Fertility of Pedicellate Spikelets in Sorghum Is Controlled by a Jasmonic Acid Regulatory Module

 ,
,  , ,
, ,  and
and 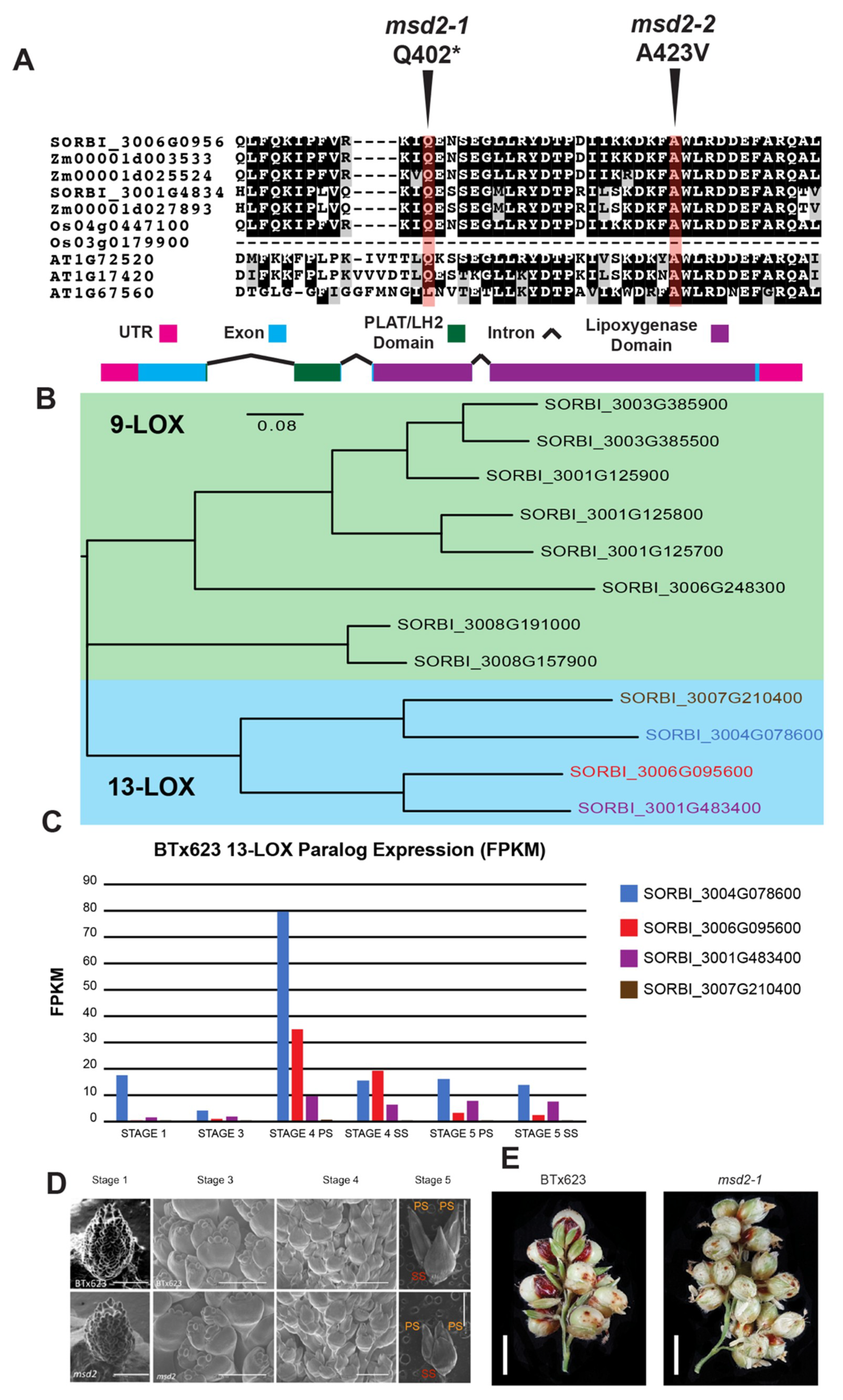{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Significance
2. Introduction
3. Results
3.1. MSD2 Encodes a Lipoxygenase in the Jasmonic Acid Biosynthetic Pathway
3.2. The msd2 Phenotype is Rescued by Exogenous Methyl-JA Treatment
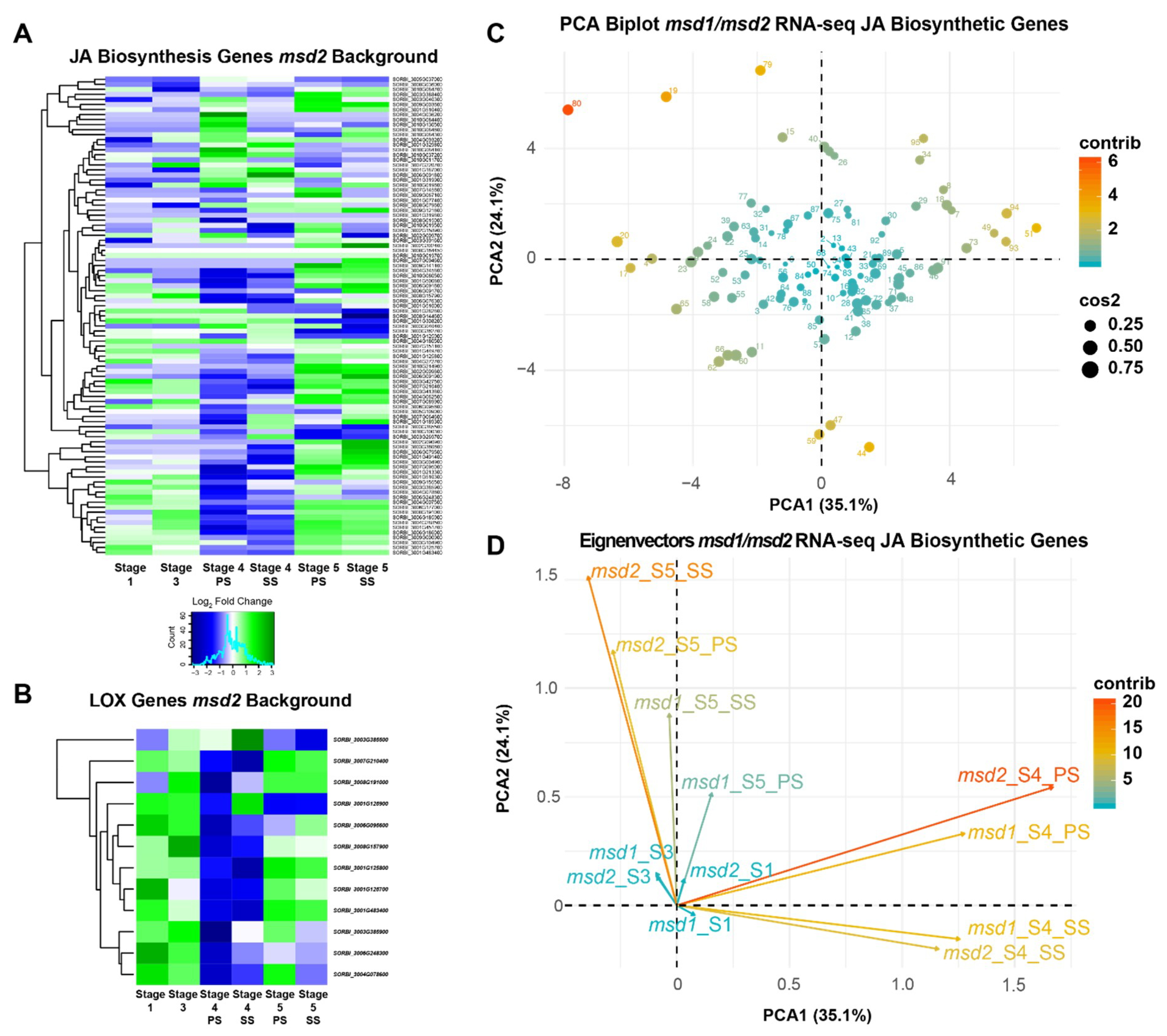
3.3. MSD2 Regulatory Networks Are Similar to MSD1
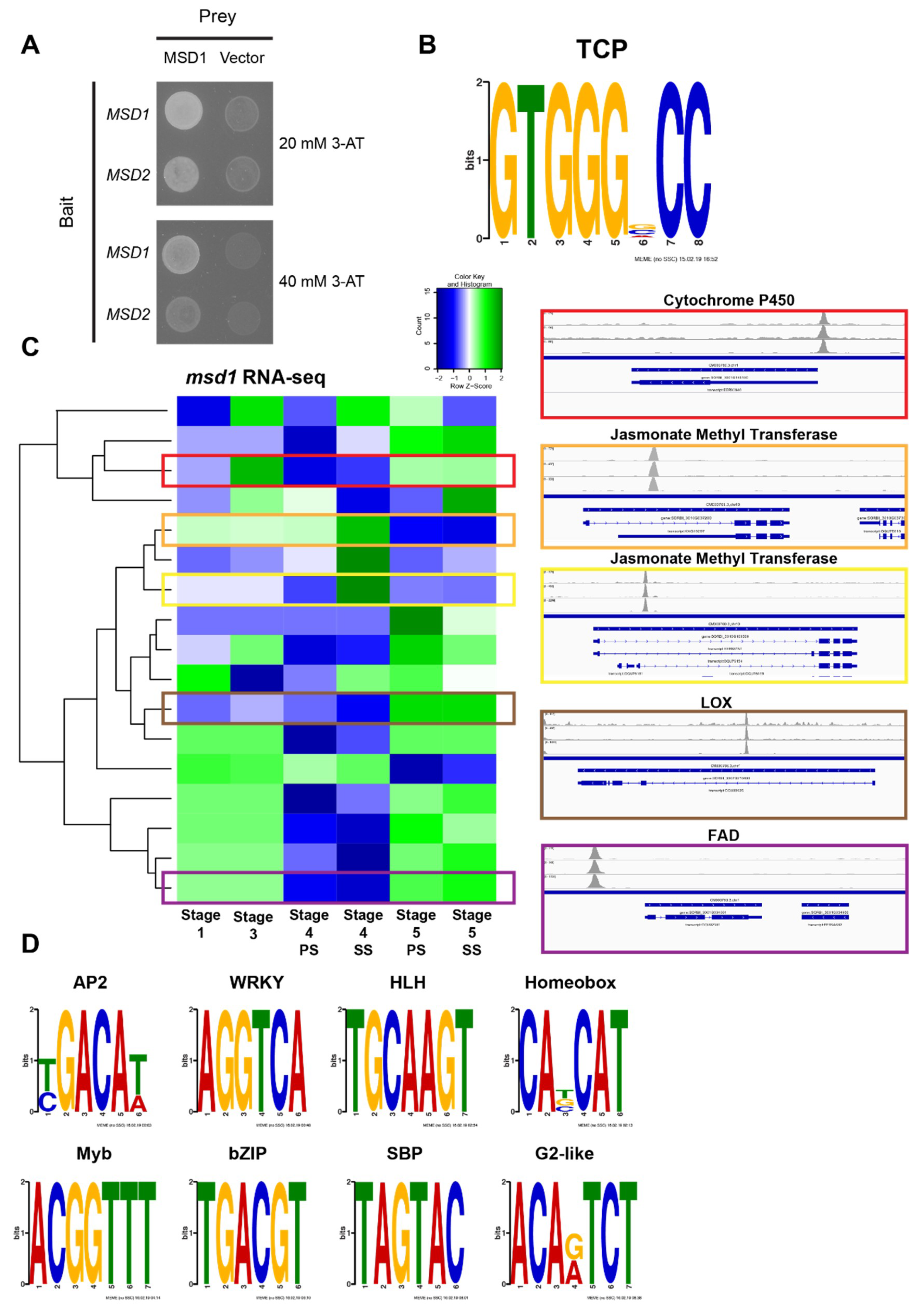
3.4. MSD2 Is Regulated by the TCP Transcription Factor MSD1
4. Discussion
5. Materials and Methods
5.1. Identification of MSD2 and Phenotypic Evaluation
5.2. Transcriptome Profiling
5.3. DAP-Seq Analysis
5.4. Phenotypic Rescue of msd2 with Exogenous Methyl-Jasmonate
6. Data Availability
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dillon:, S.L.; Shapter, F.M.; Henry, R.J.; Cordeiro, G.; Izquierdo, L.; Lee, L.S. Domestication to crop improvement: Genetic resources for Sorghum and Saccharum (Andropogoneae). Ann. Bot. 2007, 100, 975–989. [Google Scholar] [CrossRef] [PubMed]
- de Wet, J.M.J.; Huckabay, J.P. The origin of Sorghum bicolor. II. Distribution and domestication. Evolution 1967, 21, 787–802. [Google Scholar] [CrossRef] [PubMed]
- Paterson, A.H.; Bowers, J.E.; Bruggmann, R.; Dubchak, I.; Grimwood, J.; Gundlach, H.; Haberer, G.; Hellsten, U.; Mitros, T.; Poliakov, A.; et al. The Sorghum bicolor genome and the diversification of grasses. Nature 2009, 457, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Zhou, H.; Bi, H.; Fromm, M.; Yang, B.; Weeks, D.P. Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Res. 2013, 41, e188. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Li, H.; Chen, L.L.; Xie, K. Recent advances in genome editing using CRISPR/Cas9. Front. Plant Sci. 2016, 7, 703. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.J.; Klein, P.E.; Bortiri, E.; Acharya, C.B.; Rooney, W.L.; Kresovich, S. Inheritance of inflorescence architecture in sorghum. Theor. Appl. Genet. 2006, 113, 931–942. [Google Scholar] [CrossRef]
- Jiao, Y.; Lee, Y.K.; Gladman, N.; Chopra, R.; Christensen, S.A.; Regulski, M.; Burow, G.; Hayes, C.; Burke, J.; Ware, D.; et al. MSD1 regulates pedicellate spikelet fertility in sorghum through the jasmonic acid pathway. Nat. Commun. 2018, 9, 822. [Google Scholar] [CrossRef]
- Dalberg, J. Classification and Characterization of Sorghum. Sorghum: Origin, History, Technology, and Production; John Wiley & Sons, Inc.: New York, NY, USA, 2000; pp. 99–130. [Google Scholar]
- Yuan, Z.; Zhang, D. Roles of jasmonate signalling in plant inflorescence and flower development. Curr. Opin. Plant Biol. 2015, 27, 44–51. [Google Scholar] [CrossRef]
- Wasternack, C.; Hause, B. Jasmonates: Biosynthesis, perception, signal transduction and action in plant stress response, growth and development. An update to the 2007 review in annals of botany. Ann. Bot. 2013, 111, 1021–1058. [Google Scholar] [CrossRef]
- Lyons, R.; Manners, J.M.; Kazan, K. Jasmonate biosynthesis and signaling in monocots: A comparative overview. Plant Cell Rep. 2013, 32, 815–827. [Google Scholar] [CrossRef]
- Robert-Seilaniantz, A.; Grant, M.; Jones, J.D. Hormone crosstalk in plant disease and defense: More than just jasmonate-salicylate antagonism. Annu. Rev. Phytopathol. 2011, 49, 317–343. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Christensen, S.; Isakeit, T.; Engelberth, J.; Meeley, R.; Hayward, A.; Emery, R.J.; Kolomiets, M.V. Disruption of OPR7 and OPR8 reveals the versatile functions of jasmonic acid in maize development and defense. Plant Cell 2012, 24, 1420–1436. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Luo, S.M.; Xie, J.F.; Li, Y.F.; Xu, T.; Liu, Y.; Song, Y.Y.; Zhu-Salzman, K.; Zeng, R.S. Silencing COI1 in rice increases susceptibility to chewing insects and impairs inducible defense. PLoS ONE 2012, 7, e36214. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Yuan, Z.; Chen, M.; Yin, C.; Luo, Z.; Zhao, X.; Liang, W.; Hu, J.; Zhang, D. Jasmonic acid regulates spikelet development in rice. Nat. Commun. 2014, 5, 3476. [Google Scholar] [CrossRef] [PubMed]
- Acosta, I.F.; Laparra, H.; Romero, S.P.; Schmelz, E.; Hamberg, M.; Mottinger, J.P.; Moreno, M.A.; Dellaporta, S.L. Tasselseed1 is a lipoxygenase affecting jasmonic acid signaling in sex determination of maize. Science 2009, 323, 262–265. [Google Scholar] [CrossRef]
- DeLong, A.; Calderon-Urrea, A.; Dellaporta, S.L. Sex determination gene TASSELSEED2 of maize encodes a short-chain alcohol dehydrogenase required for stage-specific floral organ abortion. Cell 1993, 74, 757–768. [Google Scholar] [CrossRef]
- Schaller, F.; Biesgen, C.; Mussig, C.; Altmann, T.; Weiler, E.W. 12-Oxophytodienoate reductase 3 (OPR3) is the isoenzyme involved in jasmonate biosynthesis. Planta 2000, 210, 979–984. [Google Scholar] [CrossRef]
- Stintzi, A.; Browse, J. The Arabidopsis male-sterile mutant, opr3, lacks the 12-oxophytodienoic acid reductase required for jasmonate synthesis. Proc. Natl. Acad. Sci. USA 2000, 97, 10625–10630. [Google Scholar] [CrossRef]
- Park, J.H.; Halitschke, R.; Kim, H.B.; Baldwin, I.T.; Feldmann, K.A.; Feyereisen, R. A knock-out mutation in allene oxide synthase results in male sterility and defective wound signal transduction in arabidopsis due to a block in jasmonic acid biosynthesis. Plant J. 2002, 31, 1–12. [Google Scholar] [CrossRef]
- Koo, A.J.; Cooke, T.F.; Howe, G.A. Cytochrome P450 CYP94B3 mediates catabolism and inactivation of the plant hormone jasmonoyl-L-isoleucine. Proc. Natl. Acad. Sci. USA 2011, 108, 9298–9303. [Google Scholar] [CrossRef]
- Lunde, C.; Kimberlin, A.; Leiboff, S.; Koo, A.J.; Hake, S. Tasselseed5 overexpresses a wound-inducible enzyme, ZmCYP94B1, that affects jasmonate catabolism, sex determination, and plant architecture in maize. Commun. Biol. 2019, 2, 114. [Google Scholar] [CrossRef] [PubMed]
- Caldelari, D.; Wang, G.; Farmer, E.E.; Dong, X. Arabidopsis lox3 lox4 double mutants are male sterile and defective in global proliferative arrest. Plant Mol. Biol. 2011, 75, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Burke, J.; Chopra, R.; Burow, G.; Chen, J.; Wang, B.; Hayes, C.; Emendack, Y.; Ware, D.; Xin, Z. A sorghum mutant resource as an efficient platform for gene discovery in grasses. Plant Cell 2016, 28, 1551–1562. [Google Scholar] [CrossRef] [PubMed]
- Feussner, I.; Wasternack, C. The lipoxygenase pathway. Annu. Rev. Plant Biol. 2002, 53, 275–297. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Carpenter, R.; Vincent, C.; Copsey, L.; Coen, E. Origin of floral asymmetry in antirrhinum. Nature 1996, 383, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Doebley, J.; Stec, A.; Hubbard, L. The evolution of apical dominance in maize. Nature 1997, 386, 485–488. [Google Scholar] [CrossRef]
- Kosugi, S.; Ohashi, Y. PCF1 and PCF2 specifically bind to cis elements in the rice proliferating cell nuclear antigen gene. Plant Cell 1997, 9, 1607–1619. [Google Scholar]
- Davidson, R.M.; Gowda, M.; Moghe, G.; Lin, H.; Vaillancourt, B.; Shiu, S.H.; Jiang, N.; Robin Buell, C. Comparative transcriptomics of three poaceae species reveals patterns of gene expression evolution. Plant J. 2012, 71, 492–502. [Google Scholar] [CrossRef]
- Makita, Y.; Shimada, S.; Kawashima, M.; Kondou-Kuriyama, T.; Toyoda, T.; Matsui, M. Morokoshi: Transcriptome database in Sorghum bicolor. Plant Cell Physiol. 2015, 56, e6. [Google Scholar] [CrossRef]
- Petryszak, R.; Burdett, T.; Fiorelli, B.; Fonseca, N.A.; Gonzalez-Porta, M.; Hastings, E.; Huber, W.; Jupp, S.; Keays, M.; Kryvych, N.; et al. Expression atlas update—A database of gene and transcript expression from microarray- and sequencing-based functional genomics experiments. Nucleic Acids Res. 2014, 42, D926–D932. [Google Scholar] [CrossRef]
- Olson, A.; Klein, R.R.; Dugas, D.V.; Lu, Z.; Regulski, M.; Klein, P.E.; Ware, D. Expanding and vetting Sorghum bicolor gene annotations through transcriptome and methylome sequencing. Plant Genome 2014, 7. [Google Scholar] [CrossRef]
- Huang, H.; Liu, B.; Liu, L.; Song, S. Jasmonate action in plant growth and development. J. Exp. Bot. 2017, 68, 1349–1359. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Turner, J.G. Wound-induced endogenous jasmonates stunt plant growth by inhibiting mitosis. PLoS ONE 2008, 3, e3699. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, R.C.; Huang, S.C.; Song, L.; Lewsey, M.G.; Bartlett, A.; Nery, J.R.; Galli, M.; Gallavotti, A.; Ecker, J.R. Cistrome and epicistrome features shape the regulatory DNA landscape. Cell 2016, 166, 1598. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, D.D.; Han, L.H.; Tao, M.; Hu, Q.Q.; Wu, W.Y.; Zhang, J.B.; Li, X.B.; Huang, G.Q. Genome-wide identification and characterization of tcp transcription factor genes in upland cotton (Gossypium hirsutum). Sci. Rep. 2017, 7, 10118. [Google Scholar] [CrossRef] [PubMed]
- Weirauch, M.T.; Yang, A.; Albu, M.; Cote, A.G.; Montenegro-Montero, A.; Drewe, P.; Najafabadi, H.S.; Lambert, S.A.; Mann, I.; Cook, K.; et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 2014, 158, 1431–1443. [Google Scholar] [CrossRef] [PubMed]
- Stitz, M.; Baldwin, I.T.; Gaquerel, E. Diverting the flux of the ja pathway in Nicotiana attenuata compromises the plant’s defense metabolism and fitness in nature and glasshouse. PLoS ONE 2011, 6, e25925. [Google Scholar] [CrossRef]
- Ahmad, P.; Rasool, S.; Gul, A.; Sheikh, S.A.; Akram, N.A.; Ashraf, M.; Kazi, A.M.; Gucel, S. Jasmonates: Multifunctional roles in stress tolerance. Front. Plant Sci. 2016, 7, 813. [Google Scholar] [CrossRef]
- Li, C.; Schilmiller, A.L.; Liu, G.; Lee, G.I.; Jayanty, S.; Sageman, C.; Vrebalov, J.; Giovannoni, J.J.; Yagi, K.; Kobayashi, Y.; et al. Role of beta-oxidation in jasmonate biosynthesis and systemic wound signaling in tomato. Plant Cell 2005, 17, 971–986. [Google Scholar] [CrossRef]
- Yan, L.; Zhai, Q.; Wei, J.; Li, S.; Wang, B.; Huang, T.; Du, M.; Sun, J.; Kang, L.; Li, C.B.; et al. Role of Tomato Lipoxygenase D in Wound-Induced Jasmonate Biosynthesis and Plant Immunity to Insect Herbivores. PLoS Genet. 2013, 9, e1003964. [Google Scholar] [CrossRef]
- Dobritzsch, S.; Weyhe, M.; Schubert, R.; Dindas, J.; Hause, G.; Kopka, J.; Hause, B. Dissection of jasmonate functions in tomato stamen development by transcriptome and metabolome analyses. BMC Biol. 2015, 13, 28. [Google Scholar] [CrossRef] [PubMed]
- Niwa, T.; Suzuki, T.; Takebayashi, Y.; Ishiguro, R.; Higashiyama, T.; Sakakibara, H.; Ishiguro, S. Jasmonic acid facilitates flower opening and floral organ development through the upregulated expression of SlMYB21 transcription factor in tomato. Biosci. Biotechnol. Biochem. 2018, 82, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Bull, H.; Casao, M.C.; Zwirek, M.; Flavell, A.J.; Thomas, W.T.B.; Guo, W.; Zhang, R.; Rapazote-Flores, P.; Kyriakidis, S.; Russell, J.; et al. Barley SIX-ROWED SPIKE3 encodes a putative jumonji C-type H3K9me2/me3 demethylase that represses lateral spikelet fertility. Nat. Commun. 2017, 8, 936. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, S.; Kawai-Oda, A.; Ueda, J.; Nishida, I.; Okada, K. The defective in anther dehiscience gene encodes a novel phospholipase A1 catalyzing the initial step of jasmonic acid biosynthesis, which synchronizes pollen maturation, anther dehiscence, and flower opening in Arabidopsis. Plant Cell 2001, 13, 2191–2209. [Google Scholar] [CrossRef] [PubMed]
- Shuai, B.; Reynaga-Pena, C.G.; Springer, P.S. The lateral organ boundaries gene defines a novel, plant-specific gene family. Plant Physiol. 2002, 129, 747–761. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.Y.; Jiang, W.J.; Yu, H.J. The expression profiling of the lipoxygenase (LOX) family genes during fruit development, abiotic stress and hormonal treatments in cucumber (Cucumis sativus L.). Int. J. Mol. Sci. 2012, 13, 2481–2500. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, G.J.; Liu, Q.; Shreshtha, P.; Li, Z.; Rahman, S. RNAi-mediated down-regulation of the expression of OsFAD2-1: Effect on lipid accumulation and expression of lipid biosynthetic genes in the rice grain. BMC Plant Biol. 2016, 16, 189. [Google Scholar] [CrossRef] [PubMed]
- De Geyter, N.; Gholami, A.; Goormachtig, S.; Goossens, A. Transcriptional machineries in jasmonate-elicited plant secondary metabolism. Trends Plant Sci. 2012, 17, 349–359. [Google Scholar] [CrossRef]
- Long, H.K.; Prescott, S.L.; Wysocka, J. Ever-changing landscapes: Transcriptional enhancers in development and evolution. Cell 2016, 167, 1170–1187. [Google Scholar] [CrossRef]
- Xin, Z.; Wang, M.L.; Barkley, N.A.; Burow, G.; Franks, C.; Pederson, G.; Burke, J. Applying genotyping (TILLING) and phenotyping analyses to elucidate gene function in a chemically induced sorghum mutant population. BMC Plant Biol. 2008, 8, 103. [Google Scholar] [CrossRef] [PubMed]
- Xin, Z.; Chen, J. A high throughput DNA extraction method with high yield and quality. Plant Methods 2012, 8, 26. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Pritchard, B.; Rios, D.; Chen, Y.; Flicek, P.; Cunningham, F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 2010, 26, 2069–2070. [Google Scholar] [CrossRef] [PubMed]
- Monaco, M.K.; Stein, J.; Naithani, S.; Wei, S.; Dharmawardhana, P.; Kumari, S.; Amarasinghe, V.; Youens-Clark, K.; Thomason, J.; Preece, J.; et al. Gramene 2013: Comparative plant genomics resources. Nucleic Acids Res. 2014, 42, D1193–D1199. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. Star: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. Meme suite: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Zhou, X.; Ling, Y.; Zhang, Z.; Su, Z. AgriGo: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38, W64–W70. [Google Scholar] [CrossRef]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. Panther version 11: Expanded annotation data from gene ontology and reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef]
- RStudio Team. RStudio: Integrated Development for R; Rstudio, Inc.: Boston, MA, USA, 2015. [Google Scholar]
- Bartlett, A.; O’Malley, R.C.; Huang, S.C.; Galli, M.; Nery, J.R.; Gallavotti, A.; Ecker, J.R. Mapping genome-wide transcription-factor binding sites using DAP-seq. Nat. Protoc. 2017, 12, 1659–1672. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-seq (macs). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [PubMed]
- Tello-Ruiz, M.K.; Naithani, S.; Stein, J.C.; Gupta, P.; Campbell, M.; Olson, A.; Wei, S.; Preece, J.; Geniza, M.J.; Jiao, Y.; et al. Gramene 2018: Unifying comparative genomics and pathway resources for plant research. Nucleic Acids Res. 2018, 46, D1181–D1189. [Google Scholar] [CrossRef] [PubMed]
- McCormick, R.F.; Truong, S.K.; Sreedasyam, A.; Jenkins, J.; Shu, S.; Sims, D.; Kennedy, M.; Amirebrahimi, M.; Weers, B.D.; McKinley, B.; et al. The Sorghum bicolor reference genome: Improved assembly, gene annotations, a transcriptome atlas, and signatures of genome organization. Plant J. 2018, 93, 338–354. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gladman, N.; Jiao, Y.; Lee, Y.K.; Zhang, L.; Chopra, R.; Regulski, M.; Burow, G.; Hayes, C.; Christensen, S.A.; Dampanaboina, L.; et al. Fertility of Pedicellate Spikelets in Sorghum Is Controlled by a Jasmonic Acid Regulatory Module. Int. J. Mol. Sci. 2019, 20, 4951. https://doi.org/10.3390/ijms20194951
Gladman N, Jiao Y, Lee YK, Zhang L, Chopra R, Regulski M, Burow G, Hayes C, Christensen SA, Dampanaboina L, et al. Fertility of Pedicellate Spikelets in Sorghum Is Controlled by a Jasmonic Acid Regulatory Module. International Journal of Molecular Sciences. 2019; 20(19):4951. https://doi.org/10.3390/ijms20194951
Chicago/Turabian StyleGladman, Nicholas, Yinping Jiao, Young Koung Lee, Lifang Zhang, Ratan Chopra, Michael Regulski, Gloria Burow, Chad Hayes, Shawn A. Christensen, Lavanya Dampanaboina, and et al. 2019. "Fertility of Pedicellate Spikelets in Sorghum Is Controlled by a Jasmonic Acid Regulatory Module" International Journal of Molecular Sciences 20, no. 19: 4951. https://doi.org/10.3390/ijms20194951
APA StyleGladman, N., Jiao, Y., Lee, Y. K., Zhang, L., Chopra, R., Regulski, M., Burow, G., Hayes, C., Christensen, S. A., Dampanaboina, L., Chen, J., Burke, J., Ware, D., & Xin, Z. (2019). Fertility of Pedicellate Spikelets in Sorghum Is Controlled by a Jasmonic Acid Regulatory Module. International Journal of Molecular Sciences, 20(19), 4951. https://doi.org/10.3390/ijms20194951