The Regulatory Role of MicroRNAs in Breast Cancer

 ,
,
Abstract
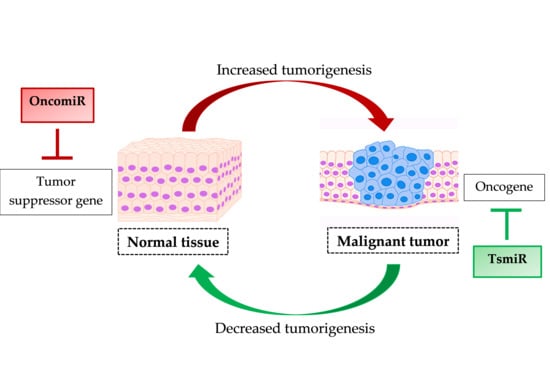
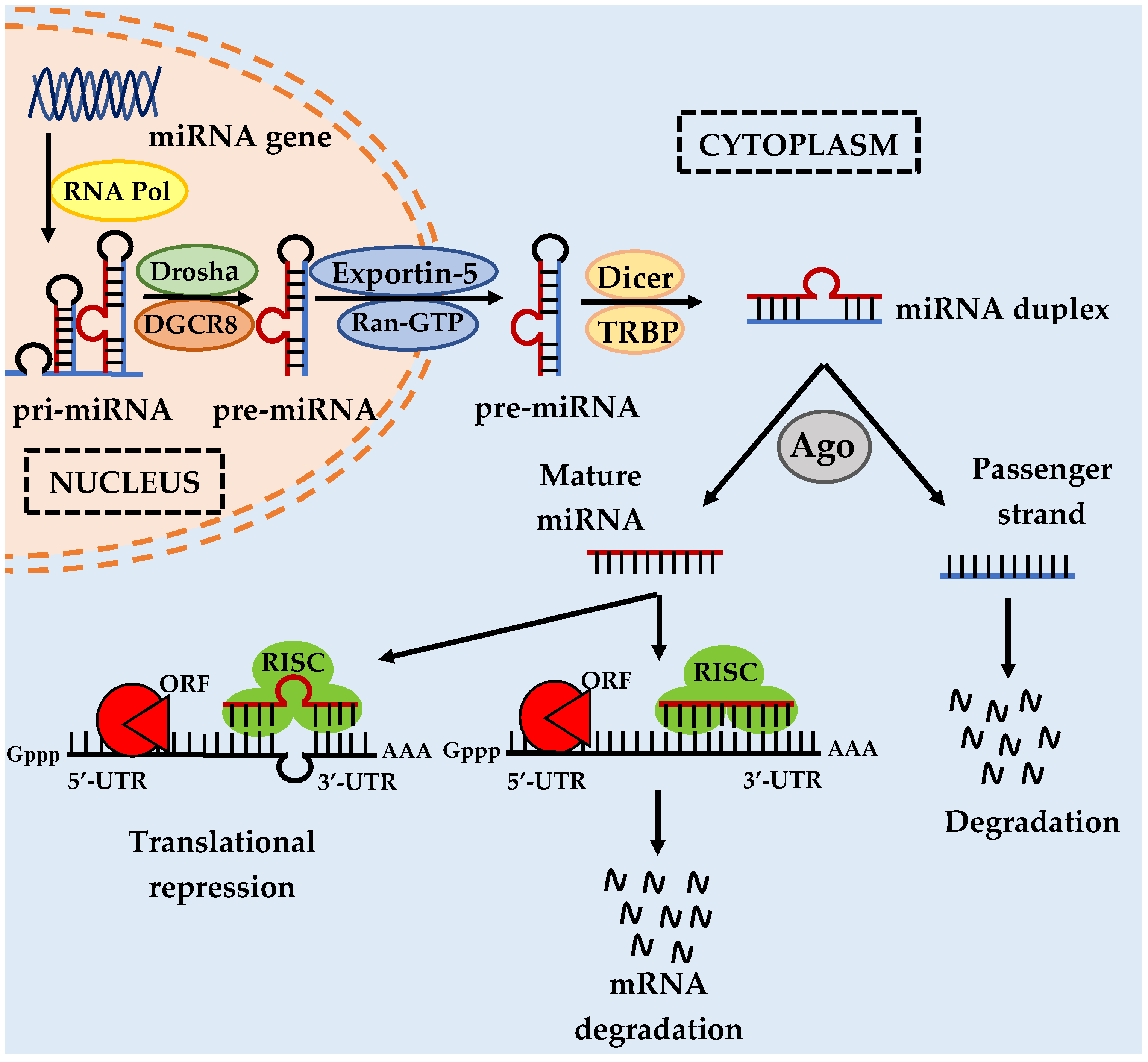
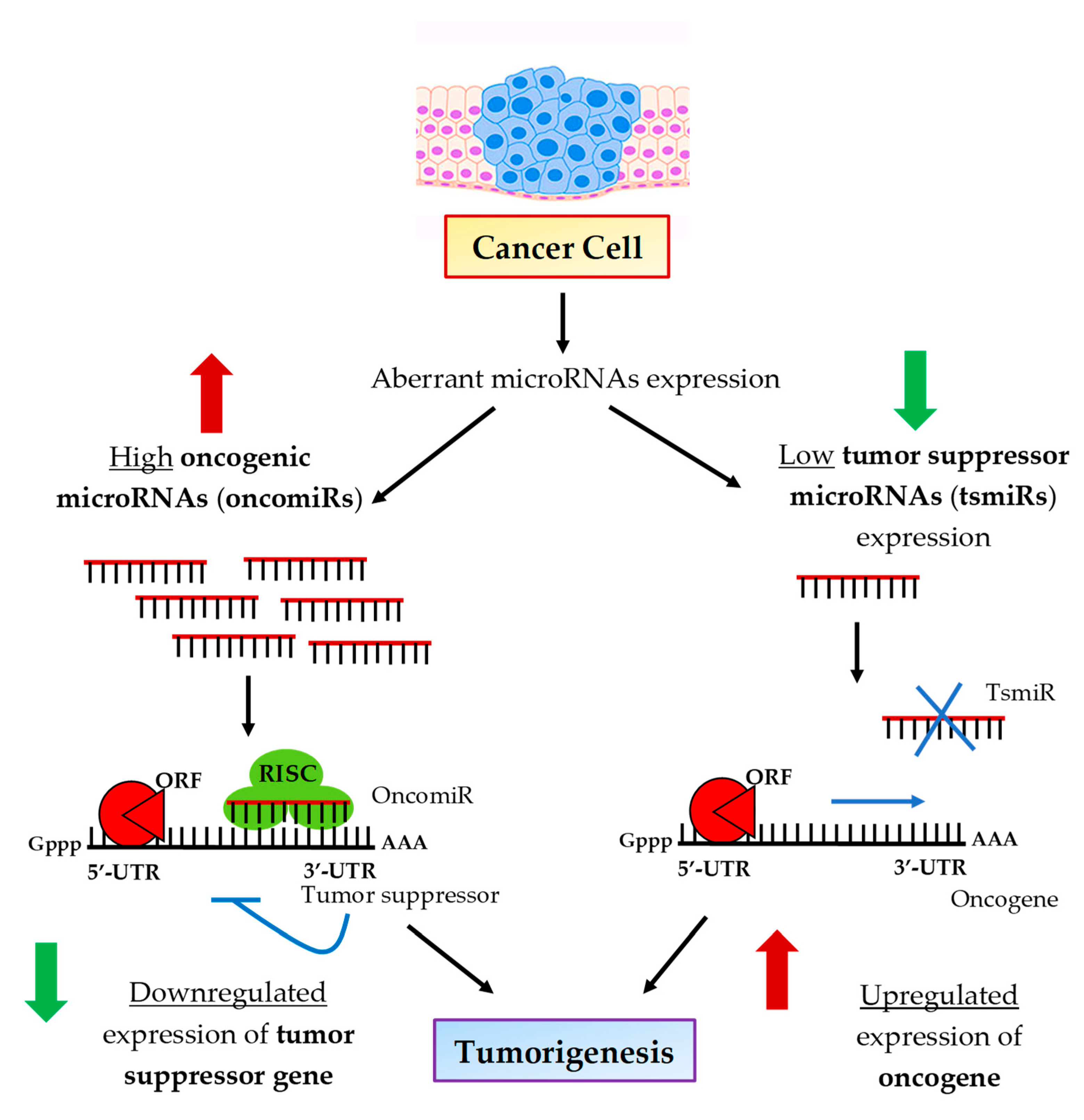
1. Introduction
2. Breast Cancer-Linked MicroRNAs
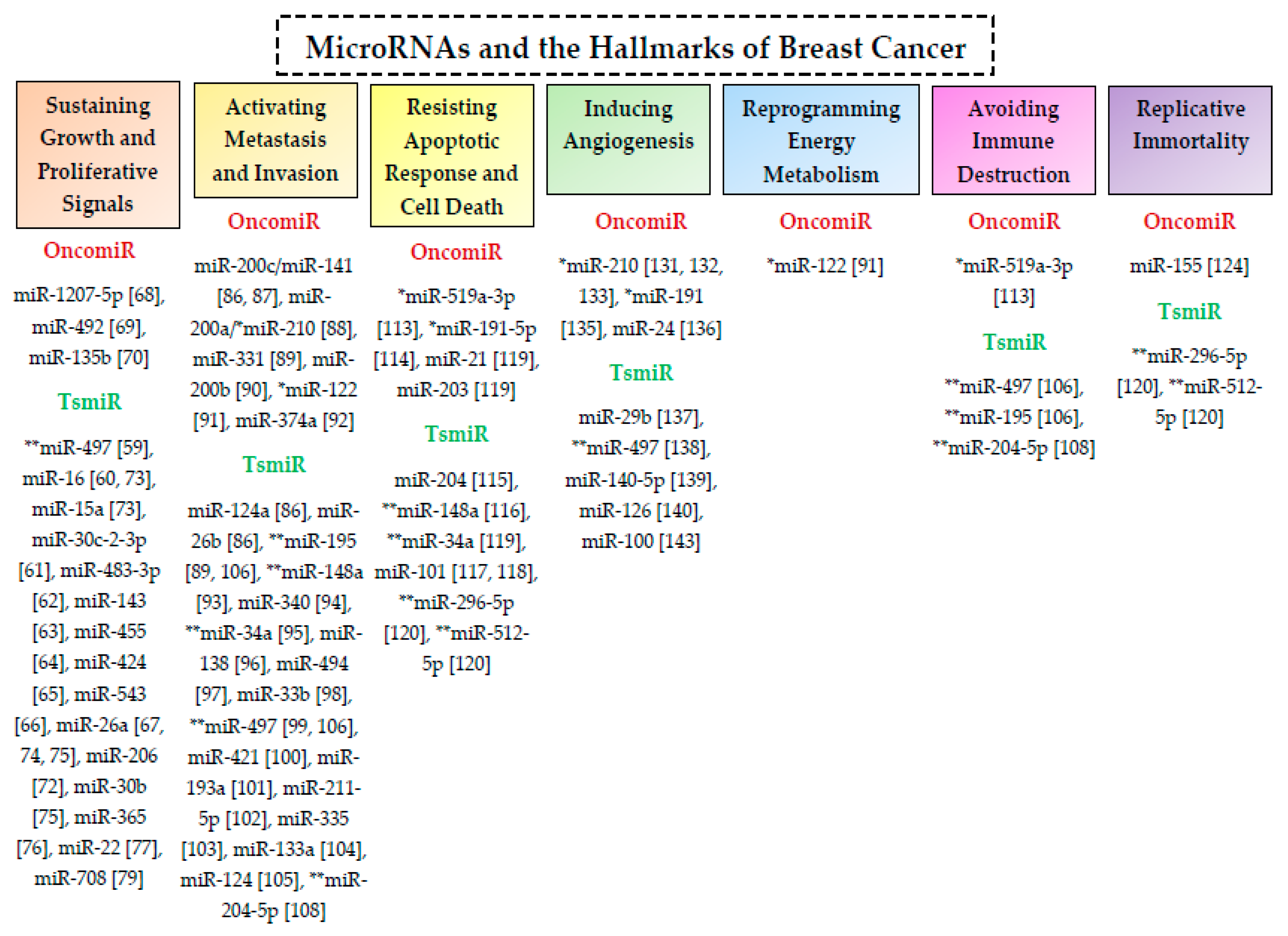
3. MicroRNAs and the Hallmarks of Breast Cancer
3.1. Cell Proliferation and Cell Cycle Regulation
3.2. Metastasis and Invasion
3.3. Apoptotic Response and Cell Death
3.4. Hypoxia and Angiogenesis
4. MicroRNAs and Breast Cancer Progression
5. MicroRNAs Act in Networks in Their Regulation of Breast Cancer
6. Therapeutic Potential and Delivery Options of MicroRNAs in Breast Cancer
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| miRNA | microRNA |
| mRNA | messenger RNA |
| oncomiR | oncogenic miRNA |
| tsmiR | tumor suppressor miRNAs |
References
- Bhaskaran, M.; Mohan, M. MicroRNAs: History, biogenesis, and their evolving role in animal development and disease. Vet. Pathol. 2014, 51, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2018, 47, D155–D162. [Google Scholar] [CrossRef]
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in control of gene expression: An overview of nuclear functions. Int. J. Mol. Sci. 2016, 17, 1712. [Google Scholar] [CrossRef] [PubMed]
- Monteys, A.M.; Spengler, R.M.; Wan, J.; Tecedor, L.; Lennox, K.A.; Xing, Y.; Davidson, B.L. Structure and activity of putative intronic miRNA promoters. RNA 2010, 16, 495–505. [Google Scholar] [CrossRef]
- Hinske, L.C.; França, G.S.; Torres, H.A.; Ohara, D.T.; Lopes-Ramos, C.M.; Heyn, J.; Galante, P.A. miRIAD–integrating microRNA inter- and intragenic data. Database J. Biol. Databases Curation 2014, 2014, bau099. [Google Scholar] [CrossRef]
- Liu, B.; Shyr, Y.; Cai, J.; Liu, Q. Interplay between miRNAs and host genes and their role in cancer. Brief. Funct. Genom. 2019, 18, 255–266. [Google Scholar] [CrossRef]
- Olena, A.F.; Patton, J.G. Genomic organization of microRNAs. J. Cell. Physiol. 2010, 222, 540–545. [Google Scholar] [CrossRef]
- Jiu, S.; Zhu, X.; Wang, J.; Zhang, C.; Mu, Q.; Wang, C.; Fang, J. Genome-wide mapping and analysis of grapevine microRNAs and their potential target genes. Plant Genome 2015, 8, 1–16. [Google Scholar] [CrossRef]
- Havens, M.A.; Reich, A.A.; Duelli, D.M.; Hastings, M.L. Biogenesis of mammalian microRNAs by a non-canonical processing pathway. Nucleic Acids Res. 2012, 40, 4626–4640. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.-H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef] [PubMed]
- Schanen, B.C.; Li, X. Transcriptional regulation of mammalian miRNA genes. Genomics 2011, 97, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Borchert, G.M.; Lanier, W.; Davidson, B.L. RNA polymerase III transcribes human microRNAs. Nat. Struct. Mol. Biol. 2006, 13, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Lee, Y.; Yeom, K.H.; Kim, Y.K.; Jin, H.; Kim, V.N. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004, 18, 3016–3027. [Google Scholar] [CrossRef] [PubMed]
- Roth, B.M.; Ishimaru, D.; Hennig, M. The core microprocessor component DiGeorge syndrome critical region 8 (DGCR8) is a nonspecific RNA-binding protein. J. Biol. Chem. 2013, 288, 26785–26799. [Google Scholar] [CrossRef] [PubMed]
- Yi, R.; Qin, Y.; Macara, I.G.; Cullen, B.R. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003, 17, 3011–3016. [Google Scholar] [CrossRef] [PubMed]
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005, 436, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Yoda, M.; Kawamata, T.; Paroo, Z.; Ye, X.; Iwasaki, S.; Liu, Q.; Tomari, Y. ATP-dependent human RISC assembly pathways. Nat. Struct. Mol. Biol. 2010, 17, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Pong, S.K.; Gullerova, M. Noncanonical functions of microRNA pathway enzymes- Drosha, DGCR8, Dicer and Ago proteins. FEBS Lett. 2018, 592, 2973–2986. [Google Scholar] [CrossRef]
- Ambrus, A.M.; Frolov, M.V. The diverse roles of RNA helicases in RNAi. Cell Cycle 2009, 8, 3500–3505. [Google Scholar] [CrossRef] [PubMed]
- Oliveto, S.; Mancino, M.; Manfrini, N.; Biffo, S. Role of microRNAs in translation regulation and cancer. World J. Biol. Chem. 2017, 8, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Fuchs Wightman, F.; Giono, L.E.; Fededa, J.P.; de la Mata, M. Target RNAs strike back on microRNAs. Front. Genet. 2018, 9, 435. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.A.; Esteller, M. Dysregulation of microRNAs in cancer: Playing with fire. FEBS Lett. 2011, 585, 2087–2099. [Google Scholar] [CrossRef] [PubMed]
- Mallanna, S.K.; Rizzino, A. Emerging roles of microRNAs in the control of embryonic stem cells and the generation of induced pluripotent stem cells. Dev. Biol. 2010, 344, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, A.; Singh, S.; Singh, A.P. MicroRNA-based cancer therapeutics: Big hope from small RNAs. Mol. Cell Pharmacol. 2010, 2, 213–219. [Google Scholar] [PubMed]
- Reddy, K.B. MicroRNA (miRNA) in cancer. Cancer Cell Int. 2015, 15, 38. [Google Scholar] [CrossRef]
- Zhou, S.; Jin, J.; Wang, J.; Zhang, Z.; Freedman, J.H.; Zheng, Y.; Cai, L. MiRNAs in cardiovascular diseases: Potential biomarkers, therapeutic targets and challenges. Acta Pharmacol. Sin. 2018, 39, 1073–1084. [Google Scholar] [CrossRef]
- Sharma, S.; Lu, H.C. MicroRNAs in neurodegeneration: Current findings and potential impacts. J. Alzheimer’s Dis. Parkinsonism 2018, 8, 420. [Google Scholar] [CrossRef]
- Long, H.; Wang, X.; Chen, Y.; Wang, L.; Zhao, M.; Lu, Q. Dysregulation of microRNAs in autoimmune diseases: Pathogenesis, biomarkers and potential therapeutic targets. Cancer Lett. 2018, 428, 90–103. [Google Scholar] [CrossRef]
- Loh, H.-Y.; Lau, Y.-Y.; Lai, K.-S.; Osman, M.A. Transcriptional and Post-Transcriptional Regulation; Kais, G., Ed.; IntechOpen: London, UK, 2018; pp. 77–100. ISBN 978-1-78923-791-7. [Google Scholar]
- Tan, W.; Liu, B.; Qu, S.; Liang, G.; Luo, W.; Gong, C. MicroRNAs and cancer: Key paradigms+ in molecular therapy. Oncol. Lett. 2018, 15, 2735–2742. [Google Scholar] [CrossRef]
- Mandujano-Tinoco, E.A.; García-Venzor, A.; Melendez-Zajgla, J.; Maldonado, V. New emerging roles of microRNAs in breast cancer. Breast Cancer Res. Treat. 2018, 171, 247–259. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA 2018, 68, 394–424. [Google Scholar]
- Makki, J. Diversity of breast carcinoma: Histological subtypes and clinical relevance. Clin. Med. Insights Pathol. 2015, 8, 23–31. [Google Scholar] [CrossRef]
- Polyak, K. Heterogeneity in breast cancer. J. Clin. Investig. 2011, 121, 3786–3788. [Google Scholar] [CrossRef]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast cancer cell line classification and its relevance with breast tumor subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef]
- Polyak, K. Breast cancer: Origins and evolution. J. Clin. Investig. 2007, 117, 3155–3163. [Google Scholar] [CrossRef]
- Karagiannis, G.S.; Goswami, S.; Jones, J.G.; Oktay, M.H.; Condeelis, J.S. Signatures of breast cancer metastasis at a glance. J. Cell Sci. 2016, 129, 1751–1758. [Google Scholar] [CrossRef]
- Castañeda-Gill, J.M.; Vishwanatha, J.K. Antiangiogenic mechanisms and factors in breast cancer treatment. J. Carcinog. 2016, 15, 1. [Google Scholar]
- Ahmad, A. Pathways to breast cancer recurrence. ISRN Oncol. 2013, 2013, 290568. [Google Scholar] [CrossRef]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar]
- Schunkert, E.M.; Zhao, W.; Zänker, K. Breast cancer recurrence risk assessment: Is non-invasive monitoring an option? Biomed. Hub. 2018, 3, 1–17. [Google Scholar] [CrossRef]
- Miller, K.D.; Nogueira, L.; Mariotto, A.B.; Rowland, J.H.; Yabroff, K.R.; Alfano, C.M.; Siegel, R.L. Cancer treatment and survivorship statistics. CA 2019, 2019, 1–23. [Google Scholar]
- Sun, Y.S.; Zhao, Z.; Yang, Z.N.; Xu, F.; Lu, H.J.; Zhu, Z.Y.; Zhu, H.P. Risk factors and preventions of breast cancer. Int. J. Biol. Sci. 2017, 13, 1387–1397. [Google Scholar] [CrossRef]
- Huszno, J.; Budryk, M.; Kołosza, Z.; Nowara, E. The risk factors of toxicity during chemotherapy and radiotherapy in breast cancer patients according to the presence of BRCA gene mutation. Contemp. Oncol. 2015, 19, 72–76. [Google Scholar] [CrossRef]
- Pavlopoulou, A.; Oktay, Y.; Vougas, K.; Louka, M.; Vorgias, C.E.; Georgakilas, A.G. Determinants of resistance to chemotherapy and ionizing radiation in breast cancer stem cells. Cancer Lett. 2016, 380, 485–493. [Google Scholar] [CrossRef]
- Kurozumi, S.; Yamaguchi, Y.; Kurosumi, M.; Ohira, M.; Matsumoto, H.; Horiguchi, J. Recent trends in microRNA research into breast cancer with particular focus on the associations between microRNAs and intrinsic subtypes. J. Hum. Genet. 2016, 62, 15–24. [Google Scholar] [CrossRef]
- Ohzawa, H.; Miki, A.; Teratani, T.; Shiba, S.; Sakuma, Y.; Nishimura, W.; Yasuda, Y. Usefulness of miRNA profiles for predicting pathological responses to neoadjuvant chemotherapy in patients with human epidermal growth factor receptor 2-positive breast cancer. Oncol. Lett. 2017, 13, 1731–1740. [Google Scholar] [CrossRef]
- Van Schooneveld, E.; Wouters, M.C.; Van der Auwera, I.; Peeters, D.J.; Wildiers, H.; Van Dam, P.A.; Van Laere, S.J. Expression profiling of cancerous and normal breast tissues identifies microRNAs that are differentially expressed in serum from patients with (metastatic) breast cancer and healthy volunteers. Breast Cancer Res. 2012, 14, R34. [Google Scholar] [CrossRef]
- Iorio, M.V.; Ferracin, M.; Liu, C.-G.; Veronese, A.; Spizzo, R.; Sabbioni, S.; Croce, C.M. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005, 65, 7065–7070. [Google Scholar] [CrossRef]
- Wang, W.; Luo, Y.P. MicroRNAs in breast cancer: Oncogene and tumor suppressors with clinical potential. J. Zhejiang Univ. Sci. B 2015, 16, 18–31. [Google Scholar] [CrossRef]
- Corcoran, C.; Friel, A.M.; Duffy, M.J.; Crown, J.; O’Driscoll, L. Intracellular and extracellular microRNAs in breast cancer. Clin. Chem. 2011, 57, 18–32. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Joshi, M.; Singh Sodhi, K.; Pandey, R.; Singh, J.; Goyal, S.; Dahal, A. MicroRNA: Biomarker for cancer diagnosis and prognosis. J. Pharm. Biomed. Sci. 2014, 4, 600–610. [Google Scholar]
- Hallstrom, T.C.; Nevins, J.R. Balancing the decision of cell proliferation and cell fate. Cell Cycle 2009, 8, 532–535. [Google Scholar] [CrossRef]
- Nahta, R.; Al-Mulla, F.; Al-Temaimi, R.; Amedei, A.; Andrade-Vieira, R.; Bay, S.N.; Bisson, W.H. Mechanisms of environmental chemicals that enable the cancer hallmark of evasion of growth suppression. Carcinogenesis 2015, 36 (Suppl. 1), S2–S18. [Google Scholar] [CrossRef]
- Karp, G. Cancer. In Cell Biology, 7th ed.; John Wiley and Sons Ltd.: Chichester, UK, 2010; pp. 651–676. ISBN 0470505761. [Google Scholar]
- Luo, Q.; Li, X.; Gao, Y.; Long, Y.; Chen, L.; Huang, Y.; Fang, L. MiRNA-497 regulates cell growth and invasion by targeting cyclin E1 in breast cancer. Cancer Cell Int. 2013, 13, 95. [Google Scholar] [CrossRef]
- Guo, X.; Connick, M.C.; Vanderhoof, J.; Ishak, M.A.; Hartley, R.S. MicroRNA-16 modulates HuR regulation of cyclin E1 in breast cancer cells. Int. J. Mol. Sci. 2015, 16, 7112–7132. [Google Scholar] [CrossRef]
- Shukla, K.; Sharma, A.K.; Ward, A.; Will, R.; Hielscher, T.; Balwierz, A.; Wiemann, S. MicroRNA-30c-2-3p negatively regulates NF-κB signaling and cell cycle progression through downregulation of TRADD and CCNE1 in breast cancer. Mol. Oncol. 2015, 9, 1106–1119. [Google Scholar] [CrossRef]
- Huang, X.; Lyu, J. Tumor suppressor function of miR-483-3p on breast cancer via targeting of the cyclin E1 gene. Exp. Ther. Med. 2018, 16, 2615–2620. [Google Scholar] [CrossRef]
- Zhou, L.L.; Dong, J.L.; Huang, G.; Sun, Z.L.; Wu, J. MicroRNA-143 inhibits cell growth by targeting ERK5 and MAP3K7 in breast cancer. Braz. J. Med. Biol. Res. 2017, 50, e5891. [Google Scholar] [CrossRef]
- Wang, B.; Zou, A.; Ma, L.; Chen, X.; Wang, L.; Zeng, X.; Tan, T. MiR-455 inhibits breast cancer cell proliferation through targeting CDK14. Eur. J. Pharmacol. 2017, 807, 138–143. [Google Scholar] [CrossRef]
- Xie, D.; Song, H.; Wu, T.; Li, D.; Hua, K.; Xu, H.; Fang, L. MicroRNA‑424 serves an anti‑oncogenic role by targeting cyclin‑dependent kinase 1 in breast cancer cells. Oncol. Rep. 2018, 40, 3416–3426. [Google Scholar] [CrossRef]
- Chen, P.; Xu, W.; Luo, Y.; Zhang, Y.; He, Y.; Yang, S.; Yuan, Z. MicroRNA 543 suppresses breast cancer cell proliferation, blocks cell cycle and induces cell apoptosis via direct targeting of ERK/MAPK. Oncotargets Ther. 2017, 10, 1423–1431. [Google Scholar] [CrossRef]
- Huang, Z.M.; Ge, H.F.; Yang, C.C.; Cai, Y.; Chen, Z.; Tian, W.Z.; Tao, J.L. MicroRNA-26a-5p inhibits breast cancer cell growth by suppressing RNF6 expression. Kaohsiung J. Med. Sci. 2019, 35, 467–473. [Google Scholar] [CrossRef]
- Yan, C.; Chen, Y.; Kong, W.; Fu, L.; Liu, Y.; Yao, Q.; Yuan, Y. PVT1-derived miR-1207-5p promotes breast cancer cell growth by targeting STAT6. Cancer Sci. 2017, 108, 868–876. [Google Scholar] [CrossRef]
- Shen, F.; Cai, W.-S.; Feng, Z.; Li, J.-L.; Chen, J.-W.; Cao, J.; Xu, B. MiR-492 contributes to cell proliferation and cell cycle of human breast cancer cells by suppressing SOX7 expression. Tumor Biol. 2015, 36, 1913–1921. [Google Scholar] [CrossRef]
- Hua, K.; Jin, J.; Zhao, J.; Song, J.; Song, H.; Li, D.; Fang, L. MiR-135b, upregulated in breast cancer, promotes cell growth and disrupts the cell cycle by regulating LATS2. Int. J. Oncol. 2016, 48, 1997–2006. [Google Scholar] [CrossRef]
- An, X.; Sarmiento, C.; Tan, T.; Zhu, H. Regulation of multidrug resistance by microRNAs in anti-cancer therapy. Acta Pharm. Sin. B 2017, 7, 38–51. [Google Scholar] [CrossRef]
- Ren, Y.; Wang, H.; Zhang, Y.; Liu, Y. WBP2 modulates G1/S transition in ER+ breast cancer cells and is a direct target of miR-206. Cancer Chemother. Pharmacol. 2017, 79, 1003–1011. [Google Scholar] [CrossRef]
- Chu, J.; Zhu, Y.; Liu, Y.; Sun, L.; Lv, X.; Wu, Y.; Liu, Q. E2F7 overexpression leads to tamoxifen resistance in breast cancer cells by competing with E2F1 at miR-15a/16 promoter. Oncotarget 2015, 6, 31944–31957. [Google Scholar] [CrossRef]
- Liu, J.; Li, X.; Wang, M.; Xiao, G.; Yang, G.; Wang, H.; Pang, Y. A miR-26a/E2F7 feedback loop contributes to tamoxifen resistance in ER-positive breast cancer. Int. J. Oncol. 2018, 53, 1601–1612. [Google Scholar] [CrossRef]
- Tormo, E.; Adam-Artigues, A.; Ballester, S.; Pineda, B.; Zazo, S.; González-Alonso, P.; Eroles, P. The role of miR-26a and miR-30b in HER2+ breast cancer trastuzumab resistance and regulation of the CCNE2 gene. Sci. Rep. 2017, 7, 41309. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Z.; Wang, Q.; Xing, X.J.; Zhao, Y. Overexpression of microRNA-365 inhibits breast cancer cell growth and chemo-resistance through GALNT4. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4710–4718. [Google Scholar]
- Song, Y.K.; Wang, Y.; Wen, Y.Y.; Zhao, P.; Bian, Z.J. MicroRNA-22 suppresses breast cancer cell growth and increases paclitaxel sensitivity by targeting NRAS. Technol. Cancer Res. Treat. 2018, 17. [Google Scholar] [CrossRef]
- Lin, K.T.; Wang, L.H. New dimension of glucocorticoids in cancer treatment. Steroids 2016, 111, 84–88. [Google Scholar] [CrossRef]
- Senthil Kumar, K.J.; Gokila Vani, M.; Hsieh, H.W.; Lin, C.C.; Liao, J.W.; Chueh, P.J.; Wang, S.Y. MicroRNA-708 activation by glucocorticoid receptor agonists regulate breast cancer tumorigenesis and metastasis via downregulation of NF-κB signaling. Carcinogenesis 2019, 40, 335–348. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Huysentruyt, L.C. On the origin of cancer metastasis. Crit. Rev. Oncog. 2013, 18, 43–73. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Jeanes, A.; Gottardi, C.J.; Yap, A.S. Cadherins and cancer: How does cadherin dysfunction promote tumor progression? Oncogene 2008, 27, 6920–6929. [Google Scholar] [CrossRef]
- Tian, X.; Liu, Z.; Niu, B.; Zhang, J.; Tan, T.K.; Lee, S.R.; Zheng, G. E-Cadherin/β-Catenin complex and the epithelial barrier. J. Biomed. Biotechnol. 2011, 2011, 1–6. [Google Scholar] [CrossRef]
- Liu, C.Y.; Lin, H.H.; Tang, M.J.; Wang, Y.K. Vimentin contributes to epithelial-mesenchymal transition cancer cell mechanics by mediating cytoskeletal organization and focal adhesion maturation. Oncotarget 2015, 6, 15966–15983. [Google Scholar] [CrossRef]
- Wheelock, M.J.; Shintani, Y.; Maeda, M.; Fukumoto, Y.; Johnson, K.R. Cadherin switching. J. Cell Sci. 2008, 121, 727–735. [Google Scholar] [CrossRef]
- Jin, T.; Suk Kim, H.; Ki Choi, S.; Hye Hwang, E.; Woo, J.; Suk Ryu, H.; … Kyung Moon, W. MicroRNA-200c/141 upregulates SerpinB2 to promote breast cancer cell metastasis and reduce patient survival. Oncotarget 2017, 8, 32769–32782. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, W.; Li, B.; Stringer-Reasor, E.; Chu, C.; Sun, L.; Wang, L. MicroRNA-200c and microRNA-141 are regulated by a FOXP3-KAT2B axis and associated with tumor metastasis in breast cancer. Breast Cancer Res. 2017, 19, 73. [Google Scholar] [CrossRef]
- Shao, B.; Wang, X.; Zhang, L.; Li, D.; Liu, X.; Song, G.; Li, H. Plasma microRNAs predict chemoresistance in patients with metastatic breast cancer. Technol. Cancer Res. Treat. 2019, 18. [Google Scholar] [CrossRef]
- McAnena, P.; Tanriverdi, K.; Curran, C.; Gilligan, K.; Freedman, J.E.; Brown, J.; Kerin, M.J. Circulating microRNAs miR-331 and miR-195 differentiate local luminal a from metastatic breast cancer. BMC Cancer 2019, 19, 436. [Google Scholar] [CrossRef]
- Hong, H.; Yu, H.; Yuan, J.; Guo, C.; Cao, H.; Li, W.; Xiao, C. MicroRNA-200b impacts breast cancer cell migration and invasion by regulating Ezrin-Radixin-Moesin. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2016, 22, 1946–1952. [Google Scholar] [CrossRef]
- Fong, M.Y.; Zhou, W.; Liu, L.; Alontaga, A.Y.; Chandra, M.; Ashby, J.; Wang, S.E. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 2015, 17, 183–194. [Google Scholar] [CrossRef]
- Cai, J.; Guan, H.; Fang, L.; Yang, Y.; Zhu, X.; Yuan, J.; Li, M. MicroRNA-374a activates Wnt/β-catenin signaling to promote breast cancer metastasis. J. Clin. Investig. 2013, 123, 566–579. [Google Scholar] [CrossRef]
- Jiang, Q.; He, M.; Ma, M.T.; Wu, H.Z.; Yu, Z.J.; Guan, S.; Wei, M.J. MicroRNA-148a inhibits breast cancer migration and invasion by directly targeting WNT-1. Oncol. Rep. 2015, 35, 1425–1432. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi-Yeganeh, S.; Paryan, M.; Arefian, E.; Vasei, M.; Ghanbarian, H.; Mahdian, R.; Soleimani, M. MicroRNA-340 inhibits the migration, invasion, and metastasis of breast cancer cells by targeting Wnt pathway. Tumor Biol. 2016, 37, 8993–9000. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yao, L.; Zhang, J.; Li, X.; Dang, S.; Zeng, K.; Gao, F. Tumor-suppressive microRNA-34a inhibits breast cancer cell migration and invasion via targeting oncogenic TPD52. Tumor Biol. 2016, 37, 7481–7491. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, D.; Feng, Z.; Mao, J.; Zhang, C.; Lu, Y.; Li, L. MicroRNA-138 modulates metastasis and EMT in breast cancer cells by targeting vimentin. Biomed. Pharmacother. 2016, 77, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Zhan, M.N.; Yu, X.T.; Tang, J.; Zhou, C.X.; Wang, C.L.; Yin, Q.Q.; Zhao, Q. MicroRNA-494 inhibits breast cancer progression by directly targeting PAK1. Cell Death Dis. 2017, 8, e2529. [Google Scholar] [CrossRef]
- Lin, Y.; Liu, A.Y.; Fan, C.; Zheng, H.; Li, Y.; Zhang, C.; Ouyang, G. MicroRNA-33b inhibits breast cancer metastasis by targeting HMGA2, SALL4 and Twist1. Sci. Rep. 2015, 5, 9995. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, Y.; Shi, Z.; Hu, Y.; Meng, T.; Zhang, X.; Zhang, J. MicroRNA-497 modulates breast cancer cell proliferation, invasion, and survival by targeting SMAD7. DNA Cell Biol. 2016, 35, 521–529. [Google Scholar] [CrossRef]
- Pan, Y.; Jiao, G.; Wang, C.; Yang, J.; Yang, W. MicroRNA-421 inhibits breast cancer metastasis by targeting metastasis associated 1. Biomed. Pharmacother. 2016, 83, 1398–1406. [Google Scholar] [CrossRef]
- Xie, F.; Hosany, S.; Zhong, S.; Jiang, Y.; Zhang, F.; Lin, L.; Hu, X. MicroRNA-193a inhibits breast cancer proliferation and metastasis by downregulating WT1. PLoS ONE 2017, 12, e0185565. [Google Scholar] [CrossRef]
- Chen, L.L.; Zhang, Z.J.; Yi, Z.B.; Li, J.J. MicroRNA-211-5p suppresses tumour cell proliferation, invasion, migration and metastasis in triple-negative breast cancer by directly targeting SETBP1. Br. J. Cancer 2017, 117, 78–88. [Google Scholar] [CrossRef]
- Dong, Y.; Liu, Y.; Jiang, A.; Li, R.; Yin, M.; Wang, Y. MicroRNA-335 suppresses the proliferation, migration, and invasion of breast cancer cells by targeting EphA4. Mol. Cell. Biochem. 2018, 439, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Sui, Y.; Zhang, X.; Yang, H.; Wei, W.; Wang, M. MicroRNA-133a acts as a tumour suppressor in breast cancer through targeting LASP1. Oncol. Rep. 2017, 39, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.; Chen, C.; Li, X.; Wei, Z.; Liu, Z.; Liu, Y. MicroRNA 124 suppresses cell proliferation and invasion of triple negative breast cancer cells by targeting STAT3. Mol. Med. Rep. 2019, 19, 3667–3675. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Cai, Y.; Zhang, D.; Sun, J.; Xu, C.; Zhao, W.; Pan, C. MiR-195/miR-497 regulate cd274 expression of immune regulatory ligands in triple-negative breast cancer. J. Breast Cancer 2018, 21, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Manson, Q.F.; Schrijver, W.; Ter Hoeve, N.D.; Moelans, C.B.; van Diest, P.J. Frequent discordance in PD-1 and PD-L1 expression between primary breast tumors and their matched distant metastases. Clin. Exp. Metastasis 2019, 36, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.S.; Ryu, H.S.; Kim, N.; Kim, J.; Lee, E.; Moon, H.; Moon, H.G. Tumor suppressor microRNA-204-5p regulates growth, metastasis, and immune microenvironment remodeling in breast cancer. Cancer Res. 2019, 79, 1520–1534. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Tiwari, M. Apoptosis and survival. Indian J. Hum. Genet. 2011, 17, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef]
- Su, Z.; Yang, Z.; Xu, Y.; Chen, Y.; Yu, Q. MicroRNAs in apoptosis, autophagy and necroptosis. Oncotarget 2015, 6, 8474–8490. [Google Scholar] [CrossRef]
- Breunig, C.; Pahl, J.; Küblbeck, M.; Miller, M.; Antonelli, D.; Erdem, N.; Wiemann, S. MicroRNA-519a-3p mediates apoptosis resistance in breast cancer cells and their escape from recognition by natural killer cells. Cell Death Dis. 2017, 8, e2973. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Nagpal, N.; Ghosh, P.C.; Kulshreshtha, R. P53-miR-191-SOX4 regulatory loop affects apoptosis in breast cancer. RNA 2017, 23, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Qiu, W.; Zhang, G.; Xu, S.; Gao, Q.; Yang, Z. MicroRNA-204 targets JAK2 in breast cancer and induces cell apoptosis through the STAT3/BCl-2/survivin pathway. Int. J. Clin. Exp. Pathol. 2015, 8, 5017–5025. [Google Scholar] [PubMed]
- Li, Q.; Ren, P.; Shi, P.; Chen, Y.; Xiang, F.; Zhang, L.; Xie, M. MicroRNA-148a promotes apoptosis and suppresses growth of breast cancer cells by targeting B-cell lymphoma 2. Anti-Cancer Drugs 2017, 28, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.; Dai, Z.; Ma, Y.; Wang, Z.; Liu, X.; Wang, X. MicroRNA-101 inhibits cell proliferation and induces apoptosis by targeting EYA1 in breast cancer. Int. J. Mol. Med. 2016, 37, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zeng, H.; Li, H.; Chen, T.; Wang, L.; Zhang, K.; Wang, S. MicroRNA-101 inhibits growth, proliferation and migration and induces apoptosis of breast cancer cells by targeting Sex-Determining Region Y-Box 2. Cell. Physiol. Biochem. 2017, 43, 717–732. [Google Scholar] [CrossRef]
- Li, P.; Guo, Y.; Bledsoe, G.; Yang, Z.; Chao, L.; Chao, J. Kallistatin induces breast cancer cell apoptosis and autophagy by modulating Wnt signaling and microRNA synthesis. Exp. Cell Res. 2016, 340, 305–314. [Google Scholar] [CrossRef]
- Muraki, K.; Nyhan, K.; Han, L.; Murnane, J.P. Mechanisms of telomere loss and their consequences for chromosome instability. Front. Oncol. 2012, 2, 135. [Google Scholar] [CrossRef]
- Jafri, M.A.; Ansari, S.A.; Alqahtani, M.H.; Shay, J.W. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med. 2016, 8, 69. [Google Scholar] [CrossRef]
- Ludlow, A.T.; Slusher, A.L.; Sayed, M.E. Insights into Telomerase/hTERT alternative splicing regulation using bioinformatics and network analysis in cancer. Cancers 2019, 11, 666. [Google Scholar] [CrossRef]
- Dinami, R.; Buemi, V.; Sestito, R.; Zappone, A.; Ciani, Y.; Mano, M.; Schoeftner, S. Epigenetic silencing of miR-296 and miR-512 ensures hTERT dependent apoptosis protection and telomere maintenance in basal-type breast cancer cells. Oncotarget 2017, 8, 95674–95691. [Google Scholar] [CrossRef] [PubMed]
- Dinami, R.; Ercolani, C.; Petti, E.; Piazza, S.; Ciani, Y.; Sestito, R.; Schoeftner, S. MiR-155 drives telomere fragility in human breast cancer by targeting TRF1. Cancer Res. 2014, 74, 4145–4156. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Collins, K. Control of telomerase action at human telomeres. Nat. Struct. Mol. Biol. 2015, 22, 848–852. [Google Scholar] [CrossRef] [PubMed]
- Horsman, M.R.; Vaupel, P. Pathophysiological basis for the formation of the tumor microenvironment. Front. Oncol. 2016, 6, 66. [Google Scholar] [CrossRef]
- Hillen, F.; Griffioen, A.W. Tumour vascularization: Sprouting angiogenesis and beyond. Cancer Metastasis Rev. 2007, 26, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Langley, R.R.; Fidler, I.J. Tumor cell-organ microenvironment interactions in the pathogenesis of cancer metastasis. Endocr. Rev. 2007, 28, 297–321. [Google Scholar] [CrossRef]
- Gilkes, D.M.; Semenza, G.L. Role of hypoxia-inducible factors in breast cancer metastasis. Future Oncol. 2013, 9, 1623–1636. [Google Scholar] [CrossRef]
- Johnson, K.E.; Wilgus, T.A. Vascular endothelial growth factor and angiogenesis in the regulation of cutaneous wound repair. Adv. Wound Care 2014, 3, 647–661. [Google Scholar] [CrossRef]
- Camps, C.; Saini, H.K.; Mole, D.R.; Choudhry, H.; Reczko, M.; Guerra-Assunção, J.A.; Ragoussis, J. Integrated analysis of microRNA and mRNA expression and association with HIF binding reveals the complexity of microRNA expression regulation under hypoxia. Mol. Cancer 2014, 13, 28. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, J.; Wang, L.; Dai, H.; Li, N.; Hu, W.; Cai, H. HIF-1α promotes breast cancer cell MCF-7 proliferation and invasion through regulating miR-210. Cancer Biother. Radiopharm. 2017, 32, 297–301. [Google Scholar] [CrossRef]
- Costales, M.G.; Haga, C.L.; Velagapudi, S.P.; Childs-Disney, J.L.; Phinney, D.G.; Disney, M.D. Small molecule inhibition of microRNA-210 reprograms an oncogenic hypoxic circuit. J. Am. Chem. Soc. 2017, 139, 3446–3455. [Google Scholar] [CrossRef] [PubMed]
- Harquail, J.; LeBlanc, N.; Ouellette, R.J.; Robichaud, G.A. MiRNAs 484 and 210 regulate Pax-5 expression and function in breast cancer cells. Carcinogenesis 2019, 40, 1010–1020. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, N.; Ahmad, H.M.; Chameettachal, S.; Sundar, D.; Ghosh, S.; Kulshreshtha, R. HIF-inducible miR-191 promotes migration in breast cancer through complex regulation of TGFβ-signaling in hypoxic microenvironment. Sci. Rep. 2015, 5, 9650. [Google Scholar] [CrossRef] [PubMed]
- Roscigno, G.; Puoti, I.; Giordano, I.; Donnarumma, E.; Russo, V.; Affinito, A.; Condorelli, G. MiR-24 induces chemotherapy resistance and hypoxic advantage in breast cancer. Oncotarget 2017, 8, 19507–19521. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cai, B.; Shen, L.; Dong, Y.; Lu, Q.; Sun, S.; Chen, J. MiRNA-29b suppresses tumor growth through simultaneously inhibiting angiogenesis and tumorigenesis by targeting Akt3. Cancer Lett. 2017, 397, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Cai, X.; Huang, C.; Xu, J.; Liu, A. MiR-497 suppresses angiogenesis in breast carcinoma by targeting HIF-1α. Oncol. Rep. 2015, 35, 1696–1702. [Google Scholar] [CrossRef]
- Lu, Y.; Qin, T.; Li, J.; Wang, L.; Zhang, Q.; Jiang, Z.; Mao, J. MicroRNA-140-5p inhibits invasion and angiogenesis through targeting VEGF-A in breast cancer. Cancer Gene Ther. 2017, 24, 386–392. [Google Scholar] [CrossRef]
- Alhasan, L. MiR-126 modulates angiogenesis in breast cancer by targeting VEGF-A -mRNA. Asian Pac. J. Cancer Prev. APJCP 2019, 20, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Trivanović, D.; Krstić, J.; Djordjević, I.O.; Mojsilović, S.; Santibanez, J.F.; Bugarski, D.; Jauković, A. The roles of mesenchymal stromal/stem cells in tumor microenvironment associated with inflammation. Mediat. Inflamm. 2016, 2016, 7314016. [Google Scholar] [CrossRef]
- Zhou, J.; Tan, X.; Tan, Y.; Li, Q.; Ma, J.; Wang, G. Mesenchymal stem cell derived exosomes in cancer progression, metastasis and drug delivery: A comprehensive review. J. Cancer 2018, 9, 3129–3137. [Google Scholar] [CrossRef]
- Pakravan, K.; Babashah, S.; Sadeghizadeh, M.; Mowla, S.J.; Mossahebi-Mohammadi, M.; Ataei, F.; Javan, M. MicroRNA-100 shuttled by mesenchymal stem cell-derived exosomes suppresses in vitro angiogenesis through modulating the mTOR/HIF-1α/VEGF signaling axis in breast cancer cells. Cell. Oncol. 2017, 40, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.C.; Aroner, S.A.; Connolly, J.L.; Colditz, G.A.; Schnitt, S.J.; Tamimi, R.M. Breast cancer risk by extent and type of atypical hyperplasia: An update from the Nurses’ Health Studies. Cancer 2015, 122, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Van Loo, P.; Wedge, D.C.; Alexandrov, L.B.; Greenman, C.D.; Lau, K.W.; Breast cancer working group of the international cancer genome consortium. The life history of 21 breast cancers. Cell 2012, 149, 994–1007. [Google Scholar] [CrossRef]
- Kader, T.; Hill, P.; Rakha, E.A.; Campbell, I.G.; Gorringe, K.L. Atypical ductal hyperplasia: Update on diagnosis, management, and molecular landscape. Breast Cancer Res. 2018, 20, 39. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Quan, H.; Lv, J.; Meng, L.; Wang, C.; Yu, Z.; Han, J. Serum microRNA as potential biomarker to detect breast atypical hyperplasia and early-stage breast cancer. Future Oncol. 2018, 14, 3145–3161. [Google Scholar] [CrossRef]
- Stankevicins, L.; Barat, A.; Dessen, P.; Vassetzky, Y.; de Moura Gallo, C.V. The microRNA-205-5p is correlated to metastatic potential of 21T series: A breast cancer progression model. PLoS ONE 2017, 12, e0173756. [Google Scholar] [CrossRef]
- Chai, C.; Wu, H.; Wang, B.; Eisenstat, D.D.; Leng, R.P. MicroRNA-498 promotes proliferation and migration by targeting the tumor suppressor PTEN in breast cancer cells. Carcinogenesis 2018, 39, 1185–1196. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, X.; Ge, G.; Zang, X.; Shao, M.; Zou, S.; Xu, W. MiR‑498 inhibits the growth and metastasis of liver cancer by targeting ZEB2. Oncol. Rep. 2018, 41, 1638–1648. [Google Scholar] [CrossRef]
- Liu, C.; Liu, Z.; Li, X.; Tang, X.; He, J.; Lu, S. MicroRNA-1297 contributes to tumor growth of human breast cancer by targeting PTEN/PI3K/AKT signaling. Oncol. Rep. 2018, 38, 2435–2443. [Google Scholar] [CrossRef]
- Miao, Y.; Zheng, W.; Li, N.; Su, Z.; Zhao, L.; Zhou, H.; Jia, L. MicroRNA-130b targets PTEN to mediate drug resistance and proliferation of breast cancer cells via the PI3K/Akt signaling pathway. Sci. Rep. 2017, 7, 41942. [Google Scholar] [CrossRef]
- Wang, N.; Zhu, C.; Xu, Y.; Qian, W.; Zheng, M. Negative Regulation of PTEN by microRNA-221 and its association with drug resistance and cellular senescence in lung cancer cells. Biomed. Res. Int. 2018, 2018, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.M.; Yu, X.N.; Liu, T.T.; Zhu, H.R.; Shi, X.; Bilegsaikhan, E.; Zhu, J.M. microRNA-19a-3p promotes tumor metastasis and chemoresistance through the PTEN/Akt pathway in hepatocellular carcinoma. Biomed. Pharmacother. 2018, 105, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- De Anda-Jáuregui, G.; Espinal-Enríquez, J.; Drago-García, D.; Hernández-Lemus, E. Nonredundant, highly connected microRNAs control functionality in breast cancer networks. Int. J. Genom. 2018, 2018, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Simonson, B.; Das, S. MicroRNA therapeutics: The next magic bullet? Mini Rev. Med. Chem. 2015, 15, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.Y.; Ferrajoli, A.; Sood, A.K.; Lopez-Berstein, G.; Calin, G.A. MicroRNA therapeutics in cancer-an emerging concept. EBioMedicine 2016, 12, 34–42. [Google Scholar] [CrossRef] [PubMed]
- van Rooij, E.; Kauppinen, S. Development of microRNA therapeutics is coming of age. EMBO Mol. Med. 2014, 6, 851–864. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.S.; Sharp, P.A. MicroRNA sponges: Progress and possibilities. RNA 2010, 16, 2043–2050. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Miyagishi, M. Tiny masking locked nucleic acids effectively bind to mRNA and inhibit binding of microRNAs in relation to thermodynamic stability. Biomed. Rep. 2014, 2, 509–512. [Google Scholar] [CrossRef]
- Ni, S.; Yao, H.; Wang, L.; Lu, J.; Jiang, F.; Lu, A.; Zhang, G. Chemical modifications of nucleic acid aptamers for therapeutic purposes. Int. J. Mol. Sci. 2017, 18, 1683. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Carrillo, E.; Liu, Y.P.; Berkhout, B. Improving miRNA delivery by optimizing mirna expression cassettes in diverse virus vectors. Hum. Gene Ther. Methods 2017, 28, 177–190. [Google Scholar] [CrossRef]
- Wang, H.; Liu, S.; Jia, L.; Chu, F.; Zhou, Y.; He, Z.; Xu, L. Nanostructured lipid carriers for MicroRNA delivery in tumor gene therapy. Cancer Cell Int. 2018, 18, 101. [Google Scholar] [CrossRef] [PubMed]
- Naidu, S.; Magee, P.; Garofalo, M. MiRNA-based therapeutic intervention in cancer. J. Hematol. Oncol. 2015, 8, 68. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.K.; Chow, M.Y.; Zhang, Y.; Leung, S.W. siRNA versus miRNA as therapeutics for gene silencing. Molecular Therapy. Nucleic Acids 2015, 4, e252. [Google Scholar] [CrossRef] [PubMed]
- Adli, M. The CRISPR tool kit for genome editing and beyond. Nat. Commun. 2018, 9, 1911. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, W. CRISPR-mediated targeting of HER2 inhibits cell proliferation through a dominant negative mutation. Cancer Lett. 2017, 385, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Van Treuren, T.; Vishwanatha, J.K. CRISPR deletion of MIEN1 in breast cancer cells. PLoS ONE 2018, 13, e0204976. [Google Scholar] [CrossRef] [PubMed]
- Thomson, D.W.; Bracken, C.P.; Szubert, J.M.; Goodall, G.J. On measuring miRNAs after transient transfection of mimics or antisense inhibitors. PLoS ONE 2013, 8, e55214. [Google Scholar] [CrossRef]
- Kim, T.K.; Eberwine, J.H. Mammalian cell transfection: The present and the future. Anal. Bioanal. Chem. 2010, 397, 3173–3178. [Google Scholar] [CrossRef]
- Hannafon, B.N.; Cai, A.; Calloway, C.L.; Xu, Y.F.; Zhang, R.; Fung, K.M.; Ding, W.Q. MiR-23b and miR-27b are oncogenic microRNAs in breast cancer: Evidence from a CRISPR/Cas9 deletion study. BMC Cancer 2019, 19, 642. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MicroRNA | Interacted/Correlated Gene(s) and Protein(s) | Associated Events | Reference |
|---|---|---|---|
| Major oncogenic microRNAs in breast cancer | |||
| miR-1207-5p | STAT2, CDKN1A, CDKN1B | Promotion of cell proliferation and G2 cell cycle progression | [68] |
| miR-492 | SOX7, cyclin D1, c-MYC | Promotion of cell proliferation and G1–S cell cycle progression | [69] |
| miR-135b | LATS2, CDK2, p-YAP | Promotion of cell proliferation and S–G2/M cell cycle progression | [70] |
| miR-200c and miR-141 | SerpinB2, c-Jun, c-Fos, FosB, FOXP3, KAT2B | Promote metastasis and elevated in serum of metastatic mouse model and breast cancer patients | [86,87] |
| miR-331 | HER2, HOTAIR, E2F1, DOHH, PHLPP | Promotion of metastasis and invasion by elevation in plasma of metastatic breast cancer patients | [89] |
| miR-200b | Ezrin/Radixin/Moesin (ERM) | Promotion of metastasis and invasion | [90] |
| miR-122 | pyruvate kinase (PK) and citrate synthase (CS) | Promotion of metastasis by reprogrammed glucose metabolism | [91] |
| miR-374a | E-cadherin, γ-catenin, CK18, vimentin, N-cadherin, Β-catenin, WIF1, PTEN, WNT5A | Promotion of metastasis by regulating EMT and Wnt/β-catenin signaling | [92] |
| miR-519a-3p | TRAIL-R2 (TNFRSF10B), caspase-8, caspase-7, MICA, ULBP2 | Promotion of apoptosis resistance and escape from natural killer cell recognition | [113] |
| miR-191-5p | SOX4, caspase-3, caspase-7, p53 | Promotion of apoptosis resistance and doxorubicin resistance | [114] |
| miR-21 | Akt, BCL-2, BAX | Pro-survival effect can be overcome by kallistatin | [119] |
| miR-203 | PKC-ERK, SOCS3 | Pro-survival effect can be overcome by kallistatin | [119] |
| miR-155 | TRF1 | Telomere fragility and genomic instability | [124] |
| miR-210 | HIFs, GPD1L, Pax-5 | Hypoxia-inducible miRNA | [131,132,133] |
| miR-191 | HuR, TGFβ2, SMAD3, BMP4, JUN, FOS, PTGS2, CTGF, VEGFA | Hypoxia-inducible miRNA and stimulator of TGFβ-signaling pathways | [135] |
| miR-24 | Nanog, Oct-3/4, BimL, F1H1, HIF-1α, Snail, VEGFA | Hypoxia-inducible miRNA | [136] |
| Major tumor suppressor miRNAs in breast cancer | |||
| miR-497 | Cyclin E1 | Anti-proliferative and G1-S cell cycle arrest | [59] |
| SMAD7 | Anti-metastasis and anti-invasion | [99] | |
| CD274 | Anti-metastasis, anti-tumorigenic and inhibition of immune response or tumor immune escape | [106] | |
| VEGF, HIF-1α | Anti-angiogenesis and anti-tumorigenic | [138] | |
| miR-16 | Cyclin E1, E2F7 | Anti-proliferative and G1–S cell cycle arrest, restores tamoxifen sensitivity | [60,73] |
| miR-30c-2-3p | Cyclin E1 | Anti-proliferative and G1–S cell cycle arrest | [61] |
| miR-483-3p | Cyclin E1, p-NPAT, CDK2 | Anti-proliferative and G1–S cell cycle arrest | [62] |
| miR-143 | ERK5, MAP3K7, Cyclin D1 | Anti-proliferative | [63] |
| miR-455 | CDK14, Cyclin D1, p21 | Anti-proliferative | [64] |
| miR-424 | CDK1, YAP, p-ERK1/2 | Anti-proliferative and G2–M cell cycle arrest | [65] |
| miR-543 | ERK/MAPK | Anti-proliferative, cell cycle arrest and apoptosis | [66] |
| miR-26a | Cyclin D1, CDK4, CDK6, p21, p27, p53, RNF6/ERα/BCL-xL, E2F7, MYC, cyclin E2 | Anti-proliferative, G1 cell cycle arrest and restores sensitivity to tamoxifen and trastuzumab treatment | [67,74,75] |
| miR-206 | WBP2, p21, CDK4, cyclin D1 | Anti-proliferative, cell cycle arrest and restores sensitivity to tamoxifen treatment | [72] |
| miR-15a | Cyclin E1, E2F7 | Anti-proliferative and G1–S cell cycle arrest, restores tamoxifen sensitivity | [73] |
| miR-30b | Cyclin E2 | Anti-proliferative, G1 cell cycle arrest and restores sensitivity to trastuzumab treatment | [75] |
| miR-365 | GALNT4 | Anti-proliferative and restores sensitivity to Fluorouracil chemotherapeutic treatment | [76] |
| miR-22 | KRAS | Anti-proliferative and restores sensitivity to Paclitaxel chemotherapeutic treatment | [77] |
| miR-708 | IKKβ, COX-2, c-MYC | Anti-proliferative and regulates cell cycle arrest upon induction of glucocorticoid agonists, DEX and ATA | [79] |
| miR-124a and miR-26b | SerpinB2 | Anti-metastasis and anti-invasion | [86] |
| miR-195 | FASN, HMGCR, ACACA, CYP27B1 | Anti-metastasis and anti-invasion by underregulation in plasma of metastatic breast cancer patients | [89] |
| CD274 | Anti-metastasis, anti-tumorigenic and inhibits immune response or tumor immune escape | [106] | |
| miR-148a | WNT-1, β-catenin, MMP-7, TCF-4 | Anti-metastasis and anti-invasion by regulating Wnt/β-catenin signaling pathway | [93] |
| BCL-2, caspases | Promotes apoptotic response and overcomes chemoresistance | [116] | |
| miR-340 | c-MYC, CTNNB1, ROCK1 | Anti-metastasis and anti-invasion by regulating Wnt/β-catenin and Rho/Rho-associated kinase (ROCK) signaling pathways | [94] |
| miR-34a | TPD52, E-cadherin, TGF-β, N-cadherin | Anti-metastasis and anti-invasion by regulating EMT | [95] |
| P53 | Pro-apoptotic effect can be induced by kallistatin | [119] | |
| miR-138 | E-cadherin, vimentin, N-cadherin, Snail | Anti-metastasis and anti-invasion by regulating EMT | [96] |
| miR-494 | PAK1, E-cadherin | Anti-metastasis and anti-invasion | [97] |
| miR-33b | HMGA2, SALL4, Twist 1 | Anti-metastasis and anti-invasion | [98] |
| miR-421 | MTA1 | Anti-metastasis and anti-invasion | [100] |
| miR-193a | WT1 | Anti-metastasis and anti-invasion | [101] |
| miR-211-5p | SETBP1 | Anti-metastasis and anti-invasion | [102] |
| miR-335 | EphA4 | Anti-metastasis and anti-invasion | [103] |
| miR-133a | LASP1 | Anti-metastasis and anti-invasion | [104] |
| miR-124 | STAT3 | Anti-metastasis and anti-invasion | [105] |
| miR-204-5p | PIK3CB | Anti-metastasis, anti-tumorigenic, restores sensitivity towards PIK3CB inhibitors and chemotherapeutic drugs (i.e., doxorubicin, taxanes and bortezomib), and involved in tumor immune microenvironment remodeling | [108] |
| miR-204 | JAK2, BCL-2, survivin | Promotion of apoptotic response | [115] |
| miR-101 | EYA1, jagged1, Hes1, Hey1, SOX2 | Promotion of apoptotic response by negatively regulating Notch pathway | [117,118] |
| miR-296-5p and miR-512-5p | hTERT | Reduction of telomerase activity, impairment of telomere maintenance and activation of replicative senescence and apoptosis programs | [120] |
| miR-29b | Akt3, VEGF, c-MYC | Anti-angiogenesis and anti-tumorigenesis | [137] |
| miR-140-5p | VEGFA, CD31, Ki-67, MMP-9 | Anti-angiogenesis and anti-tumorigenesis | [139] |
| miR-126 | VEGFA | Anti-angiogenesis and anti-tumorigenesis | [140] |
| miR-100 | VEGF, mTOR/HIF-1α | Shuttling of miRNA enriched in MSC-derived exosomes, anti-angiogenesis and anti-tumorigenesis | [143] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loh, H.-Y.; Norman, B.P.; Lai, K.-S.; Rahman, N.M.A.N.A.; Alitheen, N.B.M.; Osman, M.A. The Regulatory Role of MicroRNAs in Breast Cancer. Int. J. Mol. Sci. 2019, 20, 4940. https://doi.org/10.3390/ijms20194940
Loh H-Y, Norman BP, Lai K-S, Rahman NMANA, Alitheen NBM, Osman MA. The Regulatory Role of MicroRNAs in Breast Cancer. International Journal of Molecular Sciences. 2019; 20(19):4940. https://doi.org/10.3390/ijms20194940
Chicago/Turabian StyleLoh, Hui-Yi, Brendan P. Norman, Kok-Song Lai, Nik Mohd Afizan Nik Abd. Rahman, Noorjahan Banu Mohamed Alitheen, and Mohd Azuraidi Osman. 2019. "The Regulatory Role of MicroRNAs in Breast Cancer" International Journal of Molecular Sciences 20, no. 19: 4940. https://doi.org/10.3390/ijms20194940
APA StyleLoh, H.-Y., Norman, B. P., Lai, K.-S., Rahman, N. M. A. N. A., Alitheen, N. B. M., & Osman, M. A. (2019). The Regulatory Role of MicroRNAs in Breast Cancer. International Journal of Molecular Sciences, 20(19), 4940. https://doi.org/10.3390/ijms20194940