Molecular Mechanisms of the Acute Kidney Injury to Chronic Kidney Disease Transition: An Updated View

 ,
, {kind=link}
{kind=link}
Abstract
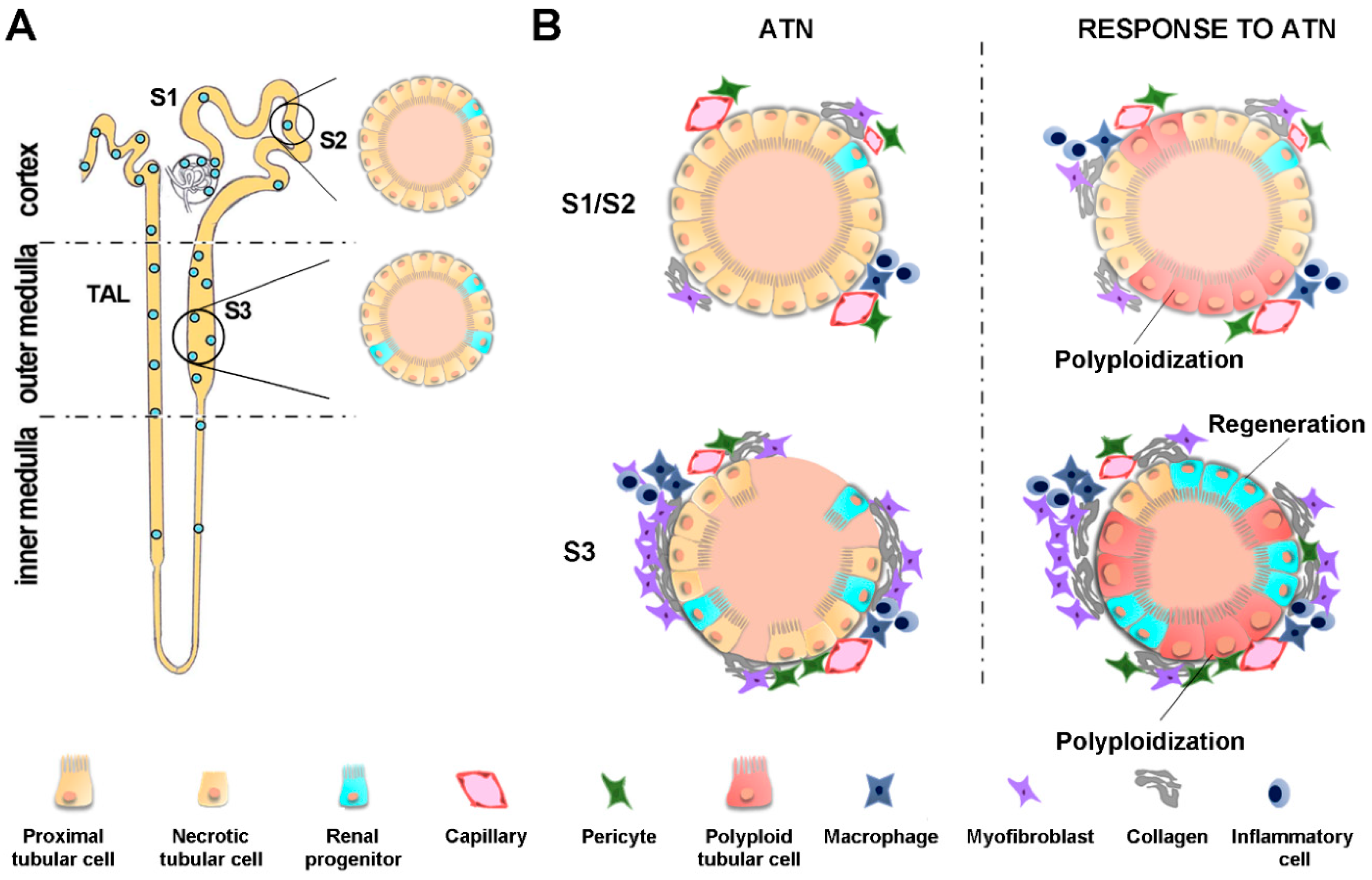
1. AKI Is Not a Self-Limiting Event
2. Pathophysiology of the AKI-to-CKD Transition
2.1. Endothelial Dysfunction
2.2. Interstitial Inflammation
2.3. Fibrosis
2.4. Tubular Epithelial Injury
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AKI | Acute kidney injury |
| Akt | Protein kinase B |
| Ang-1 | Angiopoietin-1 |
| ATN | Acute tubular necrosis |
| CKD | Chronic kidney disease |
| CSF-1 | Colony-stimulating factor-1 |
| CTGF | Connective tissue growth factor |
| DAMPs | Damage-associated molecular patterns |
| ECM | Extracellular matrix |
| EMT | Epithelial-mesenchymal transition |
| EndoMT | Endothelial to mesenchymal transition |
| EPC | Endothelial progenitor cells |
| ESRD | End stage renal disease |
| FUCCI | Fluorescent ubiquitination-based cell cycle indicator |
| Hif | Hypoxia-inducible factor |
| ICAM-1 | Intercellular adhesion molecule-1 |
| IL | Interleukin |
| IRAK-M | IL-1 receptor-associated kinase-M |
| IRI | Ischemia-reperfusion injury |
| PCNA | Proliferating cell nuclear antigen |
| PDGF | Platelet-derived growth factor |
| PDGFR-β | Platelet-derived growth factor receptor-β |
| RA | Retinoic acid |
| RAS | Renin-angiotensin system |
| S1-pr1 | Sphingosine 1-phosphate receptor 1 |
| Sirt 1 | Sirtuin 1 |
| Snail1 | Snail family zinc finger 1 |
| STAT3 | Signal transducer and transcription factor 3 |
| TEC | Tubular epithelial cell |
| TGF-β | Transforming growth factor β |
| TLR | Toll-like receptor |
| TNF-α | Tumor necrosis factor-α |
| VEGF | Vascular endothelial growth factor |
| α-SMA | α-smooth muscle actin |
References
- Ponte, B.; Felipe, C.; Muriel, A.; Tenorio, M.T.; Liaño, F. Long-term functional evolution after an acute kidney injury: A 10-year study. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.—Eur. Ren. Assoc. 2008, 23, 3859–3866. [Google Scholar] [CrossRef] [PubMed]
- Coca, S.G.; Singanamala, S.; Parikh, C.R. Chronic kidney disease after acute kidney injury: A systematic review and meta-analysis. Kidney Int. 2012, 81, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Chawla, L.S.; Eggers, P.W.; Star, R.A.; Kimmel, P.L. Acute kidney injury and chronic kidney disease as interconnected syndromes. N. Engl. J. Med. 2014, 371, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Lowe, K.G. The late prognosis in acute tubular necrosis. Lancet 1952, 259, 1086–1088. [Google Scholar] [CrossRef]
- Liaño, F.; Felipe, C.; Tenorio, M.-T.; Rivera, M.; Abraira, V.; Sáez-de-Urturi, J.-M.; Ocaña, J.; Fuentes, C.; Severiano, S. Long-term outcome of acute tubular necrosis: A contribution to its natural history. Kidney Int. 2007, 71, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Ricci, Z.; Cruz, D.N.; Ronco, C. Classification and staging of acute kidney injury: Beyond the RIFLE and AKIN criteria. Nat. Rev. Nephrol. 2011, 7, 201–208. [Google Scholar] [CrossRef] [PubMed]
- See, E.J.; Jayasinghe, K.; Glassford, N.; Bailey, M.; Johnson, D.W.; Polkinghorne, K.R.; Toussaint, N.D.; Bellomo, R. Long-term risk of adverse outcomes after acute kidney injury: A systematic review and meta-analysis of cohort studies using consensus definitions of exposure. Kidney Int. 2019, 95, 160–172. [Google Scholar] [CrossRef]
- Ishani, A.; Xue, J.L.; Himmelfarb, J.; Eggers, P.W.; Kimmel, P.L.; Molitoris, B.A.; Collins, A.J. Acute kidney injury increases risk of ESRD among elderly. J. Am. Soc. Nephrol. 2009, 20, 223–228. [Google Scholar] [CrossRef]
- Amdur, R.L.; Chawla, L.S.; Amodeo, S.; Kimmel, P.L.; Palant, C.E. Outcomes following diagnosis of acute renal failure in U.S. veterans: Focus on acute tubular necrosis. Kidney Int. 2009, 76, 1089–1097. [Google Scholar] [CrossRef]
- Ishani, A.; Nelson, D.; Clothier, B.; Schult, T.; Nugent, S.; Greer, N.; Slinin, Y.; Ensrud, K.E. The Magnitude of Acute Serum Creatinine Increase After Cardiac Surgery and the Risk of Chronic Kidney Disease, Progression of Kidney Disease, and Death. Arch. Intern. Med. 2011, 171, 226–233. [Google Scholar] [CrossRef]
- Mehta, S.; Chauhan, K.; Patel, A.; Patel, S.; Pinotti, R.; Nadkarni, G.N.; Parikh, C.R.; Coca, S.G. The prognostic importance of duration of AKI: A systematic review and meta-analysis. BMC Nephrol. 2018, 19, 91. [Google Scholar] [CrossRef] [PubMed]
- Thakar, C.V.; Christianson, A.; Himmelfarb, J.; Leonard, A.C. Acute kidney injury episodes and chronic kidney disease risk in diabetes mellitus. Clin. J. Am. Soc. Nephrol. 2011, 6, 2567–2572. [Google Scholar] [CrossRef] [PubMed]
- Devarajan, P.; Jefferies, J.L. Progression of chronic kidney disease after acute kidney injury. Prog. Pediatr. Cardiol. 2016, 41, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Uber, A.M.; Sutherland, S.M. Acute kidney injury in hospitalized children: Consequences and outcomes. Pediatr. Nephrol. Berl. Ger. 2018, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sigurjonsdottir, V.K.; Chaturvedi, S.; Mammen, C.; Sutherland, S.M. Pediatric acute kidney injury and the subsequent risk for chronic kidney disease: Is there cause for alarm? Pediatr. Nephrol. Berl. Ger. 2018, 33, 2047–2055. [Google Scholar] [CrossRef]
- Basile, D.P.; Bonventre, J.V.; Mehta, R.; Nangaku, M.; Unwin, R.; Rosner, M.H.; Kellum, J.A.; Ronco, C.; ADQI XIII Work Group. Progression after AKI: Understanding Maladaptive Repair Processes to Predict and Identify Therapeutic Treatments. J. Am. Soc. Nephrol. 2016, 27, 687–697. [Google Scholar] [CrossRef]
- Bellomo, R.; Kellum, J.A.; Ronco, C. Acute kidney injury. Lancet 2012, 380, 756–766. [Google Scholar] [CrossRef]
- Venkatachalam, M.A.; Griffin, K.A.; Lan, R.; Geng, H.; Saikumar, P.; Bidani, A.K. Acute kidney injury: A springboard for progression in chronic kidney disease. Am. J. Physiol. Renal Physiol. 2010, 298, F1078–F1094. [Google Scholar] [CrossRef]
- Rodriguez-Romo, R.; Benitez, K.; Barrera-Chimal, J.; Perez-Villalva, R.; Gomez, A.; Aguilar-Leon, D.; Rangel-Santiago, J.F.; Huerta, S.; Gamba, G.; Uribe, N.; et al. AT1 receptor antagonism before ischemia prevents the transition of acute kidney injury to chronic kidney disease. Kidney Int. 2016, 89, 363–373. [Google Scholar] [CrossRef]
- Chou, Y.-H.; Chu, T.-S.; Lin, S.-L. Role of renin-angiotensin system in acute kidney injury-chronic kidney disease transition. Nephrol. Carlton Vic 2018, 23, 121–125. [Google Scholar] [CrossRef]
- Grgic, I.; Campanholle, G.; Bijol, V.; Wang, C.; Sabbisetti, V.S.; Ichimura, T.; Humphreys, B.D.; Bonventre, J.V. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int. 2012, 82, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Ferenbach, D.A.; Bonventre, J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, M.A.; Weinberg, J.M.; Kriz, W.; Bidani, A.K. Failed Tubule Recovery, AKI-CKD Transition, and Kidney Disease Progression. J. Am. Soc. Nephrol. 2015, 26, 1765–1776. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P. The endothelial cell in ischemic acute kidney injury: Implications for acute and chronic function. Kidney Int. 2007, 72, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P.; Collett, J.A.; Yoder, M.C. Endothelial colony-forming cells and pro-angiogenic cells: Clarifying definitions and their potential role in mitigating acute kidney injury. Acta. Physiol. Oxf. Engl. 2018, 222, e12914. [Google Scholar] [CrossRef] [PubMed]
- Lech, M.; Gröbmayr, R.; Ryu, M.; Lorenz, G.; Hartter, I.; Mulay, S.R.; Susanti, H.E.; Kobayashi, K.S.; Flavell, R.A.; Anders, H.-J. Macrophage phenotype controls long-term AKI outcomes—Kidney regeneration versus atrophy. J. Am. Soc. Nephrol. 2014, 25, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.-J.; Schaefer, L. Beyond Tissue Injury—Damage-Associated Molecular Patterns, Toll-Like Receptors, and Inflammasomes Also Drive Regeneration and Fibrosis. J. Am. Soc. Nephrol. 2014, 25, 1387–1400. [Google Scholar] [CrossRef]
- Belliere, J.; Casemayou, A.; Ducasse, L.; Zakaroff-Girard, A.; Martins, F.; Iacovoni, J.S.; Guilbeau-Frugier, C.; Buffin-Meyer, B.; Pipy, B.; Chauveau, D.; et al. Specific Macrophage Subtypes Influence the Progression of Rhabdomyolysis-Induced Kidney Injury. J. Am. Soc. Nephrol. 2015, 26, 1363–1377. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.R.; Rabb, H. Immune cells in experimental acute kidney injury. Nat. Rev. Nephrol. 2015, 11, 88–101. [Google Scholar] [CrossRef]
- Sato, Y.; Yanagita, M. Immune cells and inflammation in AKI to CKD progression. Am. J. Physiol. Renal Physiol. 2018, 315, 1501–1512. [Google Scholar] [CrossRef]
- Gomez, I.G.; Duffield, J.S. The FOXD1 lineage of kidney perivascular cells and myofibroblasts: Functions and responses to injury. Kidney Int. Suppl. 2014, 4, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Mack, M.; Yanagita, M. Origin of myofibroblasts and cellular events triggering fibrosis. Kidney Int. 2015, 87, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, J.; Sato, Y.; Kitai, Y.; Wajima, S.; Yamamoto, S.; Oguchi, A.; Yamada, R.; Kaneko, K.; Kondo, M.; Uchino, E.; et al. Myofibroblasts acquire retinoic acid–producing ability during fibroblast-to-myofibroblast transition following kidney injury. Kidney Int. 2019, 95, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Kaissling, B.; Lehir, M.; Kriz, W. Renal epithelial injury and fibrosis. Biochim. Biophys. Acta 2013, 1832, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.-C.; Tang, T.-T.; Lv, L.-L.; Lan, H.-Y. Renal tubule injury: A driving force toward chronic kidney disease. Kidney Int. 2018, 93, 568–579. [Google Scholar] [CrossRef]
- Patschan, D.; Kribben, A.; Müller, G.A. Postischemic microvasculopathy and endothelial progenitor cell-based therapy in ischemic AKI: Update and perspectives. Am. J. Physiol.-Ren. Physiol. 2016, 311, 382–394. [Google Scholar] [CrossRef]
- Sradnick, J.; Rong, S.; Luedemann, A.; Parmentier, S.P.; Bartaun, C.; Todorov, V.T.; Gueler, F.; Hugo, C.P.; Hohenstein, B. Extrarenal Progenitor Cells Do Not Contribute to Renal Endothelial Repair. J. Am. Soc. Nephrol. 2016, 27, 1714–1726. [Google Scholar] [CrossRef]
- Kumar, S. Cellular and molecular pathways of renal repair after acute kidney injury. Kidney Int. 2018, 93, 27–40. [Google Scholar] [CrossRef]
- Tanaka, S.; Tanaka, T.; Nangaku, M. Hypoxia as a key player in the AKI-to-CKD transition. Am. J. Physiol. Renal Physiol. 2014, 307, 1187–1195. [Google Scholar] [CrossRef]
- Chade, A.R. Vascular Endothelial Growth Factor Therapy for the Kidney: Are We There Yet? J. Am. Soc. Nephrol. 2016, 27, 1–3. [Google Scholar] [CrossRef]
- Chade, A.R.; Tullos, N.A.; Harvey, T.W.; Mahdi, F.; Bidwell, G.L. Renal Therapeutic Angiogenesis Using a Bioengineered Polymer-Stabilized Vascular Endothelial Growth Factor Construct. J. Am. Soc. Nephrol. 2016, 27, 1741–1752. [Google Scholar] [CrossRef] [PubMed]
- Hörbelt, M.; Lee, S.-Y.; Mang, H.E.; Knipe, N.L.; Sado, Y.; Kribben, A.; Sutton, T.A. Acute and chronic microvascular alterations in a mouse model of ischemic acute kidney injury. Am. J. Physiol.-Ren. Physiol. 2007, 293, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Kapitsinou, P.P.; Sano, H.; Michael, M.; Kobayashi, H.; Davidoff, O.; Bian, A.; Yao, B.; Zhang, M.-Z.; Harris, R.C.; Duffy, K.J.; et al. Endothelial HIF-2 mediates protection and recovery from ischemic kidney injury. J. Clin. Invest. 2014, 124, 2396–2409. [Google Scholar] [CrossRef] [PubMed]
- Perry, H.M.; Huang, L.; Ye, H.; Liu, C.; Sung, S.J.; Lynch, K.R.; Rosin, D.L.; Bajwa, A.; Okusa, M.D. Endothelial Sphingosine 1‑Phosphate Receptor‑1 Mediates Protection and Recovery from Acute Kidney Injury. J. Am. Soc. Nephrol. 2016, 27, 3383–3393. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Wiesener, M.; Bernhardt, W.; Eckardt, K.-U.; Warnecke, C. The human HIF (hypoxia-inducible factor)-3α gene is a HIF-1 target gene and may modulate hypoxic gene induction. Biochem. J. 2009, 424, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Vasko, R.; Xavier, S.; Chen, J.; Lin, C.H.S.; Ratliff, B.; Rabadi, M.; Maizel, J.; Tanokuchi, R.; Zhang, F.; Cao, J.; et al. Endothelial Sirtuin 1 Deficiency Perpetrates Nephrosclerosis through Downregulation of Matrix Metalloproteinase-14: Relevance to Fibrosis of Vascular Senescence. J. Am. Soc. Nephrol. 2014, 25, 276–291. [Google Scholar] [CrossRef] [PubMed]
- Kida, Y.; Zullo, J.A.; Goligorsky, M.S. Endothelial sirtuin 1 inactivation enhances capillary rarefaction and fibrosis following kidney injury through Notch activation. Biochem. Biophys. Res. Commun. 2016, 478, 1074–1079. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P.; Friedrich, J.L.; Spahic, J.; Knipe, N.; Mang, H.; Leonard, E.C.; Changizi-Ashtiyani, S.; Bacallao, R.L.; Molitoris, B.A.; Sutton, T.A. Impaired endothelial proliferation and mesenchymal transition contribute to vascular rarefaction following acute kidney injury. Am. J. Physiol.-Ren. Physiol. 2011, 300, 721–733. [Google Scholar] [CrossRef]
- Xavier, S.; Vasko, R.; Matsumoto, K.; Zullo, J.A.; Chen, R.; Maizel, J.; Chander, P.N.; Goligorsky, M.S. Curtailing Endothelial TGF-β Signaling Is Sufficient to Reduce Endothelial-Mesenchymal Transition and Fibrosis in CKD. J. Am. Soc. Nephrol. 2015, 26, 817–829. [Google Scholar] [CrossRef]
- Kramann, R.; Wongboonsin, J.; Chang-Panesso, M.; Machado, F.G.; Humphreys, B.D. Gli1+ Pericyte Loss Induces Capillary Rarefaction and Proximal Tubular Injury. J. Am. Soc. Nephrol. 2017, 28, 776–784. [Google Scholar] [CrossRef]
- Betsholtz, C. Insight into the physiological functions of PDGF through genetic studies in mice. Cytokine Growth Factor Rev. 2004, 15, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Sundberg, C.; Kowanetz, M.; Brown, L.F.; Detmar, M.; Dvorak, H.F. Stable expression of angiopoietin-1 and other markers by cultured pericytes: Phenotypic similarities to a subpopulation of cells in maturing vessels during later stages of angiogenesis in vivo. Lab. Investig. J. Tech. Methods Pathol. 2002, 82, 387–401. [Google Scholar] [CrossRef]
- Carvalho, R.L.C. Defective paracrine signalling by TGF in yolk sac vasculature of endoglin mutant mice: A paradigm for hereditary haemorrhagic telangiectasia. Development 2004, 131, 6237–6247. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, L.E.; Hemo, I.; Keshet, E. A plasticity window for blood vessel remodelling is defined by pericyte coverage of the preformed endothelial network and is regulated by PDGF-B and VEGF. Dev. Camb. Engl. 1998, 125, 1591–1598. [Google Scholar]
- Chae, S.-S.; Paik, J.-H.; Allende, M.L.; Proia, R.L.; Hla, T. Regulation of limb development by the sphingosine 1-phosphate receptor S1p1/EDG-1 occurs via the hypoxia/VEGF axis. Dev. Biol. 2004, 268, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.-J. Immune system modulation of kidney regeneration—mechanisms and implications. Nat. Rev. Nephrol. 2014, 10, 347–358. [Google Scholar] [CrossRef]
- Stamatiades, E.G.; Tremblay, M.-E.; Bohm, M.; Crozet, L.; Bisht, K.; Kao, D.; Coelho, C.; Fan, X.; Yewdell, W.T.; Davidson, A.; et al. Immune Monitoring of Trans-endothelial Transport by Kidney-Resident Macrophages. Cell 2016, 166, 991–1003. [Google Scholar] [CrossRef]
- Karasawa, K.; Asano, K.; Moriyama, S.; Ushiki, M.; Monya, M.; Iida, M.; Kuboki, E.; Yagita, H.; Uchida, K.; Nitta, K.; et al. Vascular-Resident CD169-Positive Monocytes and Macrophages Control Neutrophil Accumulation in the Kidney with Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. 2015, 26, 896–906. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef]
- Clements, M.; Gershenovich, M.; Chaber, C.; Campos-Rivera, J.; Du, P.; Zhang, M.; Ledbetter, S.; Zuk, A. Differential Ly6C Expression after Renal Ischemia-Reperfusion Identifies Unique Macrophage Populations. J. Am. Soc. Nephrol. 2016, 27, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.-S.; Kim, J.; Park, Y.-K.; Park, K.M. Infiltrated Macrophages Contribute to Recovery after Ischemic Injury But Not to Ischemic Preconditioning in Kidneys. Transplantation 2008, 85, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.-J.; Feng, D.; Wang, H.; Guan, Y.; Yan, X.; Gao, B. IL-22 Ameliorates Renal Ischemia-Reperfusion Injury by Targeting Proximal Tubule Epithelium. J. Am. Soc. Nephrol. 2014, 25, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Skrypnyk, N.I.; Skvarca, L.B.; Penchev, R.; Zhang, K.X.; Rochon, E.R.; Fall, J.L.; Paueksakon, P.; Yang, H.; Alford, C.E.; et al. Retinoic Acid Signaling Coordinates Macrophage-Dependent Injury and Repair after AKI. J. Am. Soc. Nephrol. 2016, 27, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-L.; Li, B.; Rao, S.; Yeo, E.-J.; Hudson, T.E.; Nowlin, B.T.; Pei, H.; Chen, L.; Zheng, J.J.; Carroll, T.J.; et al. Macrophage Wnt7b is critical for kidney repair and regeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 4194–4199. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Tan, R.J.; Fu, H.; Liu, Y. Wnt/β-catenin signaling in kidney injury and repair: A double-edged sword. Lab. Invest. 2016, 96, 156–167. [Google Scholar] [CrossRef]
- Tan, R.J.; Zhou, D.; Zhou, L.; Liu, Y. Wnt/β-catenin signaling and kidney fibrosis. Kidney Int. Suppl. 2014, 4, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.-Z.; Yao, B.; Yang, S.; Jiang, L.; Wang, S.; Fan, X.; Yin, H.; Wong, K.; Miyazawa, T.; Chen, J.; et al. CSF-1 signaling mediates recovery from acute kidney injury. J. Clin. Invest. 2012, 122, 4519–4532. [Google Scholar] [CrossRef]
- Baek, J.-H.; Zeng, R.; Weinmann-Menke, J.; Valerius, M.T.; Wada, Y.; Ajay, A.K.; Colonna, M.; Kelley, V.R. IL-34 mediates acute kidney injury and worsens subsequent chronic kidney disease. J. Clin. Invest. 2015, 125, 3198–3214. [Google Scholar] [CrossRef]
- Lee, S.A.; Noel, S.; Sadasivam, M.; Hamad, A.R.A.; Rabb, H. Role of Immune Cells in Acute Kidney Injury and Repair. Nephron 2017, 137, 282–286. [Google Scholar] [CrossRef]
- Kim, M.-G.; Koo, T.Y.; Yan, J.-J.; Lee, E.; Han, K.H.; Jeong, J.C.; Ro, H.; Kim, B.S.; Jo, S.-K.; Oh, K.H.; et al. IL-2/Anti-IL-2 Complex Attenuates Renal Ischemia-Reperfusion Injury through Expansion of Regulatory T Cells. J. Am. Soc. Nephrol. 2013, 24, 1529–1536. [Google Scholar] [CrossRef] [PubMed]
- Gandolfo, M.T.; Jang, H.R.; Bagnasco, S.M.; Ko, G.-J.; Agreda, P.; Satpute, S.R.; Crow, M.T.; King, L.S.; Rabb, H. Foxp3+ regulatory T cells participate in repair of ischemic acute kidney injury. Kidney Int. 2009, 76, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Baudoux, T.; Husson, C.; De Prez, E.; Jadot, I.; Antoine, M.-H.; Nortier, J.L.; Hougardy, J.-M. CD4+ and CD8+ T Cells Exert Regulatory Properties During Experimental Acute Aristolochic Acid Nephropathy. Sci. Rep. 2018, 8, 5334. [Google Scholar] [CrossRef] [PubMed]
- Renner, B.; Strassheim, D.; Amura, C.R.; Kulik, L.; Ljubanovic, D.; Glogowska, M.J.; Takahashi, K.; Carroll, M.C.; Holers, V.M.; Thurman, J.M. B Cell Subsets Contribute to Renal Injury and Renal Protection after Ischemia/Reperfusion. J. Immunol. 2010, 185, 4393–4400. [Google Scholar] [CrossRef] [PubMed]
- Klingberg, F.; Hinz, B.; White, E.S. The myofibroblast matrix: Implications for tissue repair and fibrosis: The myofibroblast matrix. J. Pathol. 2013, 229, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Mackensen-Haen, S.; Bader, R.; Grund, K.E.; Bohle, A. Correlations between renal cortical interstitial fibrosis, atrophy of the proximal tubules and impairment of the glomerular filtration rate. Clin. Nephrol. 1981, 15, 167–171. [Google Scholar] [PubMed]
- Picken, M.; Long, J.; Williamson, G.A.; Polichnowski, A.J. Progression of Chronic Kidney Disease After Acute Kidney Injury: Role of Self-Perpetuating Versus Hemodynamic-Induced Fibrosis. Hypertens. Dallas Tex 1979 2016, 68, 921–928. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Takaori, K.; Nakamura, J.; Yamamoto, S.; Nakata, H.; Sato, Y.; Takase, M.; Nameta, M.; Yamamoto, T.; Economides, A.N.; Kohno, K.; et al. Severity and Frequency of Proximal Tubule Injury Determines Renal Prognosis. J. Am. Soc. Nephrol. 2016, 27, 2393–2406. [Google Scholar] [CrossRef]
- Sato, Y.; Mii, A.; Hamazaki, Y.; Fujita, H.; Nakata, H.; Masuda, K.; Nishiyama, S.; Shibuya, S.; Haga, H.; Ogawa, O.; et al. Heterogeneous fibroblasts underlie age-dependent tertiary lymphoid tissues in the kidney. JCI Insight 2016, 1, e87680. [Google Scholar] [CrossRef]
- Allinovi, M.; de Chiara, L.; Angelotti, M.L.; Becherucci, F.; Romagnani, P. Anti-fibrotic treatments: A review of clinical evidence. Matrix Biol. 2018, 68–69, 333–354. [Google Scholar] [CrossRef]
- De Chiara, L.; Romagnani, P. Tubule repair: With a little help from my “unexpected” friends. Kidney Int. 2019, 95, 487–489. [Google Scholar] [CrossRef] [PubMed]
- Sharfuddin, A.A.; Molitoris, B.A. Pathophysiology of ischemic acute kidney injury. Nat. Rev. Nephrol. 2011, 7, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Funk, J.A.; Schnellmann, R.G. Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am. J. Physiol. Renal Physiol. 2012, 302, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chang, J.; Yao, B.; Niu, A.; Kelly, E.; Breeggemann, M.C.; Abboud Werner, S.L.; Harris, R.C.; Zhang, M.-Z. Proximal tubule-derived colony stimulating factor-1 mediates polarization of renal macrophages and dendritic cells, and recovery in acute kidney injury. Kidney Int. 2015, 88, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Huen, S.C.; Huynh, L.; Marlier, A.; Lee, Y.; Moeckel, G.W.; Cantley, L.G. GM-CSF Promotes Macrophage Alternative Activation after Renal Ischemia/Reperfusion Injury. J. Am. Soc. Nephrol. 2015, 26, 1334–1345. [Google Scholar] [CrossRef]
- Kramann, R.; Kusaba, T.; Humphreys, B.D. Who regenerates the kidney tubule? Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. -Eur. Ren. Assoc. 2015, 30, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Chang-Panesso, M.; Humphreys, B.D. Cellular plasticity in kidney injury and repair. Nat. Rev. Nephrol. 2017, 13, 39–46. [Google Scholar] [CrossRef]
- Witzgall, R.; Brown, D.; Schwarz, C.; Bonventre, J.V. Localization of proliferating cell nuclear antigen, vimentin, c-Fos, and clusterin in the postischemic kidney. Evidence for a heterogenous genetic response among nephron segments, and a large pool of mitotically active and dedifferentiated cells. J. Clin. Invest. 1994, 93, 2175–2188. [Google Scholar] [CrossRef]
- Grande, M.T.; Sánchez-Laorden, B.; López-Blau, C.; De Frutos, C.A.; Boutet, A.; Arévalo, M.; Rowe, R.G.; Weiss, S.J.; López-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef]
- Lovisa, S.; LeBleu, V.S.; Tampe, B.; Sugimoto, H.; Vadnagara, K.; Carstens, J.L.; Wu, C.-C.; Hagos, Y.; Burckhardt, B.C.; Pentcheva-Hoang, T.; et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015, 21, 998–1009. [Google Scholar] [CrossRef]
- Vogetseder, A.; Karadeniz, A.; Kaissling, B.; Le Hir, M. Tubular cell proliferation in the healthy rat kidney. Histochem. Cell Biol. 2005, 124, 97–104. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vogetseder, A.; Picard, N.; Gaspert, A.; Walch, M.; Kaissling, B.; le Hir, M. Proliferation capacity of the renal proximal tubule involves the bulk of differentiated epithelial cells. Am. J. Physiol.-Cell Physiol. 2008, 294, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010, 16, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Canaud, G.; Bonventre, J.V. Cell cycle arrest and the evolution of chronic kidney disease from acute kidney injury. Nephrol. Dial. Transplant. 2015, 30, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Lazzeri, E.; Angelotti, M.L.; Peired, A.; Conte, C.; Marschner, J.A.; Maggi, L.; Mazzinghi, B.; Lombardi, D.; Melica, M.E.; Nardi, S.; et al. Endocycle-related tubular cell hypertrophy and progenitor proliferation recover renal function after acute kidney injury. Nat. Commun. 2018, 9, 1344. [Google Scholar] [CrossRef] [PubMed]
- Lazzeri, E.; Angelotti, M.L.; Conte, C.; Anders, H.-J.; Romagnani, P. Surviving Acute Organ Failure: Cell Polyploidization and Progenitor Proliferation. Trends Mol. Med. 2019, 25, 366–381. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, D.; Becherucci, F.; Romagnani, P. How much can the tubule regenerate and who does it? An open question. Nephrol. Dial. Transplant. 2016, 31, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, D.; Boström, A.-K.; Nilsson, K.; Hansson, J.; Sjölund, J.; Möller, C.; Jirström, K.; Nilsson, E.; Landberg, G.; Axelson, H.; et al. Isolation and characterization of progenitor-like cells from human renal proximal tubules. Am. J. Pathol. 2011, 178, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Angelotti, M.L.; Ronconi, E.; Ballerini, L.; Peired, A.; Mazzinghi, B.; Sagrinati, C.; Parente, E.; Gacci, M.; Carini, M.; Rotondi, M.; et al. Characterization of renal progenitors committed toward tubular lineage and their regenerative potential in renal tubular injury. Stem Cells Dayt. Ohio 2012, 30, 1714–1725. [Google Scholar] [CrossRef]
- Smeets, B.; Boor, P.; Dijkman, H.; Sharma, S.V.; Jirak, P.; Mooren, F.; Berger, K.; Bornemann, J.; Gelman, I.H.; Floege, J.; et al. Proximal tubular cells contain a phenotypically distinct, scattered cell population involved in tubular regeneration: Phenotypically distinct proximal tubular cells. J. Pathol. 2013, 229, 645–659. [Google Scholar] [CrossRef]
- Kumar, S.; Liu, J.; Pang, P.; Krautzberger, A.M.; Reginensi, A.; Akiyama, H.; Schedl, A.; Humphreys, B.D.; McMahon, A.P. Sox9 Activation Highlights a Cellular Pathway of Renal Repair in the Acutely Injured Mammalian Kidney. Cell Rep. 2015, 12, 1325–1338. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Huang, S.; Reidy, K.; Han, S.H.; Chinga, F.; Susztak, K. Sox9-Positive Progenitor Cells Play a Key Role in Renal Tubule Epithelial Regeneration in Mice. Cell Rep. 2016, 14, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Berger, K.; Bangen, J.-M.; Hammerich, L.; Liedtke, C.; Floege, J.; Smeets, B.; Moeller, M.J. Origin of regenerating tubular cells after acute kidney injury. Proc. Natl. Acad. Sci. USA 2014, 111, 1533–1538. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, P.; Lasagni, L.; Remuzzi, G. Renal progenitors: An evolutionary conserved strategy for kidney regeneration. Nat. Rev. Nephrol. 2013, 9, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Rinkevich, Y.; Montoro, D.T.; Contreras-Trujillo, H.; Harari-Steinberg, O.; Newman, A.M.; Tsai, J.M.; Lim, X.; Van-Amerongen, R.; Bowman, A.; Januszyk, M.; et al. In Vivo Clonal Analysis Reveals Lineage-Restricted Progenitor Characteristics in Mammalian Kidney Development, Maintenance, and Regeneration. Cell Rep. 2014, 7, 1270–1283. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D.; Valerius, M.T.; Kobayashi, A.; Mugford, J.W.; Soeung, S.; Duffield, J.S.; McMahon, A.P.; Bonventre, J.V. Intrinsic Epithelial Cells Repair the Kidney after Injury. Cell Stem Cell 2008, 2, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Shu, Z.; Row, S.; Deng, W.-M. Endoreplication: The Good, the Bad, and the Ugly. Trends Cell Biol. 2018, 28, 465–474. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guzzi, F.; Cirillo, L.; Roperto, R.M.; Romagnani, P.; Lazzeri, E. Molecular Mechanisms of the Acute Kidney Injury to Chronic Kidney Disease Transition: An Updated View. Int. J. Mol. Sci. 2019, 20, 4941. https://doi.org/10.3390/ijms20194941
Guzzi F, Cirillo L, Roperto RM, Romagnani P, Lazzeri E. Molecular Mechanisms of the Acute Kidney Injury to Chronic Kidney Disease Transition: An Updated View. International Journal of Molecular Sciences. 2019; 20(19):4941. https://doi.org/10.3390/ijms20194941
Chicago/Turabian StyleGuzzi, Francesco, Luigi Cirillo, Rosa Maria Roperto, Paola Romagnani, and Elena Lazzeri. 2019. "Molecular Mechanisms of the Acute Kidney Injury to Chronic Kidney Disease Transition: An Updated View" International Journal of Molecular Sciences 20, no. 19: 4941. https://doi.org/10.3390/ijms20194941
APA StyleGuzzi, F., Cirillo, L., Roperto, R. M., Romagnani, P., & Lazzeri, E. (2019). Molecular Mechanisms of the Acute Kidney Injury to Chronic Kidney Disease Transition: An Updated View. International Journal of Molecular Sciences, 20(19), 4941. https://doi.org/10.3390/ijms20194941