CDDO-Me Attenuates Vasogenic Edema and Astroglial Death by Regulating NF-κB p65 Phosphorylations and Nrf2 Expression Following Status Epilepticus

Abstract
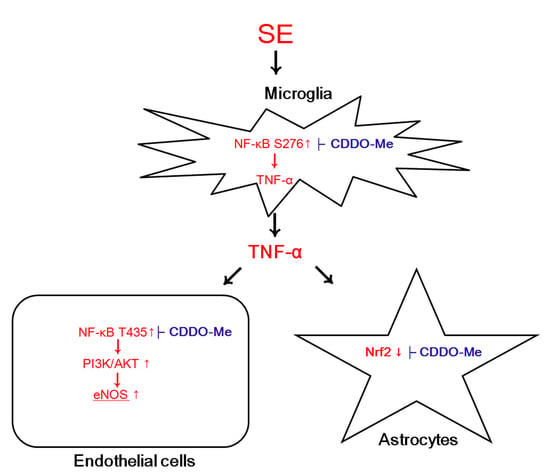
:
1. Introduction
2. Results
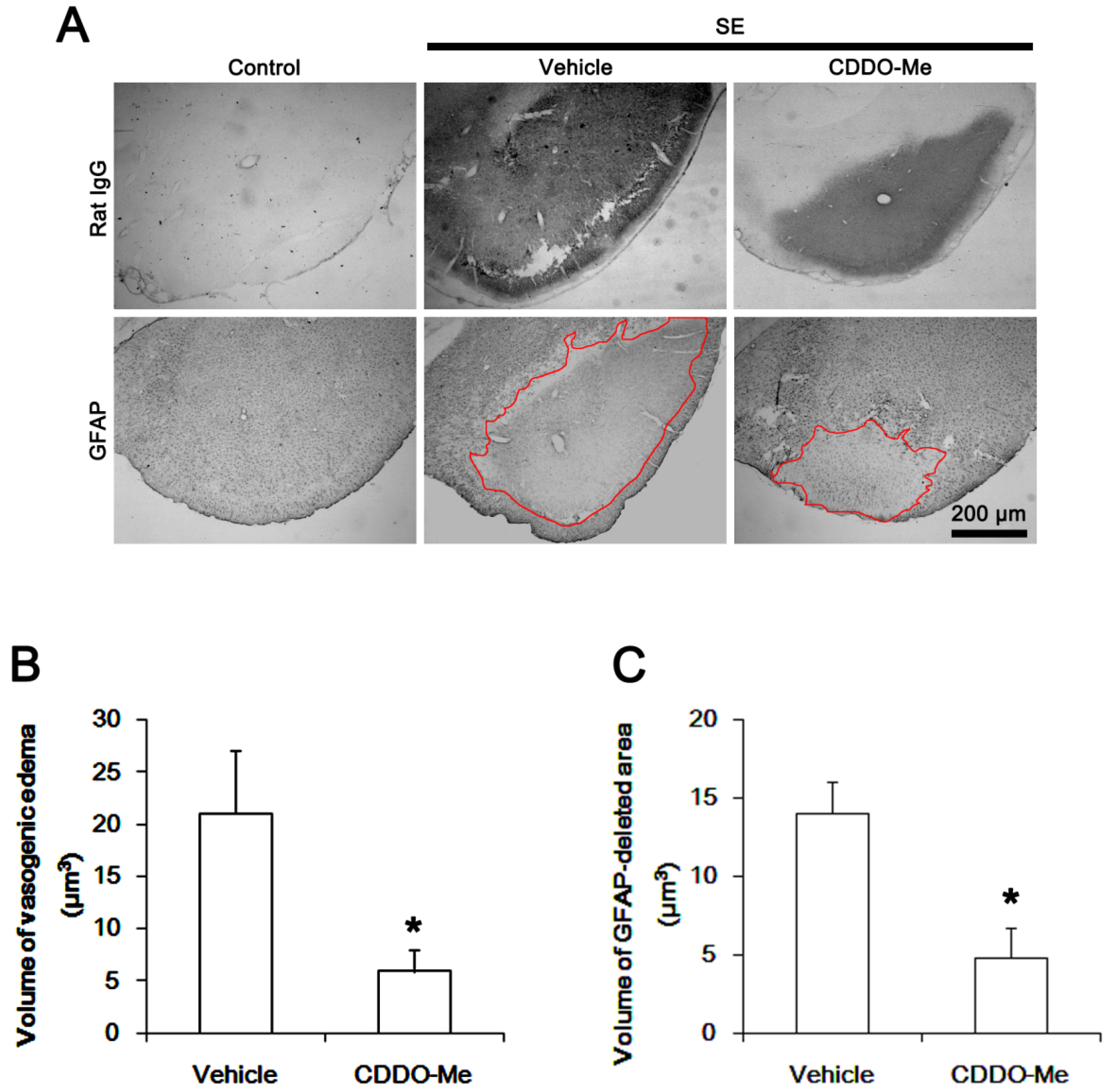
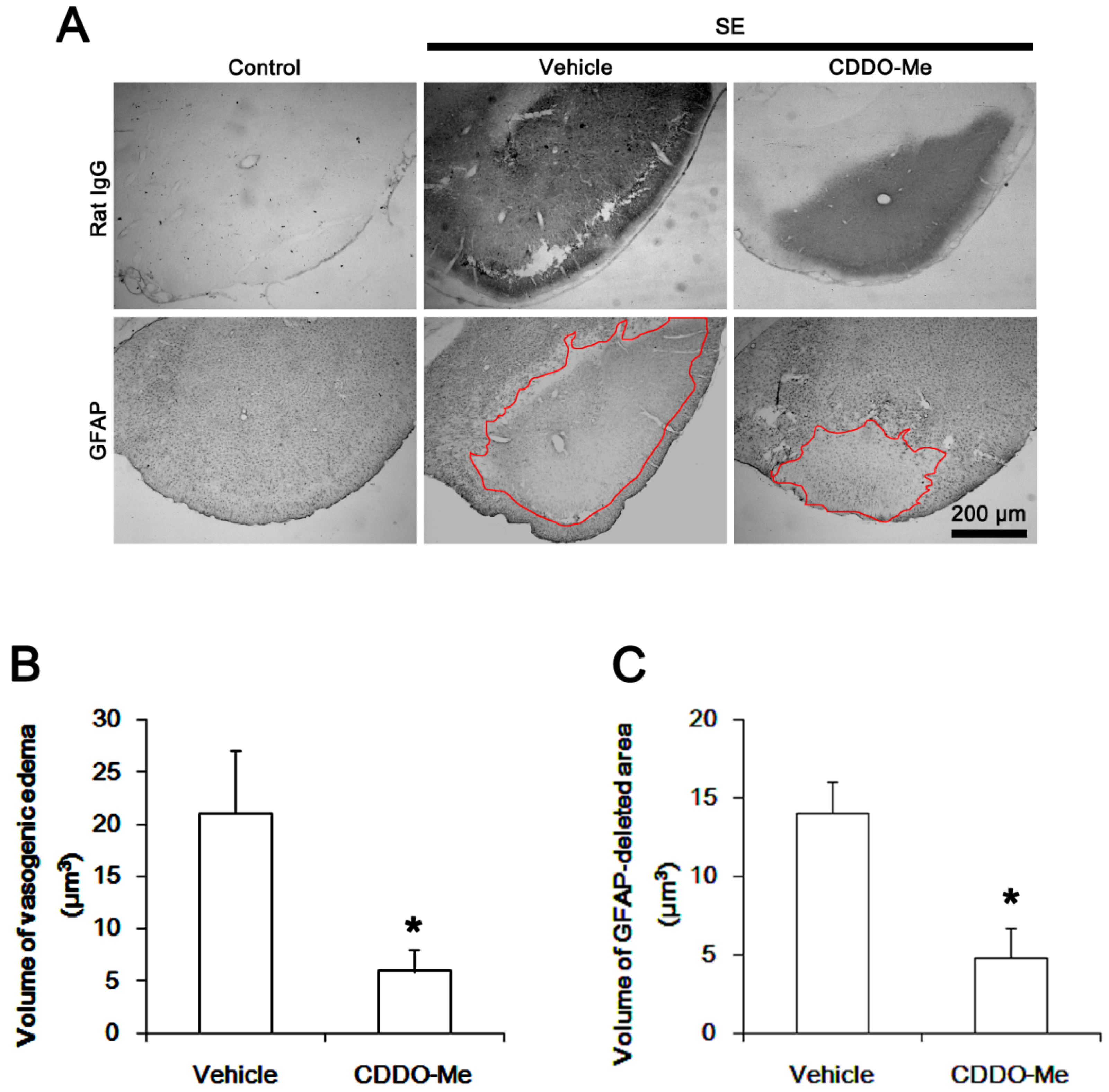
2.1. CDDO-Me Effectively Attenuates SE-Induced Vasogenic Edema in the PC
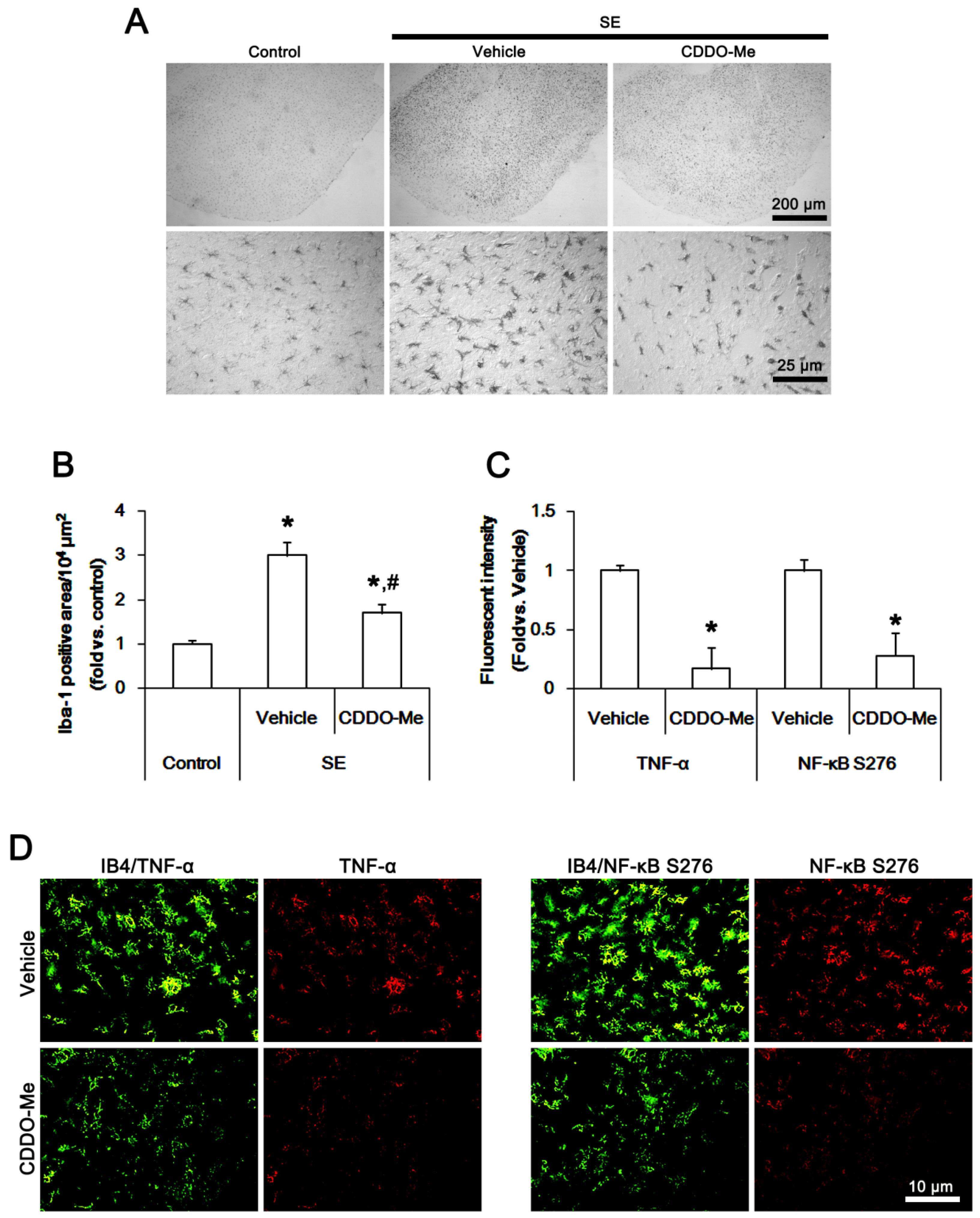
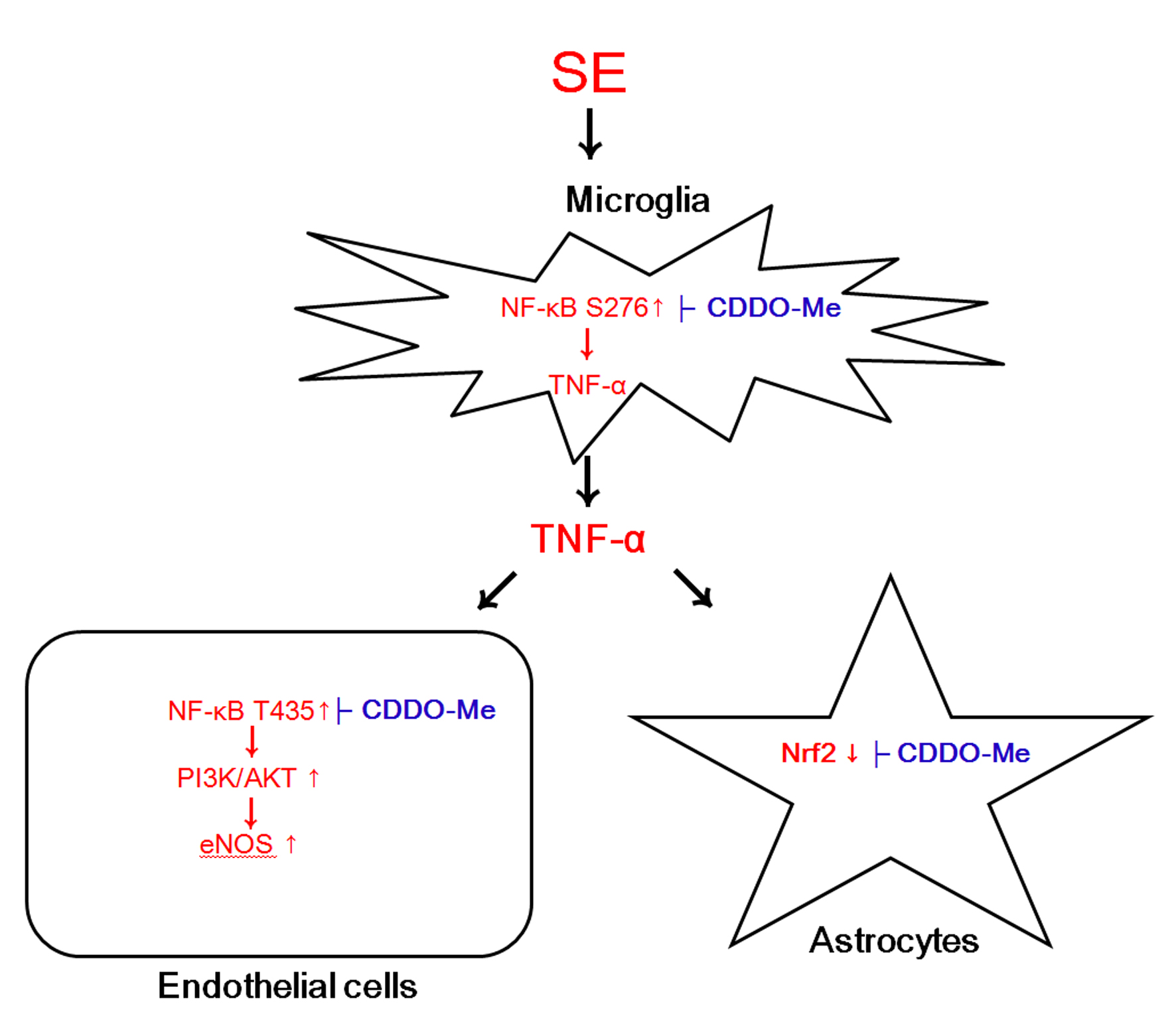
2.2. CDDO-Me Inhibits Microglial Activation and TNF-α Synthesis by Abrogating NF-κB S276 Phosphorylation Following SE
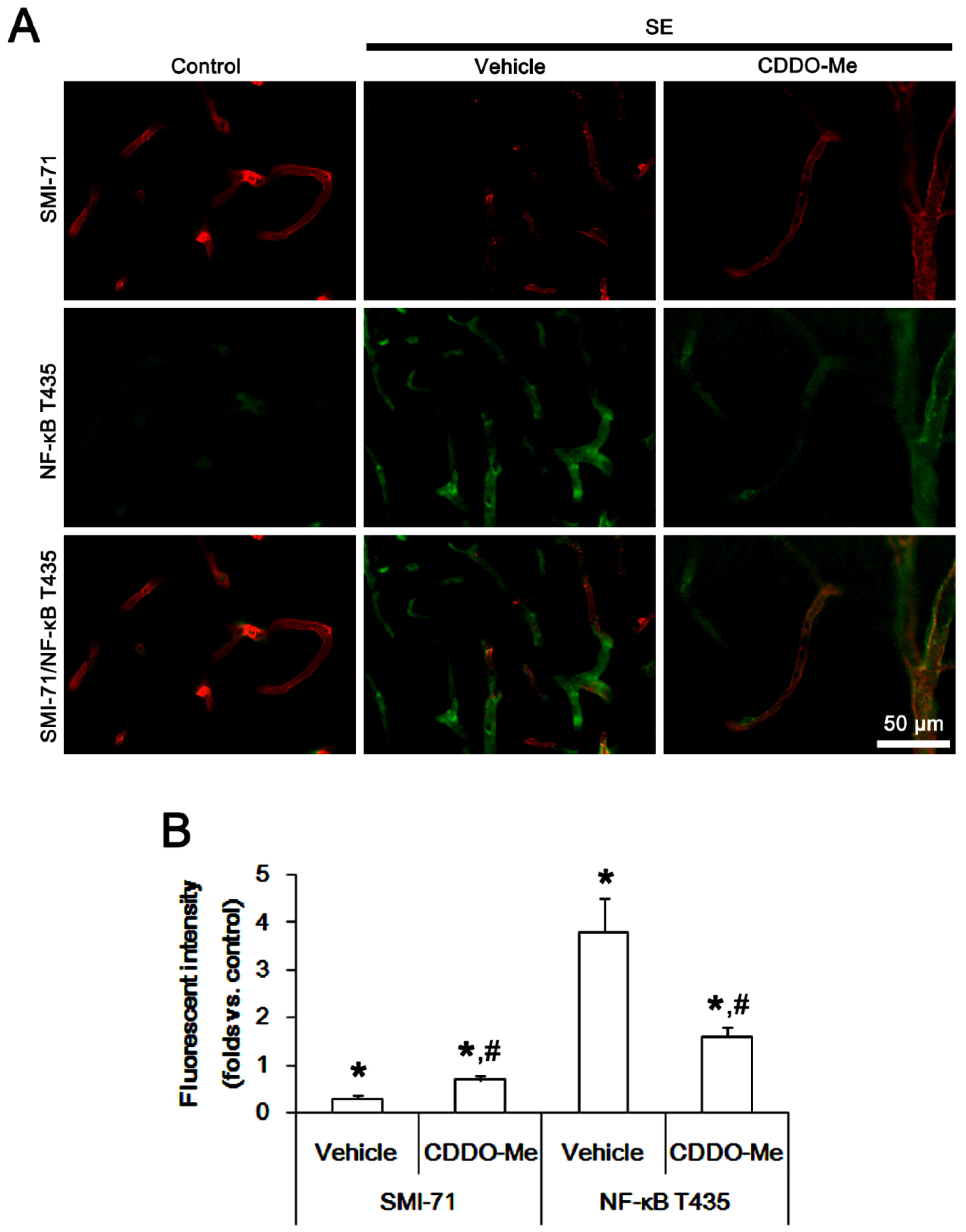
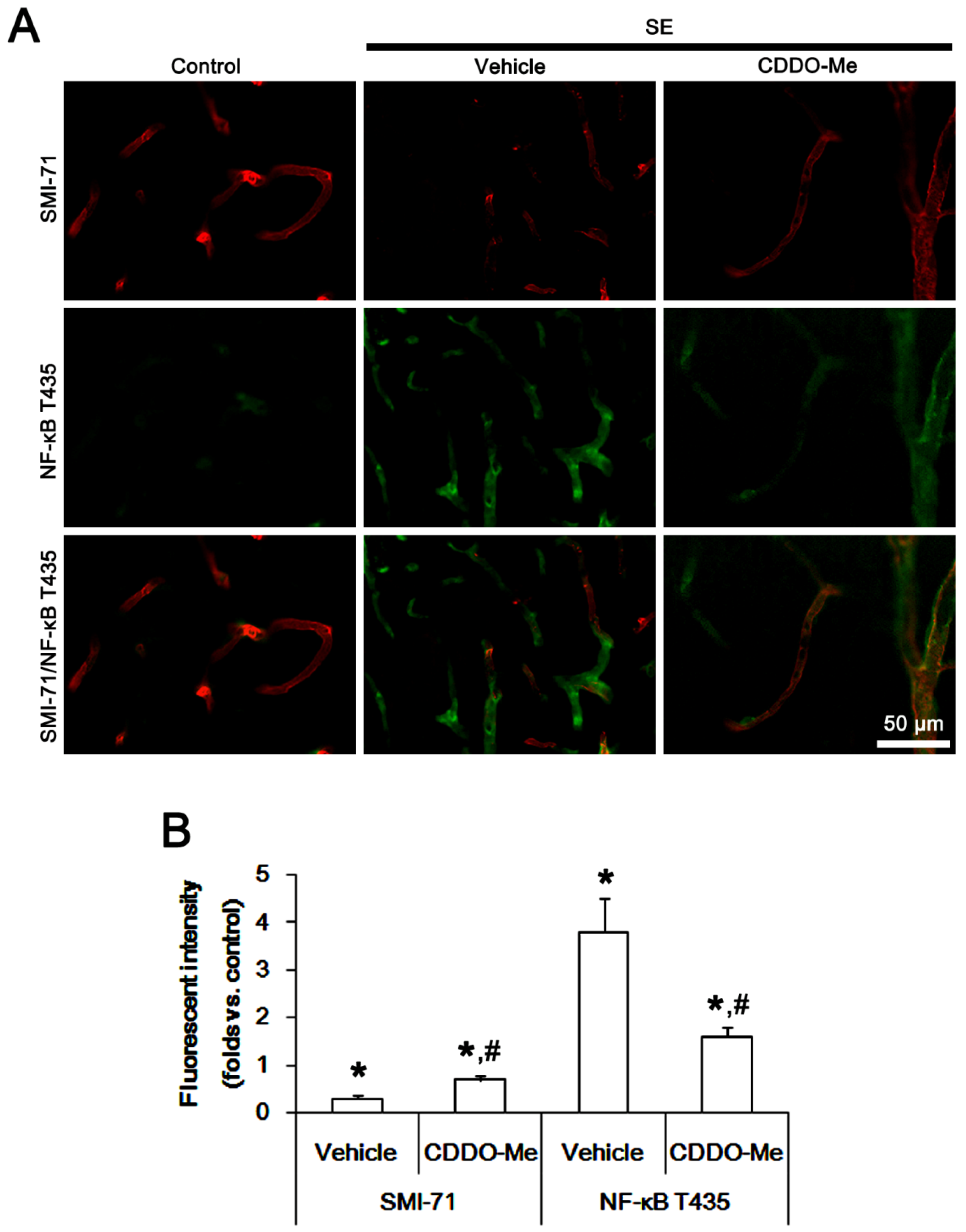
2.3. CDDO-Me Decreases Endothelial NF-κB T435 Phosphorylation Following SE
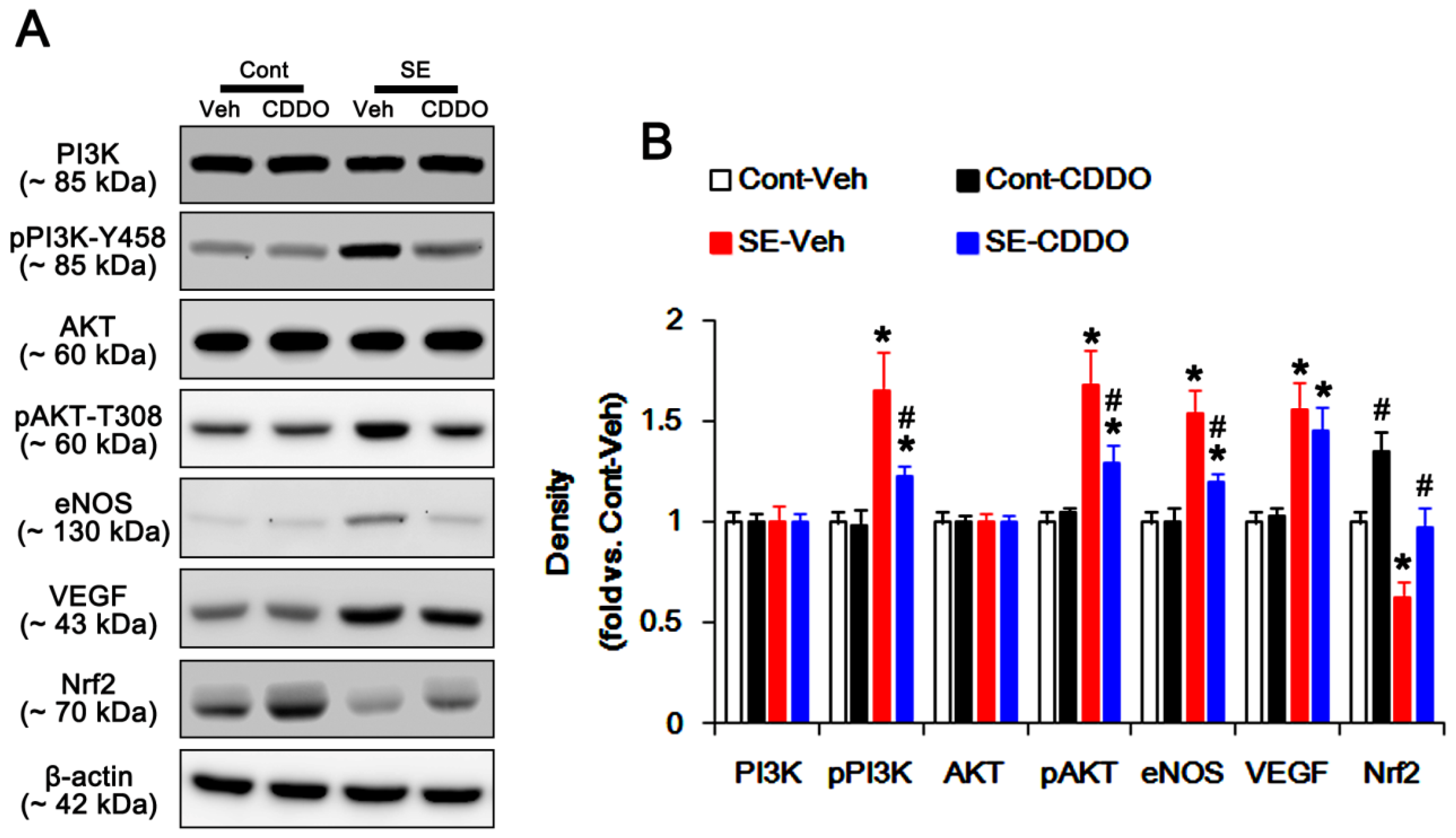
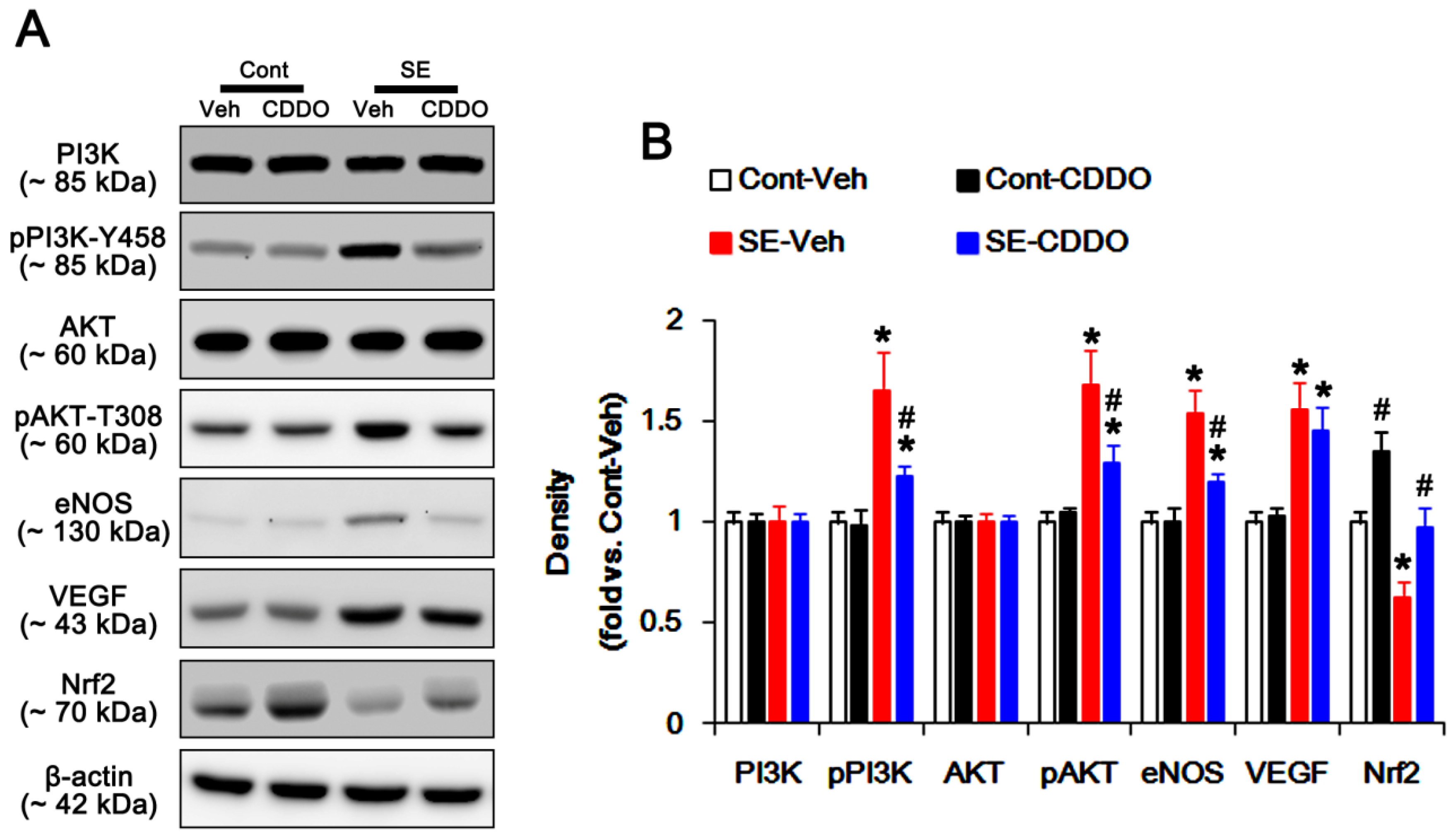
2.4. CDDO-Me Inhibits PI3K/AKT/eNOS Signaling Pathway Following SE
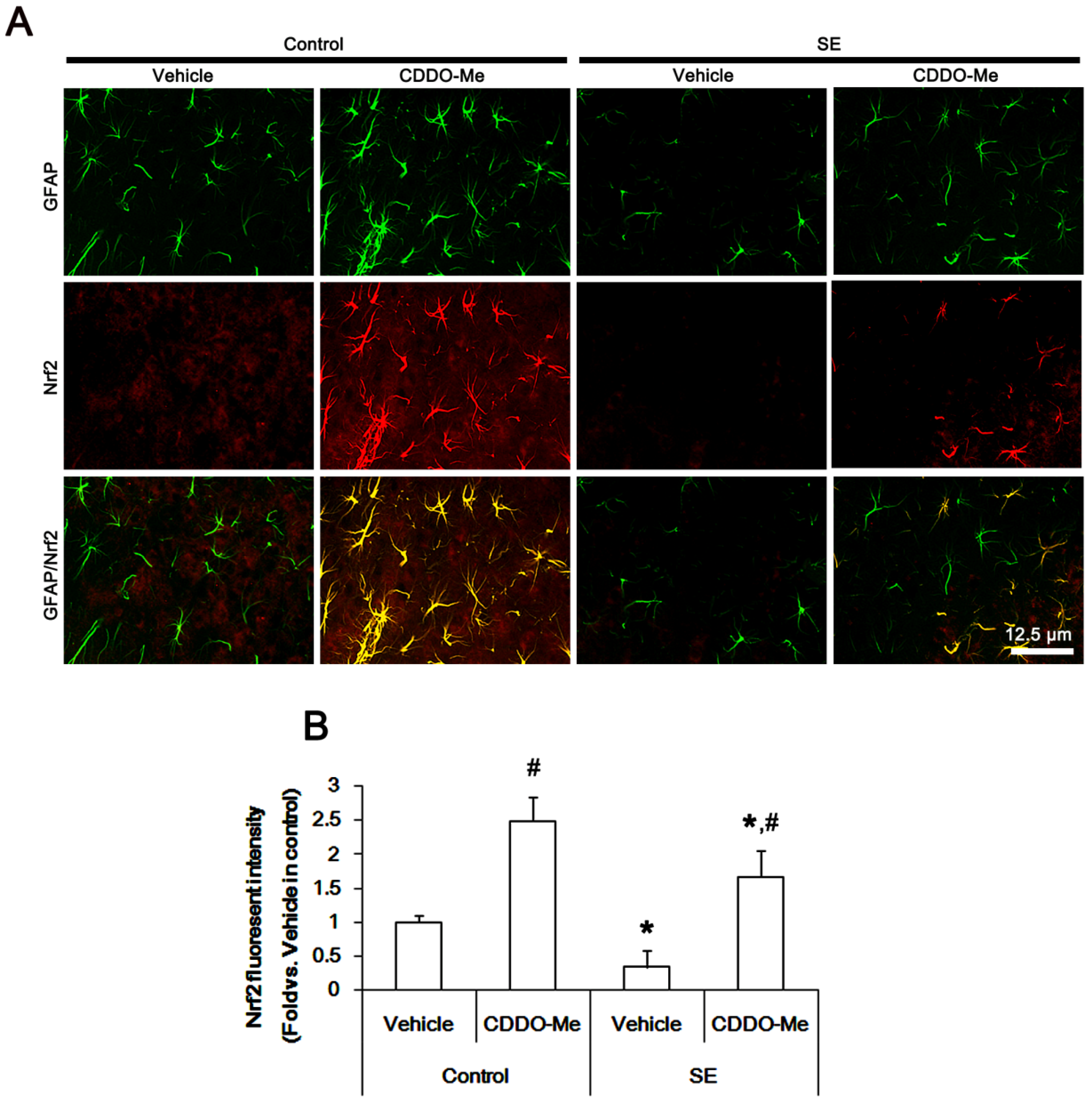
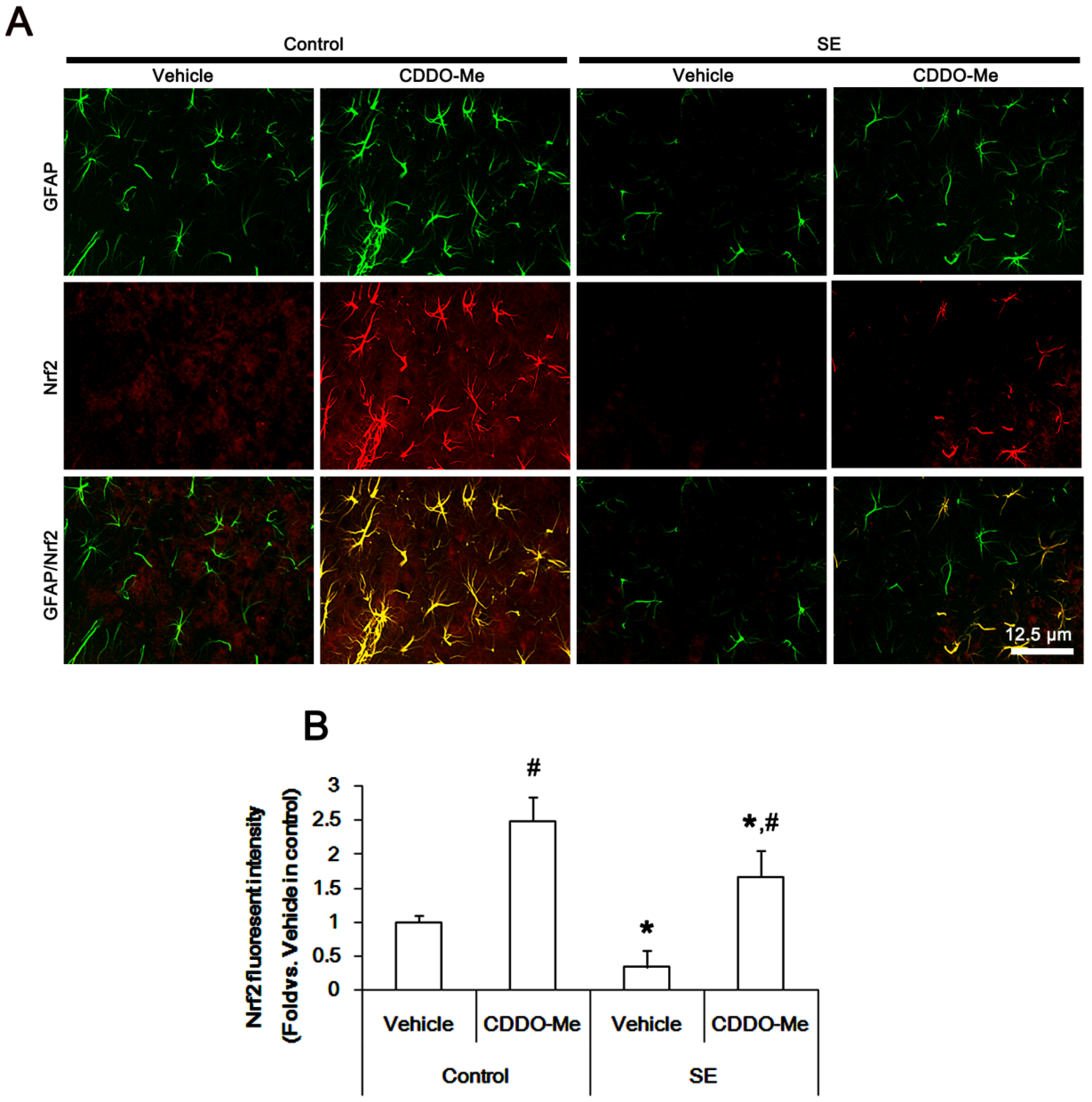
2.5. CDDO-Me Mitigates SE-Induced Astroglial Loss by Enhancing Nrf2 Expression
3. Discussion
4. Materials and Methods
4.1. Experimental Animals and Chemicals
4.2. Surgery and Drug Infusion
4.3. SE Induction
4.4. Tissue Processing
4.5. Immunohistochemistry
4.6. Measurements of Volumes of Vasogenic Edema and GFAP-Deleted Lesion, Iba-1 Positive Area, and Fluorescent Intensities
4.7. Western Blot
4.8. Data Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kim, J.E.; Ryu, H.J.; Choi, S.Y.; Kang, T.C. Tumor necrosis factor-α-mediated threonine 435 phosphorylation of p65 nuclear factor-κB subunit in endothelial cells induces vasogenic edema and neutrophil infiltration in the rat piriform cortex following status epilepticus. J. Neuroinflamm. 2012, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Ko, A.R.; Hyun, H.W.; Kang, T.C. ETB receptor-mediated MMP-9 activation induces vasogenic edema via ZO-1 protein degradation following status epilepticus. Neuroscience 2015, 304, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Lippmann, K.; Kamintsky, L.; Kim, S.Y.; Lublinsky, S.; Prager, O.; Nichtweiss, J.F.; Salar, S.; Kaufer, D.; Heinemann, U.; Friedman, A. Epileptiform activity and spreading depolarization in the blood-brain barrier-disrupted peri-infarct hippocampus are associated with impaired GABAergic inhibition and synaptic plasticity. J. Cereb. Blood Flow Metab. 2017, 37, 1803–1819. [Google Scholar] [CrossRef] [PubMed]
- Ralay Ranaivo, H.; Wainwright, M.S. Albumin activates astrocytes and microglia through mitogen-activated protein kinase pathways. Brain Res. 2010, 1313, 222–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.L.; Zhou, Y.; Tian, W.D.; Li, H.J.; Li, K.C.; Miao, X.; An, G.Z.; Wang, X.W.; Guo, G.Z.; Ding, G.R. Electromagnetic pulse activated brain microglia via the p38 MAPK pathway. Neurotoxicology 2016, 52, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Vinet, J.; Vainchtein, I.D.; Spano, C.; Giordano, C.; Bordini, D.; Curia, G.; Dominici, M.; Boddeke, H.W.; Eggen, B.J.; Biagini, G. Microglia are less pro-inflammatory than myeloid infiltrates in the hippocampus of mice exposed to status epilepticus. Glia 2016, 64, 1350–1362. [Google Scholar] [CrossRef]
- Varvel, N.H.; Neher, J.J.; Bosch, A.; Wang, W.; Ransohoff, R.M.; Miller, R.J.; Dingledine, R. Infiltrating monocytes promote brain inflammation and exacerbate neuronal damage after status epilepticus. Proc. Natl. Acad. Sci. USA 2016, 113, E5665–E5674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigau, V.; Morin, M.; Rousset, M.C.; de Bock, F.; Lebrun, A.; Coubes, P.; Picot, M.C.; Baldy-Moulinier, M.; Bockaert, J.; Crespel, A.; et al. Angiogenesis is associated with blood-brain barrier permeability in temporal lobe epilepsy. Brain 2007, 130, 1942–1956. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J.; Kim, J.E.; Choi, H.C.; Song, H.K.; Kang, T.C. Cellular and regional specific changes in multidrug efflux transporter expression during recovery of vasogenic edema in the rat hippocampus and piriform cortex. BMB Rep. 2015, 48, 348–353. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Kang, T.C. TRPC3- and ET(B) receptor-mediated PI3K/AKT activation induces vasogenic edema formation following status epilepticus. Brain Res. 2017, 1672, 58–64. [Google Scholar] [CrossRef]
- Jo, S.M.; Ryu, H.J.; Kim, J.E.; Yeo, S.I.; Kim, M.J.; Choi, H.C.; Song, H.K.; Kang, T.C. Up-regulation of endothelial endothelin-1 expression prior to vasogenic edema formation in the rat piriform cortex following status epilepticus. Neurosci. Lett. 2011, 501, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Ryu, H.J.; Kang, T.C. Status epilepticus induces vasogenic edema via tumor necrosis factor-α/endothelin-1-mediated two different pathways. PLoS ONE 2013, 8, e74458. [Google Scholar] [CrossRef] [PubMed]
- Min, S.J.; Kang, T.C. Positive feedback role of TRPC3 in TNF-α-mediated vasogenic edema formation induced by status epilepticus independent of ET(B) receptor activation. Neuroscience 2016, 337, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Honda, T.; Rounds, B.V.; Gribble, G.W.; Suh, N.; Wang, Y.; Sporn, M.B. Design and synthesis of 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid, a novel and highly active inhibitor of nitric oxide production in mouse macrophages. Bioorg. Med. Chem. Lett. 1998, 8, 2711–2714. [Google Scholar] [CrossRef]
- Tran, T.A.; McCoy, M.K.; Sporn, M.B.; Tansey, M.G. The synthetic triterpenoid CDDO-methyl ester modulates microglial activities, inhibits TNF production, and provides dopaminergic neuroprotection. J. Neuroinflamm. 2008, 5, 14. [Google Scholar] [CrossRef]
- Ahmad, R.; Raina, D.; Meyer, C.; Kharbanda, S.; Kufe, D. Triterpenoid CDDO-Me blocks the NF-kappaB pathway by direct inhibition of IKKbeta on Cys-179. J. Biol. Chem. 2006, 281, 35764–35769. [Google Scholar] [CrossRef]
- Yore, M.M.; Liby, K.T.; Honda, T.; Gribble, G.W.; Sporn, M.B. The synthetic triterpenoid 1-[2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole blocks nuclear factor-kappaB activation through direct inhibition of IkappaB kinase beta. Mol. Cancer Ther. 2006, 5, 3232–3239. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Liby, K.T.; Stephenson, K.K.; Holtzclaw, W.D.; Gao, X.; Suh, N.; Williams, C.; Risingsong, R.; Honda, T.; Gribble, G.W.; et al. Extremely potent triterpenoid inducers of the phase 2 response: Correlations of protection against oxidant and inflammatory stress. Proc. Natl. Acad. Sci. USA 2005, 102, 4584–4589. [Google Scholar] [CrossRef] [Green Version]
- Asehnoune, K.; Strassheim, D.; Mitra, S.; Kim, J.Y.; Abraham, E. Involvement of reactive oxygen species in Toll-like receptor 4-dependent activation of NF-kappa B. J. Immunol. 2004, 172, 2522–2529. [Google Scholar] [CrossRef]
- Thimmulappa, R.K.; Fuchs, R.J.; Malhotra, D.; Scollick, C.; Traore, K.; Bream, J.H.; Trush, M.A.; Liby, K.T.; Sporn, M.B.; Kensler, T.W.; et al. Preclinical evaluation of targeting the Nrf2 pathway by triterpenoids (CDDO-Im and CDDO-Me) for protection from LPS-induced inflammatory response and reactive oxygen species in human peripheral blood mononuclear cells and neutrophils. Antioxid. Redox Signal. 2007, 9, 1963–1970. [Google Scholar] [CrossRef]
- Imai, T.; Takagi, T.; Kitashoji, A.; Yamauchi, K.; Shimazawa, M.; Hara, H. Nrf2 activator ameliorates hemorrhagic transformation in focal cerebral ischemia under warfarin anticoagulation. Neurobiol. Dis. 2016, 89, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Yeo, S.I.; Ryu, H.J.; Kim, M.J.; Kim, D.S.; Jo, S.M.; Kang, T.C. Astroglial loss and edema formation in the rat piriform cortex and hippocampus following pilocarpine-induced status epilepticus. J. Comp. Neurol. 2010, 518, 4612–4628. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Kim, J.E.; Kim, Y.J.; Kim, M.J.; Kang, T.C. Hyperforin attenuates microglia activation and inhibits p65-Ser276 NFκB phosphorylation in the rat piriform cortex following status epilepticus. Neurosci. Res. 2014, 85, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Park, H.; Choi, S.H.; Kong, M.J.; Kang, T.C. CDDO-Me Selectively Attenuates CA1 Neuronal Death Induced by Status Epilepticus via Facilitating Mitochondrial Fission Independent of LONP1. Cells 2019, 8, 833. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.K.; Ryu, H.J.; Kim, J.E.; Jo, S.M.; Choi, H.C.; Song, H.K.; Kang, T.C. The roles of P2X7 receptor in regional-specific microglial responses in the rat brain following status epilepticus. Neurol. Sci. 2012, 33, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Park, H.; Choi, S.H.; Kong, M.J.; Kang, T.C. Roscovitine attenuates microglia activation and monocyte Infiltration via p38 MAPK Inhibition in the rat frontoparietal cortex following status epilepticus. Cells 2019, 8, 746. [Google Scholar] [CrossRef]
- Streit, W.J.; Walter, S.A.; Pennell, N.A. Reactive microgliosis. Prog. Neurobiol. 1999, 57, 563–581. [Google Scholar] [CrossRef]
- Okazaki, T.; Sakon, S.; Sasazuki, T.; Sakurai, H.; Doi, T.; Yagita, H.; Okumura, K.; Nakano, H. Phosphorylation of serine 276 is essential for p65 NF-kappaB subunit-dependent cellular responses. Biochem. Biophys. Res. Commun. 2003, 300, 807–812. [Google Scholar] [CrossRef]
- Christian, F.; Smith, E.L.; Carmody, R.J. The Regulation of NF-κB Subunits by Phosphorylation. Cells 2016, 5, 12. [Google Scholar] [CrossRef]
- Furusawa, J.; Funakoshi-Tago, M.; Tago, K.; Mashino, T.; Inoue, H.; Sonoda, Y.; Kasahara, T. Licochalcone A significantly suppresses LPS signaling pathway through the inhibition of NF-κB p65 phosphorylation at serine 276. Cell Signal. 2009, 21, 778–785. [Google Scholar] [CrossRef]
- Schmidt-Kastner, R.; Ingvar, M. Loss of immunoreactivity for glial fibrillary acidic protein (GFAP) in astrocytes as a marker for profound tissue damage in substantia nigra and basal cortical areas after status epilepticus induced by pilocarpine in rat. Glia 1994, 12, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Kastner, R.; Ingvar, M. Laminar damage of neurons and astrocytes in neocortex and hippocampus of rat after long-lasting status epilepticus induced by pilocarpine. Epilepsy Res. 1996, 12, 309–316. [Google Scholar]
- Kim, D.S.; Kim, J.E.; Kwak, S.E.; Choi, K.C.; Kim, D.W.; Kwon, O.S.; Choi, S.Y.; Kang, T.C. Spatiotemporal characteristics of astroglial death in the rat hippocampo-entorhinal complex following pilocarpine-induced status epilepticus. J. Comp. Neurol. 2008, 511, 581–598. [Google Scholar] [CrossRef]
- Takagi, T.; Kitashoji, A.; Iwawaki, T.; Tsuruma, K.; Shimazawa, M.; Yoshimura, S.; Iwama, T.; Hara, H. Temporal activation of Nrf2 in the penumbra and Nrf2 activator-mediated neuroprotection in ischemia-reperfusion injury. Free Radic. Biol. Med. 2014, 72, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Viatour, P.; Merville, M.P.; Bours, V.; Chariot, A. Phosphorylation of NF-kappaB and IkappaB proteins: Implications in cancer and inflammation. Trends Biochem. Sci. 2005, 30, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Kim, Y.J.; Kim, J.Y.; Kang, T.C. PARP1 activation/expression modulates regional-specific neuronal and glial responses to seizure in a hemodynamic-independent manner. Cell Death Dis. 2014, 5, e1362. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Itoh, K.; Takahashi, S.; Sato, H.; Yanagawa, T.; Katoh, Y.; Bannai, S.; Yamamoto, M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J. Biol. Chem. 2000, 275, 16023–16029. [Google Scholar] [CrossRef]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Jiang, L.; Erb, H.; Johnson, J.A.; Murphy, T.H. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J. Neurosci. 2003, 23, 3394–3406. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef]
- Kobayashi, A.; Ohta, T.; Yamamoto, M. Unique function of the Nrf2-Keap1 pathway in the inducible expression of antioxidant and detoxifying enzymes. Methods Enzymol. 2004, 378, 273–286. [Google Scholar]
- Ichikawa, T.; Li, J.; Meyer, C.J.; Janicki, J.S.; Hannink, M.; Cui, T. Dihydro-CDDO-trifluoroethyl amide (dh404), a novel Nrf2 activator, suppresses oxidative stress in cardiomyocytes. PLoS ONE 2009, 4, e8391. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Paonessa, J.D.; Zhang, Y. Mechanism of chemical activation of Nrf2. PLoS ONE 2012, 7, e35122. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.Q.; Wang, Y.; Senitko, M.; Meyer, C.; Wigley, W.C.; Ferguson, D.A.; Grossman, E.; Chen, J.; Zhou, X.J.; Hartono, J.; et al. Bardoxolone methyl (BARD) ameliorates ischemic AKI and increases expression of protective genes Nrf2, PPARγ, and HO-1. Am. J. Physiol. Ren. Physiol. 2011, 300, F1180–F1192. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Grigoryev, D.N.; Crow, M.T.; Haas, M.; Yamamoto, M.; Reddy, S.P.; Rabb, H. Transcription factor Nrf2 is protective during ischemic and nephrotoxic acute kidney injury in mice. Kidney Int. 2009, 76, 277–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Zeeuw, D.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Chin, M.; Christ-Schmidt, H.; Goldsberry, A.; Houser, M.; Krauth, M.; Lambers Heerspink, H.J.; et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N. Engl. J. Med. 2013, 369, 2492–2503. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Host | Manufacturer (Catalog Number) | Dilution Used |
|---|---|---|---|
| AKT | Rabbit | Cell signaling (#9272) | 1:1000 (WB) |
| eNOS | Rabbit | Abcam (#ab66127) | 1:1000 (WB) |
| GFAP | Mouse | Millipore (#MAB3402) | 1:5000 (IH) |
| IB4 | Vector (B-1205) | 1:200 (IH) | |
| Iba-1 | Rabbit | Biocare Medical (CP 290) | 1:500 (IH) |
| NFκB S276 | Rabbit | Abcam (ab106129) | 1:100 (IH) |
| NFκB T435 | Rabbit | Abcam (ab31472) | 1:100 (IH) |
| Nrf2 | Rabbit | Abcam (ab137550) | 1:1000 (WB), 1:100 (IH) |
| pAKT-T308 | Rabbit | Cell signaling (#9275) | 1:1000 (WB) |
| pPI3K-Y458 | Rabbit | Cell signaling (#4228S) | 1:1000 (WB) |
| PI3K | Rabbit | Cell signaling (#4292S) | 1:1000 (WB) |
| Rat IgG | Goat | Vector (#PI-9400) | 1:200 (IH) |
| SMI-71 | Mouse | Covance (#SMI-71R) | 1:1000 (IH) |
| TNF-α | Goat | R&D systems (AF-510-NA) | 1:1000 (IH) |
| VEGF | Rabbit | Abcam (#ab46154) | 1:1000 (WB) |
| β-actin | Mouse | Sigma (#A5316) | 1:5000 (WB) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.-J.; Park, H.; Choi, S.-H.; Kong, M.-J.; Kim, J.-E.; Kang, T.-C. CDDO-Me Attenuates Vasogenic Edema and Astroglial Death by Regulating NF-κB p65 Phosphorylations and Nrf2 Expression Following Status Epilepticus. Int. J. Mol. Sci. 2019, 20, 4862. https://doi.org/10.3390/ijms20194862
Kim M-J, Park H, Choi S-H, Kong M-J, Kim J-E, Kang T-C. CDDO-Me Attenuates Vasogenic Edema and Astroglial Death by Regulating NF-κB p65 Phosphorylations and Nrf2 Expression Following Status Epilepticus. International Journal of Molecular Sciences. 2019; 20(19):4862. https://doi.org/10.3390/ijms20194862
Chicago/Turabian StyleKim, Min-Ju, Hana Park, Seo-Hyeon Choi, Min-Jeong Kong, Ji-Eun Kim, and Tae-Cheon Kang. 2019. "CDDO-Me Attenuates Vasogenic Edema and Astroglial Death by Regulating NF-κB p65 Phosphorylations and Nrf2 Expression Following Status Epilepticus" International Journal of Molecular Sciences 20, no. 19: 4862. https://doi.org/10.3390/ijms20194862
APA StyleKim, M.-J., Park, H., Choi, S.-H., Kong, M.-J., Kim, J.-E., & Kang, T.-C. (2019). CDDO-Me Attenuates Vasogenic Edema and Astroglial Death by Regulating NF-κB p65 Phosphorylations and Nrf2 Expression Following Status Epilepticus. International Journal of Molecular Sciences, 20(19), 4862. https://doi.org/10.3390/ijms20194862