220−CD138+ plasma cells. J. Immunol. 2007, 178, 2192–2203. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Bayles, I.; Szlachta-McGinn, A.; Paul, J.; Boiko, J.; Santos, P.; Liu, J.; Wang, Z.; Borghesi, L.; Milcarek, C. Transcription elongation factor ELL2 drives immunoglobulin secretory specific mRNA production and the unfolded protein response. J. Immunol. 2014, 193, 4663–4674. [Google Scholar] [CrossRef]
- Han, D.; Lerner, A.G.; Vande Walle, L.; Upton, J.-P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. IRE1α Kinase Activation Modes Control Alternate Endoribonuclease Outputs to Determine Divergent Cell Fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef]
- Benhamron, S.; Hadar, R.; Iwawaky, T.; So, J.-S.; Lee, A.-H.; Tirosh, B. Regulated IRE-1 dependent decay participates in curtailing immunoglobulin secretion from plasma cells. Eur. J. Immunol. 2014, 44, 867–876. [Google Scholar] [CrossRef]
- Benhamron, S.; Pattanayak, S.P.; Berger, M.; Tirosh, B. mTOR activation promotes plasma cell differentiation and bypasses XBP-1 for immunoglobulin secretion. Mol. Cell. Biol. 2015, 35, 153–166. [Google Scholar] [CrossRef]
- Goldfinger, M.; Shmuel, M.; Benhamron, S.; Tirosh, B. Protein synthesis in plasma cells is regulated by crosstalk between endoplasmic reticulum stress and mTOR signaling. Eur. J. Immunol. 2011, 41, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, B.; Iwakoshi, N.N.; Glimcher, L.H.; Ploegh, H.L. XBP-1 specifically promotes IgM synthesis and secretion, but is dispensable for degradation of glycoproteins in primary B cells. J. Exp. Med. 2005, 202, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.D.; Gaudette, B.T.; Wilmore, J.R.; Chernova, I.; Bortnick, A.; Weiss, B.M.; Allman, D. mTOR has distinct functions in generating versus sustaining humoral immunity. J. Clin. Investig. 2016, 126, 4250–4261. [Google Scholar] [CrossRef] [PubMed]
- Sintes, J.; Gentile, M.; Zhang, S.; Garcia-Carmona, Y.; Magri, G.; Cassis, L.; Segura-Garzón, D.; Ciociola, A.; Grasset, E.K.; Bascones, S.; et al. mTOR intersects antibody-inducing signals from TACI in marginal zone B cells. Nat. Commun. 2017, 8, 1462. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-M.; Qiu, Y.; Yang, Z.; Kim, H.; Qian, Q.; Sun, Q.; Zhang, C.; Yin, L.; Fang, D.; Back, S.H.; et al. IRE1α prevents hepatic steatosis by processing and promoting the degradation of select microRNAs. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Herrema, H.; Zhou, Y.; Zhang, D.; Lee, J.; Salazar Hernandez, M.A.; Shulman, G.I.; Ozcan, U. XBP1s Is an Anti-lipogenic Protein. J. Biol. Chem. 2016, 291, 17394–17404. [Google Scholar] [CrossRef] [PubMed]
- So, J.-S.; Hur, K.Y.; Tarrio, M.; Ruda, V.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Lichtman, A.H.; Iwawaki, T.; Glimcher, L.H.; et al. Silencing of lipid metabolism genes through IRE1α-mediated mRNA decay lowers plasma lipids in mice. Cell Metab. 2012, 16, 487–499. [Google Scholar] [CrossRef]
- So, J.-S.; Cho, S.; Min, S.-H.; Kimball, S.R.; Lee, A.-H. IRE1α-Dependent Decay of CReP/Ppp1r15b mRNA Increases Eukaryotic Initiation Factor 2α Phosphorylation and Suppresses Protein Synthesis. Mol. Cell. Biol. 2015, 35, 2761–2770. [Google Scholar] [CrossRef]
- Hollien, J.; Weissman, J.S. Decay of Endoplasmic Reticulum-Localized mRNAs During the Unfolded Protein Response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef]
- Coelho, D.S.; Cairrão, F.; Zeng, X.; Pires, E.; Coelho, A.V.; Ron, D.; Ryoo, H.D.; Domingos, P.M. Xbp1-Independent Ire1 Signaling Is Required for Photoreceptor Differentiation and Rhabdomere Morphogenesis in Drosophila. Cell Rep. 2013, 5, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Skelly, R.H.; Schuppin, G.T.; Ishihara, H.; Oka, Y.; Rhodes, C.J. Glucose-Regulated Translational Control of Proinsulin Biosynthesis With That of the Proinsulin Endopeptidases PG2 and PC3 in the Insulin-Producing MIN6 Cell Line. Diabetes 1996, 45, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Cui, S.; Zong, C.; Gao, W.; Xu, T.; Gao, P.; Chen, J.; Qin, D.; Guan, Q.; Liu, Y.; et al. The Orphan Nuclear Receptor NR4A1 Protects Pancreatic β-Cells from Endoplasmic Reticulum (ER) Stress-mediated Apoptosis. J. Biol. Chem. 2015, 290, 20687–20699. [Google Scholar] [CrossRef]
- Gao, W.; Fu, Y.; Yu, C.; Wang, S.; Zhang, Y.; Zong, C.; Xu, T.; Liu, Y.; Li, X.; Wang, X.D. Elevation of NR4A3 expression and its possible role in modulating insulin expression in the pancreatic beta cell. PLoS ONE 2014, 9, e91462. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Kawano, J.; Takeda, K.; Yujiri, T.; Tanabe, K.; Anno, T.; Akiyama, M.; Nozaki, J.; Yoshinaga, T.; Koizumi, A.; et al. Endoplasmic reticulum stress induces Wfs1 gene expression in pancreatic β-cells via transcriptional activation. Eur. J. Endocrin. 2005, 153, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Reimold, A.M.; Iwakoshi, N.N.; Manis, J.; Vallabhajosyula, P.; Szomolanyi-Tsuda, E.; Gravallese, E.M.; Friend, D.; Grusby, M.J.; Alt, F.; Glimcher, L.H. Plasma cell differentiation requires the transcription factor XBP-1. Nature 2001, 412, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, D.; Tokuda, M.; Hosoda, A.; Iwawaki, T. Identification of a consensus element recognized and cleaved by IRE1α. Nucleic Acids Res. 2010, 38, 6265–6273. [Google Scholar] [CrossRef]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef]
- Lee, K.P.K.; Dey, M.; Neculai, D.; Cao, C.; Dever, T.E.; Sicheri, F. Structure of the Dual Enzyme Ire1 Reveals the Basis for Catalysis and Regulation in Nonconventional RNA Splicing. Cell 2008, 132, 89–100. [Google Scholar] [CrossRef]
- Sidrauski, C.; Cox, J.S.; Walter, P. tRNA Ligase Is Required for Regulated mRNA Splicing in the Unfolded Protein Response. Cell 1996, 87, 405–413. [Google Scholar] [CrossRef]
- Cox, J.S.; Walter, P. A Novel Mechanism for Regulating Activity of a Transcription Factor That Controls the Unfolded Protein Response. Cell 1996, 87, 391–404. [Google Scholar] [CrossRef]
- Niwa, M.; Patil, C.K.; DeRisi, J.; Walter, P. Genome-scale approaches for discovering novel nonconventional splicing substrates of the Ire1 nuclease. Genome Biol. 2005, 6, R3. [Google Scholar] [CrossRef] [PubMed]
- Guydosh, N.R.; Kimmig, P.; Walter, P.; Green, R. Regulated Ire1-dependent mRNA decay requires no-go mRNA degradation to maintain endoplasmic reticulum homeostasis in S. pombe. eLife 2017, 6, e29216. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.B.; Koong, A.C.; Niwa, M. Ire1 Has Distinct Catalytic Mechanisms for XBP1/HAC1 Splicing and RIDD. Cell Rep. 2014, 9, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.; Hollien, J. Ire1-mediated decay in mammalian cells relies on mRNA sequence, structure, and translational status. Mol. Biol. Cell 2015, 26, 2873–2884. [Google Scholar] [CrossRef] [PubMed]
- Tavernier, Q.; Bennana, E.; Poindessous, V.; Schaeffer, C.; Rampoldi, L.; Pietrancosta, N.; Pallet, N. Regulation of IRE1 RNase activity by the Ribonuclease inhibitor 1 (RNH1). Cell Cycle 2018, 17, 1901–1916. [Google Scholar] [CrossRef] [PubMed]
- Cross, B.C.S.; Bond, P.J.; Sadowski, P.G.; Jha, B.K.; Zak, J.; Goodman, J.M.; Silverman, R.H.; Neubert, T.A.; Baxendale, I.R.; Ron, D.; et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc. Natl. Acad. Sci. USA 2012, 109, E869–E878. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.M.; Carew, N.T.; Smith, S.M.; Milcarek, C. RNA splicing in the transition from B cells to antibody secreting cells: The influences of ELL2, snRNA and ER stress. J. Immunol. 2018, 201, 3073–3083. [Google Scholar] [CrossRef]
- Martincic, K.; Alkan, S.A.; Cheatle, A.; Borghesi, L.; Milcarek, C. Transcription elongation factor ELL2 directs immunoglobulin secretion in plasma cells by stimulating altered RNA processing. Nat. Immunol. 2009, 10, 1102–1109. [Google Scholar] [CrossRef]
- Shell, S.A.; Martincic, K.; Tran, J.; Milcarek, C. Increased phosphorylation of the carboxyl-terminal domain of RNA polymerase II and loading of polyadenylation and cotranscriptional factors contribute to regulation of the Ig heavy chain mRNA in plasma cells. J. Immunol. 2007, 179, 7663–7673. [Google Scholar] [CrossRef]
- Milcarek, C.; Albring, M.; Langer, C.; Park, K.S. The Eleven-Nineteen Lysine-rich Leukemia gene (ELL2) influences the histone H3 modifications accompanying the shift to secretory Immunoglobulin heavy chain mRNA production. J. Biol. Chem. 2011, 286, 33795–33803. [Google Scholar] [CrossRef] [PubMed]
- He, N.; Liu, M.; Hsu, J.; Xue, Y.; Chou, S.; Burlingame, A.; Krogan, N.J.; Alber, T.; Zhou, Q. HIV-1 Tat and Host AFF4 Recruit Two Transcription Elongation Factors into a Bifunctional Complex for Coordinated Activation of HIV-1 Transcription. Mol. Cell 2010, 38, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.; Li, Z.; Schulze-Gahmen, U.; Stjepanovic, G.; Zhou, Q.; Hurley, J.H. Structural basis for ELL2 and AFF4 activation of HIV-1 proviral transcription. Nat. Commun. 2017, 8, 14076. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Yoon, J.B.; Gerster, T.; Roeder, R.G. Oct-1 and Oct-2 potentiate functional interactions of a transcription factor with the proximal sequence element of small nuclear RNA genes. Mol. Cell. Biol. 1992, 12, 3247–3261. [Google Scholar] [CrossRef] [PubMed]
- Henry, R.W.; Mittal, V.; Ma, B.; Kobayashi, R.; Hernandez, N. SNAP19 mediates the assembly of a functional core promoter complex (SNAP(c)) shared by RNA polymerases II and III. Genes Dev. 1998, 12, 2664–2672. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.R.; Lin, C.; Garrett, A.S.; Thornton, J.; Mohaghegh, N.; Hu, D.; Jackson, J.; Saraf, A.; Swanson, S.K.; Seidel, C.; et al. The little elongation complex regulates small nuclear RNA transcription. Mol. Cell 2011, 44, 954–965. [Google Scholar] [CrossRef] [PubMed]
- Martincic, K.; Campbell, R.; Edwalds-Gilbert, G.; Souan, L.; Lotze, M.T.; Milcarek, C. Increase in the 64-kDa subunit of the polyadenylation/cleavage stimulatory factor during the Go to S phase transition. Proc. Natl. Acad. Sci. USA 1998, 95, 11095–11100. [Google Scholar] [CrossRef] [PubMed]
- Kuncio, G.S.; Goldstein, L. Small nuclear rnas in cellular growth and differentiation. I: Metabolic alterations seen in friend erythroleukemic cells. J. Cell. Physiol. 1981, 109, 235–241. [Google Scholar] [CrossRef]
- Ray, R.; Ray, K.; Panda, C.K. Differential alterations in metabolic pattern of the six major U snRNAs during development. Mol. Cell. Biochem. 1997, 177, 79–88. [Google Scholar] [CrossRef]
- Cheng, Z.; Du, Z.; Shang, Y.; Zhang, Y.; Zhang, T. A preliminary study: PS1 increases U1 snRNA expression associated with AD. J. Mol. Neurosci. 2017, 62, 269–275. [Google Scholar] [CrossRef]
- Singh, R.K.; Cooper, T.A. Pre-mRNA splicing in disease and therapeutics. Trends Mol. Med. 2012, 18, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Milcarek, C.; Martincic, K.; Chung-Ganster, L.-H.; Lutz, C.S. The snRNP-associated U1A levels change following IL-6 stimulation of human B-cells. Mol. Immunol. 2003, 39, 809–814. [Google Scholar] [CrossRef]
- Ma, J.; Gunderson, S.I.; Phillips, C. Non-snRNP U1A levels decrease during mammalian B-cell differentiation and release the IgM secretory poly(A) site from repression. RNA 2006, 12, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.; Pachikara, N.; Gunderson, S.I. U1A Inhibits Cleavage at the Immunoglobulin M Heavy-Chain Secretory Poly(A) Site by Binding between the Two Downstream GU-Rich Regions. Mol. Cell. Biol. 2004, 24, 6162–6171. [Google Scholar] [CrossRef] [PubMed]
- Berg, M.G.; Singh, L.N.; Younis, I.; Liu, Q.; Pinto, A.M.; Kaida, D.; Zhang, Z.; Cho, S.; Sherrill-Mix, S.; Wan, L.; et al. U1 snRNP determines mRNA length and regulates isoform expression. Cell 2012, 150, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Kaida, D.; Berg, M.G.; Younis, I.; Kasim, M.; Singh, L.N.; Wan, L.; Dreyfuss, G. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature 2010, 468, 664–668. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, M.; Lemoine, C.; Al Seesi, S.; Karunakaran, D.K.P.; Sturrock, N.; Banday, A.R.; Kilcollins, A.M.; Mandoiu, I.; Kanadia, R.N. Minor splicing snRNAs are enriched in the developing mouse CNS and are crucial for survival of differentiating retinal neurons. Dev. Neurobiol. 2014, 75, 895–907. [Google Scholar] [CrossRef]
- Barboric, M.; Lenasi, T.; Chen, H.; Johansen, E.B.; Guo, S.; Peterlin, B.M. 7SK snRNP/pTEFb couples transcription elongation with alternative splicing and is essential for vertebrate development. Proc. Natl. Acad. Sci. USA 2009, 106, 7798–7803. [Google Scholar] [CrossRef]
- Lis, J.T.; Mason, P.; Peng, J.; Price, D.H.; Werner, J. P-TEFb kinase recruitment and function at heat shock loci. Genes Dev. 2000, 14, 792–803. [Google Scholar] [CrossRef]
- Chen, R.; Yik, J.H.N.; Lew, Q.J.; Chao, S.-H. Brd4 and HEXIM1: Multiple Roles in P-TEFb Regulation and Cancer. BioMed Res. Int. 2014, 2014, 232870. [Google Scholar] [CrossRef]
- Smith, E.; Lin, C.; Shilatifard, A. The super elongation complex (SEC) and MLL in development and disease. Genes Dev. 2011, 25, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Yik, J.H.N. The Yin and Yang of P-TEFb Regulation: Implications for Human Immunodeficiency Virus Gene Expression and Global Control of Cell Growth and Differentiation. Microbiol. Mol. Biol. Rev. 2006, 70, 646–659. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.L.; Fogley, R.D.; Flynn, R.A.; Ablain, J.; Yang, S.; Saint-André, V.; Fan, Z.P.; Do, B.T.; Laga, A.C.; Fujinaga, K.; et al. Stress from Nucleotide Depletion Activates the Transcriptional Regulator HEXIM1 to Suppress Melanoma. Mol. Cell 2016, 62, 34–46. [Google Scholar] [CrossRef] [PubMed]
- De la Mata, M.; Lafaille, C.; Kornblihtt, A.R. First come, first served revisited: Factors affecting the same alternative splicing event have different effects on the relative rates of intron removal. RNA 2010, 16, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Shilatifard, A.; Duan, D.R.; Haque, D.; Florence, C.; Schubach, W.H.; Conaway, J.W.; Conaway, R.C. ELL2, a new member of an ELL family of RNA polymerase II elongation factors. Proc. Natl. Acad. Sci. USA 1997, 94, 3639–3643. [Google Scholar] [CrossRef] [PubMed]
- Arumemi, F.; Bayles, I.; Paul, J.; Milcarek, C. Shared and discrete interacting partners of ELL1 and ELL2 by yeast two-hybrid assay. Adv. Biosci. Biotechnol. 2013, 4, 774–780. [Google Scholar] [CrossRef]
- Gridasova, A.A.; Henry, R.W. The p53 Tumor Suppressor Protein Represses Human snRNA Gene Transcription by RNA Polymerases II and III Independently of Sequence-Specific DNA Binding. Mol. Cell. Biol. 2005, 25, 3247–3260. [Google Scholar] [CrossRef]
- Anwar, D.; Takahashi, H.; Watanabe, M.; Suzuki, M.; Fukuda, S.; Hatakeyama, S. p53 represses the transcription of snRNA genes by preventing the formation of little elongation complex. Biochim. Biophys. Acta 2016, 1859, 975–982. [Google Scholar] [CrossRef]
- Hu, W.; Feng, Z.; Levine, A.J. The Regulation of Multiple p53 Stress Responses is Mediated through MDM2. Genes Cancer 2012, 3, 199–208. [Google Scholar] [CrossRef]
- Ortega, A.; Roselló-Lletí, E.; Tarazón, E.; Molina-Navarro, M.M.; Martínez-Dolz, L.; González-Juanatey, J.R.; Lago, F.; Montoro-Mateos, J.D.; Salvador, A.; Rivera, M.; et al. Endoplasmic Reticulum Stress Induces Different Molecular Structural Alterations in Human Dilated and Ischemic Cardiomyopathy. PLoS ONE 2014, 9, e107635. [Google Scholar] [CrossRef]
- Santos, L.E.; Ferreira, S.T. Crosstalk between endoplasmic reticulum stress and brain inflammation in Alzheimer’s disease. Neuropharmacology 2018, 136, 350–360. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carew, N.T.; Nelson, A.M.; Liang, Z.; Smith, S.M.; Milcarek, C. Linking Endoplasmic Reticular Stress and Alternative Splicing. Int. J. Mol. Sci. 2018, 19, 3919. https://doi.org/10.3390/ijms19123919
Carew NT, Nelson AM, Liang Z, Smith SM, Milcarek C. Linking Endoplasmic Reticular Stress and Alternative Splicing. International Journal of Molecular Sciences. 2018; 19(12):3919. https://doi.org/10.3390/ijms19123919
Chicago/Turabian StyleCarew, Nolan T., Ashley M. Nelson, Zhitao Liang, Sage M. Smith, and Christine Milcarek. 2018. "Linking Endoplasmic Reticular Stress and Alternative Splicing" International Journal of Molecular Sciences 19, no. 12: 3919. https://doi.org/10.3390/ijms19123919
APA StyleCarew, N. T., Nelson, A. M., Liang, Z., Smith, S. M., & Milcarek, C. (2018). Linking Endoplasmic Reticular Stress and Alternative Splicing. International Journal of Molecular Sciences, 19(12), 3919. https://doi.org/10.3390/ijms19123919