More Than an Adipokine: The Complex Roles of Chemerin Signaling in Cancer


Abstract
1. Obesity and Cancer
2. Role of Adipokines
3. Chemerin
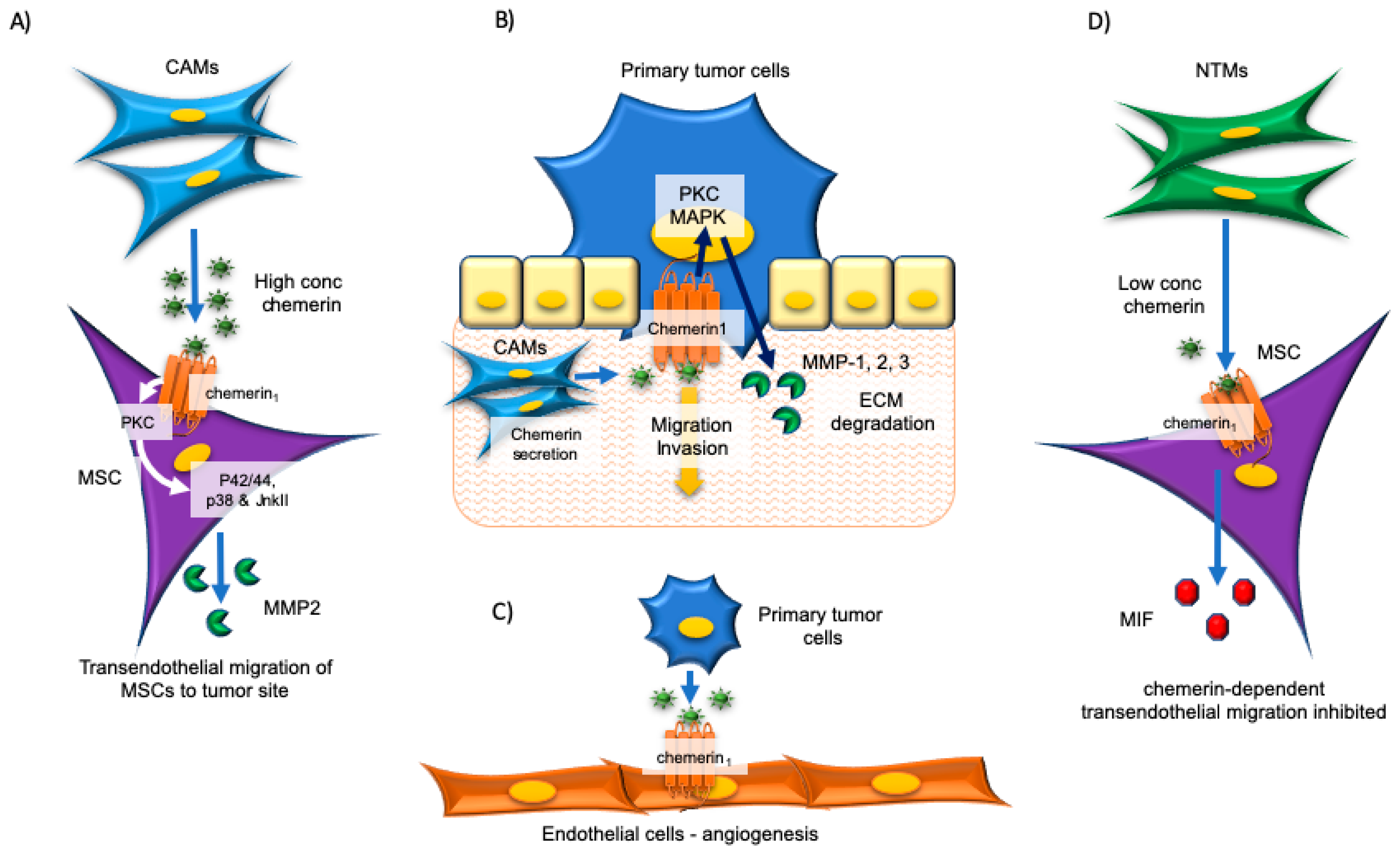
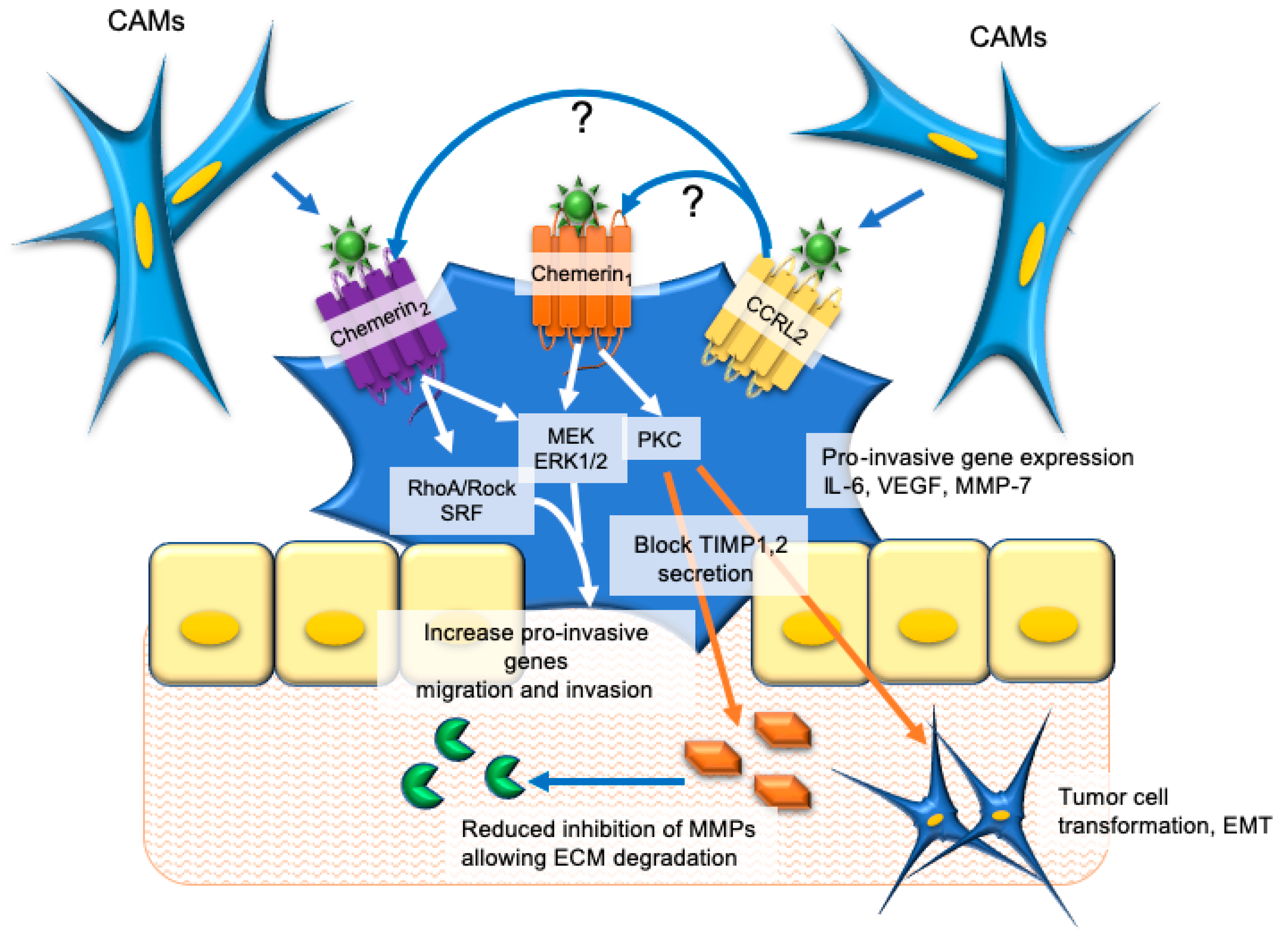
4. Esophageal and Oral Cancers
5. Colorectal and Gastric Cancer
6. Skin Cancer
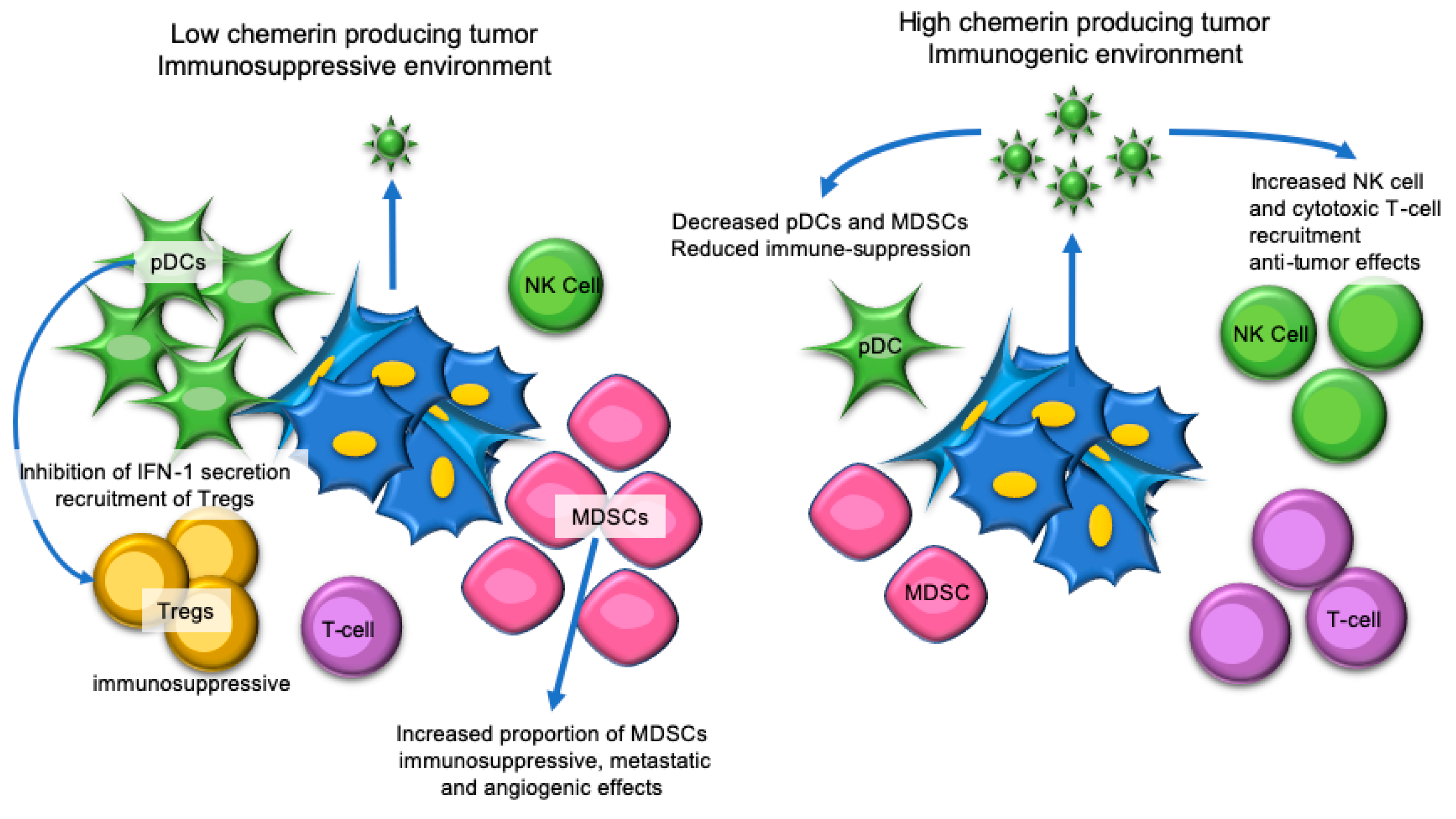
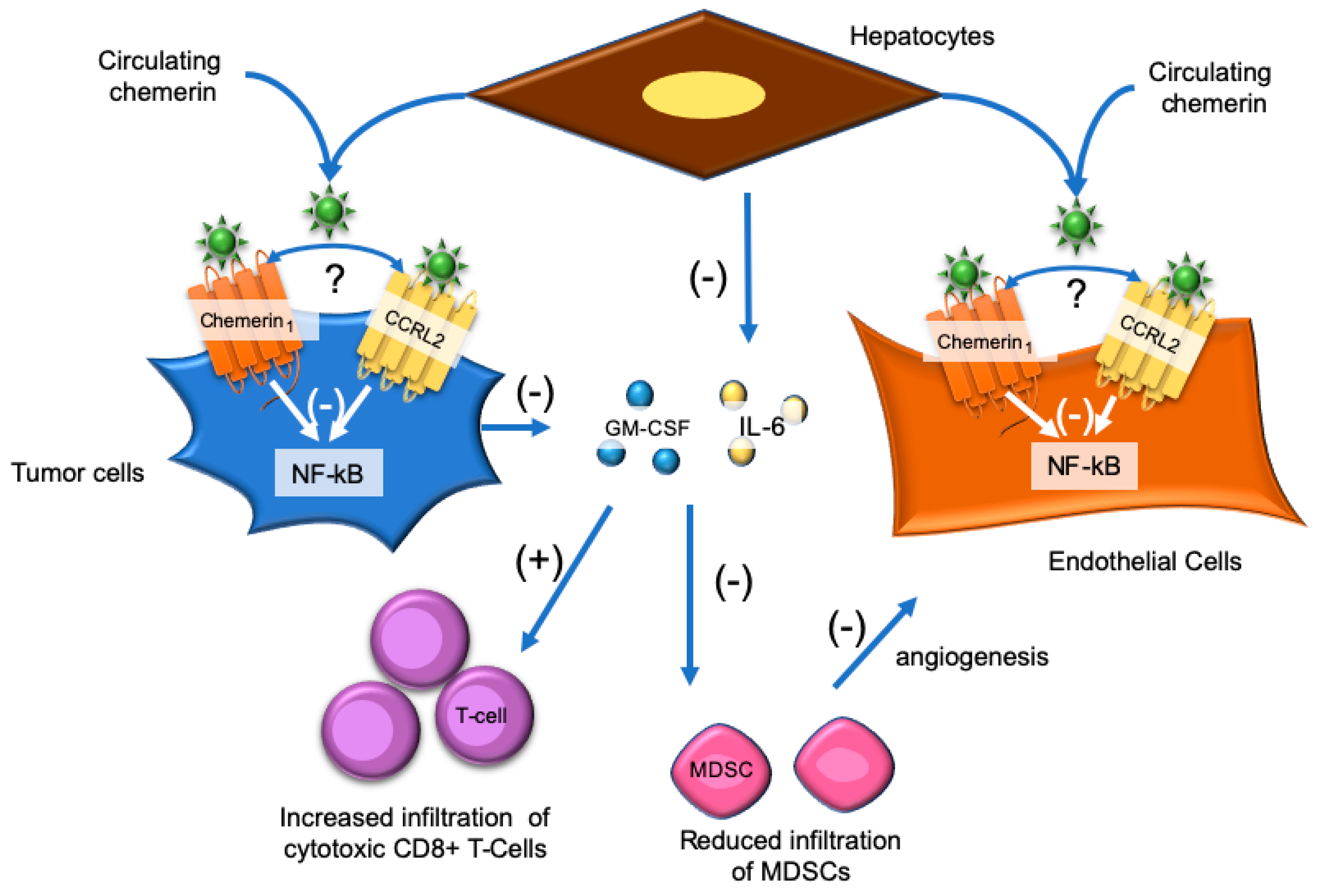
7. Hepatocellular Carcinoma
8. Adrenocortical Carcinoma
9. Renal Carcinoma
10. Thyroid Cancer
11. Breast Cancer
12. Ovarian Cancer
13. Central Nervous System Cancers
14. Lung Cancer
15. Pancreatic Cancer
16. Prostate Cancer
17. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AKT | protein kinase B |
| APC | APC regulator of Wnt signaling pathway |
| ATM | adjacent tissue myofibroblast |
| BCSC | breast cancer stem cell |
| BE | Barrett’s esophagus |
| BPH | benign prostatic hyperplasia |
| CA125 | cancer antigen 125 |
| CA 15-3 | cancer antigen 15-3 |
| CAM | cancer associated myofibroblast |
| CCRL2 | C-C Chemokine Receptor-Like 2 |
| CEA | carcinoembryonic antigen |
| chemerin1 | chemerin receptor 1 |
| chemerin2 | chemerin receptor 2 |
| CK-1 | casein kinase 1 |
| CMKLR1 | Chemokine-Like Receptor 1 |
| CRP | C-reactive peptide |
| CYFRA 21-1 | cytokeratin 19 fragment 21-1 |
| ECM | extracellular matrix |
| EMT | epithelial-to-mesenchymal transformation |
| ERK | extracellular-related kinase |
| GBM | malignant glioblastoma |
| GEPIA | Gene Expression Profiling Interactive Analysis |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| GPR1 | G Protein-coupled Receptor 1 |
| GSK3β | glycogen synthase kinase 3β |
| GTEx | Genotype-Tissue Expression |
| HCC | hepatocellular carcinoma |
| HCS | high chemerin-secreting |
| hGC | human granulosa cells |
| IFN-I | type 1 interferon |
| IFNγ | interferon gamma |
| IFNγT | interferon γ-secreting T cells |
| IL-6 | interleukin-6 |
| IL-8 | interleukin-8 |
| JNK | c-jUN N-terminal kinase |
| KGN | human ovarian granulosa-like tumor |
| LCS | low chemerin-secreting |
| MAPK | mitogen-activated protein kinase |
| MD | moderately differentiated prostate cancer (Gleason 7) |
| mDC | myeloid dendritic cell |
| MDSC | myeloid-derived suppressor cells |
| MIF | macrophage inhibitory factor |
| MIP3α | macrophage inflammatory protein-3 alpha |
| MMP | matrix metalloproteinase |
| MSC | mesenchymal stromal cell |
| NASH | non-alcoholic steatohepatitis |
| NASH-HCC | non-alcoholic steatohepatitis with dimethylnitrosamine-induced hepatocarcinoma |
| NC CNS | non-cancer CNS disease |
| NF-κB | nuclear factor kappa B |
| NK | natural killer |
| NSCLC | non-small cell lung cancer |
| NTM | normal tissue myofibroblast |
| ODC | oligodendrocytoma |
| OPL | oral pre-malignant lesion |
| OSC | oesophageal squamous cancer |
| OSCC | oral squamous cell carcinoma |
| pRCC | papillary renal cell carcinoma |
| PD | poorly differentiated prostate cancer (Gleason ≥8) |
| pDC | plasmacytoid dendritic cell |
| PI3K | phosphoinositide 3-kinase |
| PKC | protein kinase C |
| PTEN | phosphatase and tensin homolog |
| PVTT-1 | portal vein tumor thrombus cells |
| PVTT-1-Che | chemerin-overexpressing portal vein tumor thrombus cells |
| SCCOT | squamous cell carcinoma of the oral tongue |
| TAM | tumor associated macrophage |
| TCGA | The Cancer Genome Atlas |
| TIMP-1 | tissue inhibitor of metalloproteinase 1 |
| TIMP-2 | tissue inhibitor of metalloproteinase 2 |
| TME | tumor microenvironment |
| TNM | tumor-node-metastasis |
| TPM | transcripts per kilobase million |
| Treg | regulatory T cell |
| VEGF | vascular endothelial growth factor |
| WD | well differentiated prostate cancer (Gleason score ≤ 6) |
References
- Risk, N.C.D. Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 2016, 387, 1377–1396. [Google Scholar] [CrossRef]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Dalamaga, M.; Diakopoulos, K.N.; Mantzoros, C.S. The role of adiponectin in cancer: A review of current evidence. Endocr. Rev. 2012, 33, 547–594. [Google Scholar] [CrossRef] [PubMed]
- Vucenik, I.; Stains, J.P. Obesity and cancer risk: Evidence, mechanisms, and recommendations. Ann. N. Y. Acad. Sci. 2012, 1271, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-mass index and incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Lancet 2008, 371, 569–578. [Google Scholar] [CrossRef]
- Arnold, M.; Pandeya, N.; Byrnes, G.; Renehan, P.A.G.; Stevens, G.A.; Ezzati, P.M.; Ferlay, J.; Miranda, J.J.; Romieu, I.; Dikshit, R.; et al. Global burden of cancer attributable to high body-mass index in 2012: A population-based study. Lancet Oncol. 2015, 16, 36–46. [Google Scholar] [CrossRef]
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K.; International Agency for Research on Cancer Handbook Working Group. Body Fatness and Cancer—Viewpoint of the IARC Working Group. N. Engl. J. Med. 2016, 375, 794–798. [Google Scholar] [CrossRef]
- Steele, C.B.; Thomas, C.C.; Henley, S.J.; Massetti, G.M.; Galuska, D.A.; Agurs-Collins, T.; Puckett, M.; Richardson, L.C. Vital Signs: Trends in Incidence of Cancers Associated with Overweight and Obesity—United States, 2005–2014. MMWR Morb. Mortal. Wkly. Rep. 2017, 66, 1052–1058. [Google Scholar] [CrossRef]
- Ligibel, J.A.; Alfano, C.M.; Courneya, K.S.; Demark-Wahnefried, W.; Burger, R.A.; Chlebowski, R.T.; Fabian, C.J.; Gucalp, A.; Hershman, D.L.; Hudson, M.M.; et al. American Society of Clinical Oncology position statement on obesity and cancer. J. Clin. Oncol. 2014, 32, 3568–3574. [Google Scholar] [CrossRef]
- Choi, J.; Cha, Y.J.; Koo, J.S. Adipocyte biology in breast cancer: From silent bystander to active facilitator. Prog. Lipid Res. 2018, 69, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, M.; Miki, Y.; Takagi, K.; Suzuki, T.; Ishida, T.; Ohuchi, N.; Sasano, H. Interaction with adipocyte stromal cells induces breast cancer malignancy via S100A7 upregulation in breast cancer microenvironment. Breast Cancer Res. 2017, 19, 70. [Google Scholar] [CrossRef] [PubMed]
- Matafome, P.; Santos-Silva, D.; Sena, C.M.; Seica, R. Common mechanisms of dysfunctional adipose tissue and obesity-related cancers. Diabetes Metab. Res. Rev. 2013, 29, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.; Wu, Z.; Choi, C.H.J.; Nguyen, L.; Tegegne, S.; Ackerman, S.E.; Crane, A.; Marchildon, F.; Tessier-Lavigne, M.; Cohen, P. Three-Dimensional Adipose Tissue Imaging Reveals Regional Variation in Beige Fat Biogenesis and PRDM16-Dependent Sympathetic Neurite Density. Cell Metab. 2018, 27, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Lau, W.B.; Ohashi, K.; Wang, Y.; Ogawa, H.; Murohara, T.; Ma, X.L.; Ouchi, N. Role of Adipokines in Cardiovascular Disease. Circ. J. 2017, 81, 920–928. [Google Scholar] [CrossRef]
- Unamuno, X.; Gomez-Ambrosi, J.; Rodriguez, A.; Becerril, S.; Fruhbeck, G.; Catalan, V. Adipokine dysregulation and adipose tissue inflammation in human obesity. Eur. J. Clin. Investig. 2018, 48, e12997. [Google Scholar] [CrossRef] [PubMed]
- Macis, D.; Guerrieri-Gonzaga, A.; Gandini, S. Circulating adiponectin and breast cancer risk: A systematic review and meta-analysis. Int. J. Epidemiol. 2014, 43, 1226–1236. [Google Scholar] [CrossRef]
- Tworoger, S.S.; Eliassen, A.H.; Kelesidis, T.; Colditz, G.A.; Willett, W.C.; Mantzoros, C.S.; Hankinson, S.E. Plasma adiponectin concentrations and risk of incident breast cancer. J. Clin. Endocrinol. Metab. 2007, 92, 1510–1516. [Google Scholar] [CrossRef]
- Pan, H.; Deng, L.L.; Cui, J.Q.; Shi, L.; Yang, Y.C.; Luo, J.H.; Qin, D.; Wang, L. Association between serum leptin levels and breast cancer risk: An updated systematic review and meta-analysis. Medicine (Baltimore) 2018, 97, e11345. [Google Scholar] [CrossRef]
- Ishikawa, M.; Kitayama, J.; Nagawa, H. Enhanced expression of leptin and leptin receptor (OB-R) in human breast cancer. Clin. Cancer Res. 2004, 10, 4325–4331. [Google Scholar] [CrossRef]
- Garofalo, C.; Koda, M.; Cascio, S.; Sulkowska, M.; Kanczuga-Koda, L.; Golaszewska, J.; Russo, A.; Sulkowski, S.; Surmacz, E. Increased expression of leptin and the leptin receptor as a marker of breast cancer progression: Possible role of obesity-related stimuli. Clin. Cancer Res. 2006, 12, 1447–1453. [Google Scholar] [CrossRef] [PubMed]
- Jarde, T.; Caldefie-Chezet, F.; Damez, M.; Mishellany, F.; Penault-Llorca, F.; Guillot, J.; Vasson, M.P. Leptin and leptin receptor involvement in cancer development: A study on human primary breast carcinoma. Oncol. Rep. 2008, 19, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, Y.; Funahashi, T.; Tanaka, S.; Taguchi, T.; Tamaki, Y.; Shimomura, I.; Noguchi, S. High expression of leptin receptor mRNA in breast cancer tissue predicts poor prognosis for patients with high, but not low, serum leptin levels. Int. J. Cancer 2006, 118, 1414–1419. [Google Scholar] [CrossRef] [PubMed]
- Rourke, J.L.; Dranse, H.J.; Sinal, C.J. Towards an integrative approach to understanding the role of chemerin in human health and disease. Obes. Rev. 2013, 14, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Helfer, G.; Wu, Q.F. Chemerin: A multifaceted adipokine involved in metabolic disorders. J. Endocrinol. 2018, 238, R79–R94. [Google Scholar] [CrossRef] [PubMed]
- Toulany, J.; Parlee, S.D.; Sinal, C.J.; Slayter, K.; McNeil, S.; Goralski, K.B. CMKLR1 activation ex vivo does not increase proportionally to serum total chemerin in obese humans. Endocr. Connect. 2016, 5, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Parlee, S.D.; Wang, Y.; Poirier, P.; Lapointe, M.; Martin, J.; Bastien, M.; Cianflone, K.; Goralski, K.B. Biliopancreatic diversion with duodenal switch modifies plasma chemerin in early and late post-operative periods. Obesity 2015, 23, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Bozaoglu, K.; Bolton, K.; McMillan, J.; Zimmet, P.; Jowett, J.; Collier, G.; Walder, K.; Segal, D. Chemerin is a novel adipokine associated with obesity and metabolic syndrome. Endocrinology 2007, 148, 4687–4694. [Google Scholar] [CrossRef] [PubMed]
- Ernst, M.C.; Haidl, I.D.; Zuniga, L.A.; Dranse, H.J.; Rourke, J.L.; Zabel, B.A.; Butcher, E.C.; Sinal, C.J. Disruption of the chemokine-like receptor-1 (CMKLR1) gene is associated with reduced adiposity and glucose intolerance. Endocrinology 2012, 153, 672–682. [Google Scholar] [CrossRef]
- Ernst, M.C.; Issa, M.; Goralski, K.B.; Sinal, C.J. Chemerin exacerbates glucose intolerance in mouse models of obesity and diabetes. Endocrinology 2010, 151, 1998–2007. [Google Scholar] [CrossRef]
- Parlee, S.D.; Ernst, M.C.; Muruganandan, S.; Sinal, C.J.; Goralski, K.B. Serum chemerin levels vary with time of day and are modified by obesity and tumor necrosis factor-{alpha}. Endocrinology 2010, 151, 2590–2602. [Google Scholar] [CrossRef] [PubMed]
- Ress, C.; Tschoner, A.; Engl, J.; Klaus, A.; Tilg, H.; Ebenbichler, C.F.; Patsch, J.R.; Kaser, S. Effect of bariatric surgery on circulating chemerin levels. Eur. J. Clin. Investig. 2010, 40, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Sell, H.; Divoux, A.; Poitou, C.; Basdevant, A.; Bouillot, J.L.; Bedossa, P.; Tordjman, J.; Eckel, J.; Clement, K. Chemerin correlates with markers for fatty liver in morbidly obese patients and strongly decreases after weight loss induced by bariatric surgery. J. Clin. Endocrinol. Metab. 2010, 95, 2892–2896. [Google Scholar] [CrossRef] [PubMed]
- van Herpen, N.A.; Sell, H.; Eckel, J.; Schrauwen, P.; Mensink, R.P. Prolonged fasting and the effects on biomarkers of inflammation and on adipokines in healthy lean men. Horm. Metab. Res. 2013, 45, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.S.; Eisenberg, D.; Zhao, L.; Adams, C.; Leib, R.; Morser, J.; Leung, L. Chemerin activation in human obesity. Obesity 2016, 24, 1522–1529. [Google Scholar] [CrossRef] [PubMed]
- Banas, M.; Zabieglo, K.; Kasetty, G.; Kapinska-Mrowiecka, M.; Borowczyk, J.; Drukala, J.; Murzyn, K.; Zabel, B.A.; Butcher, E.C.; Schroeder, J.M.; et al. Chemerin is an antimicrobial agent in human epidermis. PLoS ONE 2013, 8, e58709. [Google Scholar] [CrossRef]
- Du, X.Y.; Zabel, B.A.; Myles, T.; Allen, S.J.; Handel, T.M.; Lee, P.P.; Butcher, E.C.; Leung, L.L. Regulation of chemerin bioactivity by plasma carboxypeptidase N, carboxypeptidase B (activated thrombin-activable fibrinolysis inhibitor), and platelets. J. Biol. Chem. 2009, 284, 751–758. [Google Scholar] [CrossRef]
- Eisinger, K.; Bauer, S.; Schaffler, A.; Walter, R.; Neumann, E.; Buechler, C.; Muller-Ladner, U.; Frommer, K.W. Chemerin induces CCL2 and TLR4 in synovial fibroblasts of patients with rheumatoid arthritis and osteoarthritis. Exp. Mol. Pathol. 2012, 92, 90–96. [Google Scholar] [CrossRef]
- Lande, R.; Gafa, V.; Serafini, B.; Giacomini, E.; Visconti, A.; Remoli, M.E.; Severa, M.; Parmentier, M.; Ristori, G.; Salvetti, M.; et al. Plasmacytoid dendritic cells in multiple sclerosis: Intracerebral recruitment and impaired maturation in response to interferon-beta. J. Neuropathol. Exp. Neurol. 2008, 67, 388–401. [Google Scholar] [CrossRef]
- Maheshwari, A.; Kurundkar, A.R.; Shaik, S.S.; Kelly, D.R.; Hartman, Y.; Zhang, W.; Dimmitt, R.; Saeed, S.; Randolph, D.A.; Aprahamian, C.; et al. Epithelial cells in fetal intestine produce chemerin to recruit macrophages. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G1–G10. [Google Scholar] [CrossRef]
- Wittamer, V.; Bondue, B.; Guillabert, A.; Vassart, G.; Parmentier, M.; Communi, D. Neutrophil-mediated maturation of chemerin: A link between innate and adaptive immunity. J. Immunol. 2005, 175, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Wittamer, V.; Franssen, J.D.; Vulcano, M.; Mirjolet, J.F.; Le Poul, E.; Migeotte, I.; Brezillon, S.; Tyldesley, R.; Blanpain, C.; Detheux, M.; et al. Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. J. Exp. Med. 2003, 198, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Du, X.Y.; Zhao, L.; Morser, J.; Leung, L.L. Proteolytic cleavage of chemerin protein is necessary for activation to the active form, Chem157S, which functions as a signaling molecule in glioblastoma. J. Biol. Chem. 2011, 286, 39510–39519. [Google Scholar] [CrossRef] [PubMed]
- Zabel, B.A.; Allen, S.J.; Kulig, P.; Allen, J.A.; Cichy, J.; Handel, T.M.; Butcher, E.C. Chemerin activation by serine proteases of the coagulation, fibrinolytic, and inflammatory cascades. J. Biol. Chem. 2005, 280, 34661–34666. [Google Scholar] [CrossRef] [PubMed]
- Meder, W.; Wendland, M.; Busmann, A.; Kutzleb, C.; Spodsberg, N.; John, H.; Richter, R.; Schleuder, D.; Meyer, M.; Forssmann, W.G. Characterization of human circulating TIG2 as a ligand for the orphan receptor ChemR23. FEBS Lett. 2003, 555, 495–499. [Google Scholar] [CrossRef]
- Zhao, L.; Yamaguchi, Y.; Sharif, S.; Du, X.Y.; Song, J.J.; Lee, D.M.; Recht, L.D.; Robinson, W.H.; Morser, J.; Leung, L.L. Chemerin158K protein is the dominant chemerin isoform in synovial and cerebrospinal fluids but not in plasma. J. Biol. Chem. 2011, 286, 39520–39527. [Google Scholar] [CrossRef]
- Guillabert, A.; Wittamer, V.; Bondue, B.; Godot, V.; Imbault, V.; Parmentier, M.; Communi, D. Role of neutrophil proteinase 3 and mast cell chymase in chemerin proteolytic regulation. J. Leukoc. Biol. 2008, 84, 1530–1538. [Google Scholar] [CrossRef]
- Kennedy, A.J.; Davenport, A.P. International Union of Basic and Clinical Pharmacology CIII: Chemerin Receptors CMKLR1 (Chemerin1) and GPR1 (Chemerin2) Nomenclature, Pharmacology, and Function. Pharmacol. Rev. 2018, 70, 174–196. [Google Scholar] [CrossRef]
- Mazzotti, C.; Gagliostro, V.; Bosisio, D.; Del Prete, A.; Tiberio, L.; Thelen, M.; Sozzani, S. The Atypical Receptor CCRL2 (C-C Chemokine Receptor-Like 2) Does Not Act As a Decoy Receptor in Endothelial Cells. Front. Immunol. 2017, 8, 1233. [Google Scholar] [CrossRef]
- Monnier, J.; Lewen, S.; O’Hara, E.; Huang, K.; Tu, H.; Butcher, E.C.; Zabel, B.A. Expression, regulation, and function of atypical chemerin receptor CCRL2 on endothelial cells. J. Immunol. 2012, 189, 956–967. [Google Scholar] [CrossRef]
- Parolini, S.; Santoro, A.; Marcenaro, E.; Luini, W.; Massardi, L.; Facchetti, F.; Communi, D.; Parmentier, M.; Majorana, A.; Sironi, M.; et al. The role of chemerin in the colocalization of NK and dendritic cell subsets into inflamed tissues. Blood 2007, 109, 3625–3632. [Google Scholar] [CrossRef] [PubMed]
- Pachynski, R.K.; Zabel, B.A.; Kohrt, H.E.; Tejeda, N.M.; Monnier, J.; Swanson, C.D.; Holzer, A.K.; Gentles, A.J.; Sperinde, G.V.; Edalati, A.; et al. The chemoattractant chemerin suppresses melanoma by recruiting natural killer cell antitumor defenses. J. Exp. Med. 2012, 209, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Treeck, O.; Buechler, C.; Ortmann, O. Chemerin and Cancer. Int. J. Mol. Sci. 2019, 20, 3750. [Google Scholar] [CrossRef] [PubMed]
- Ghallab, N.A.; Shaker, O.G. Serum and salivary levels of chemerin and MMP-9 in oral squamous cell carcinoma and oral premalignant lesions. Clin. Oral Investig. 2017, 21, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Wang, Q.J.; Feng, Y.Y.; Shang, W.; Cai, M. Overexpression of chemerin was associated with tumor angiogenesis and poor clinical outcome in squamous cell carcinoma of the oral tongue. Clin. Oral Investig. 2014, 18, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.D.; Kandola, S.; Tiszlavicz, L.; Reisz, Z.; Dockray, G.J.; Varro, A. The role of chemerin and ChemR23 in stimulating the invasion of squamous oesophageal cancer cells. Br. J. Cancer 2016, 114, 1152–1159. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.D.; Holmberg, C.; Kandola, S.; Steele, I.; Hegyi, P.; Tiszlavicz, L.; Jenkins, R.; Beynon, R.J.; Peeney, D.; Giger, O.T.; et al. Increased expression of chemerin in squamous esophageal cancer myofibroblasts and role in recruitment of mesenchymal stromal cells. PLoS ONE 2014, 9, e104877. [Google Scholar] [CrossRef] [PubMed]
- Cabia, B.; Andrade, S.; Carreira, M.C.; Casanueva, F.F.; Crujeiras, A.B. A role for novel adipose tissue-secreted factors in obesity-related carcinogenesis. Obes. Rev. 2016, 17, 361–376. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, S.; Yilmaz, F.M.; Yazici, O.; Yozgat, A.; Sezer, S.; Ozdemir, N.; Uysal, S.; Purnak, T. Inflammation and chemerin in colorectal cancer. Tumor Biol. 2016, 37, 6337–6342. [Google Scholar] [CrossRef] [PubMed]
- Eichelmann, F.; Schulze, M.B.; Wittenbecher, C.; Menzel, J.; Weikert, C.; di Giuseppe, R.; Biemann, R.; Isermann, B.; Fritche, A.; Boeing, H.; et al. Association of Chemerin Plasma Concentration With Risk of Colorectal Cancer. JAMA Netw. Open 2019, 2, e190896. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jin, H.-C.; Zhu, A.-K.; Ying, R.-C.; Wei, W.; Zhang, F.-J. Prognostic significance of plasma chemerin levels in patients with gastric cancer. Peptides 2014, 61, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Alkady, M.M.; Abdel-Messeih, P.L.; Nosseir, N.M. Assessment of serum levels of the adipocytokine chemerin in colorectal cancer patients. J. Med. Biochem. 2018, 37, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-Y.; Lee, M.-K.; Kim, N.-K.; Chu, S.-H.; Lee, D.-C.; Lee, H.-S.; Lee, J.-W.; Jeon, J.Y. Serum chemerin levels are independently associated with quality of life in colorectal cancer survivors: A pilot study. PLoS ONE 2017, 12, e0176929. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wu, W.K.K.; Liu, X.; To, K.-F.; Chen, G.G.; Yu, J.; Ng, E.K.W. Increased serum chemerin level promotes cellular invasiveness in gastric cancer: A clinical and experimental study. Peptides 2014, 51, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Takai, K.; Hanai, T.; Shiraki, M.; Suzuki, Y.; Hayashi, H.; Naiki, T.; Nishigaki, Y.; Tomita, E.; Shimizu, M.; et al. Impact of serum chemerin levels on liver functional reserves and platelet counts in patients with hepatocellular carcinoma. Int. J. Mol. Sci. 2014, 15, 11294–11306. [Google Scholar] [CrossRef]
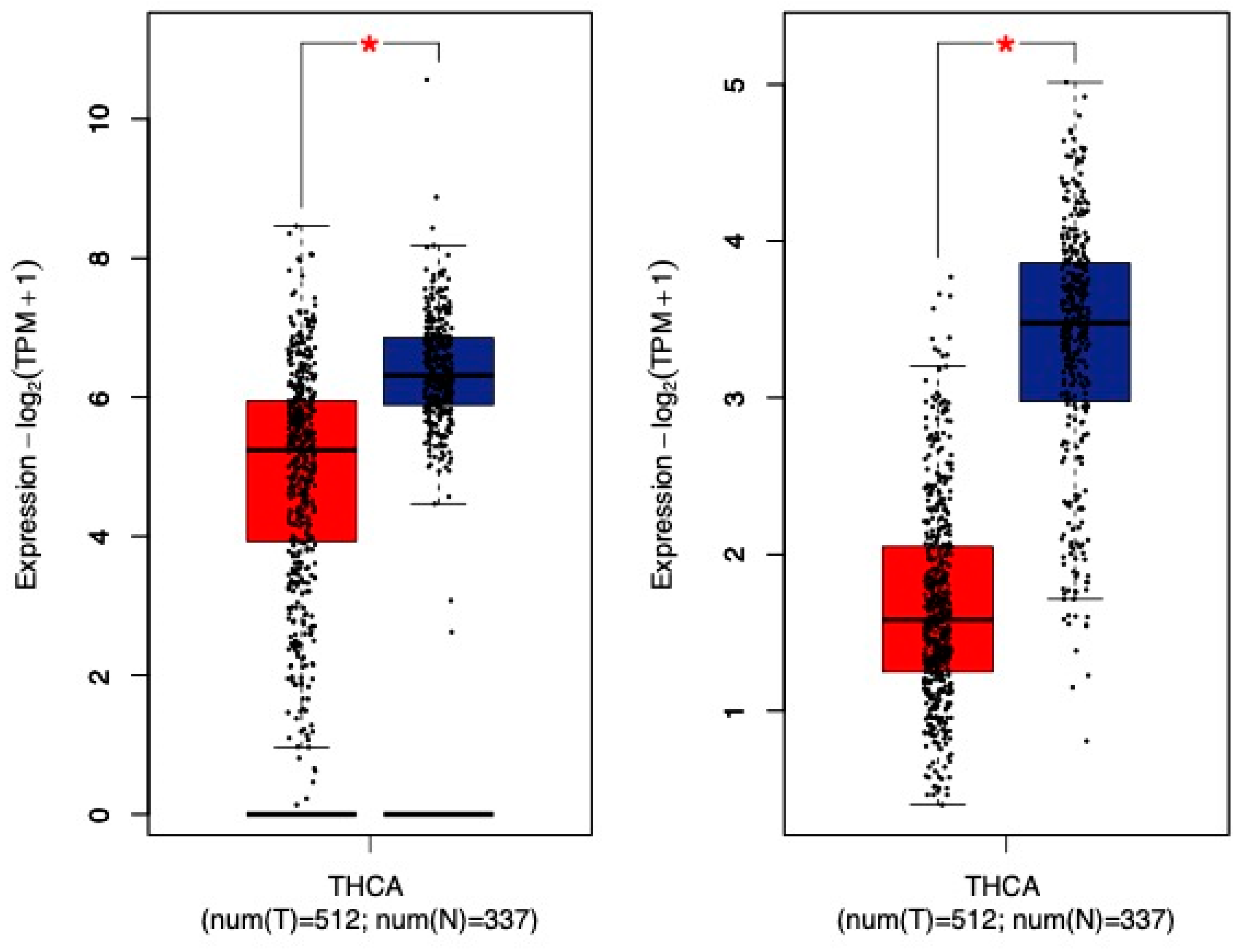
- Warakomski, J.; Romuk, E.; Jarzab, B.; Krajewska, J.; Sieminska, L. Concentrations of Selected Adipokines, Interleukin-6, and Vitamin D in Patients with Papillary Thyroid Carcinoma in Respect to Thyroid Cancer Stages. Int. J. Endocrinol. 2018, 2018, 4921803. [Google Scholar] [CrossRef] [PubMed]
- Akin, S.; Akin, S.; Gedik, E.; Haznedaroglu, E.; Dogan, A.L.; Altundag, M.K. Serum Chemerin Level in Breast Cancer. Int. J. Hematol. Oncol. 2017, 27. [Google Scholar] [CrossRef]
- Sotiropoulos, G.P.; Dalamaga, M.; Antonakos, G.; Marinou, I.; Vogiatzakis, E.; Kotopouli, M.; Karampela, I.; Christodoulatos, G.S.; Lekka, A.; Papavassiliou, A.G. Chemerin as a biomarker at the intersection of inflammation, chemotaxis, coagulation, fibrinolysis and metabolism in resectable non-small cell lung cancer. Lung Cancer 2018, 125, 291–299. [Google Scholar] [CrossRef]
- Xu, C.H.; Yang, Y.; Wang, Y.C.; Yan, J.; Qian, L.H. Prognostic significance of serum chemerin levels in patients with non-small cell lung cancer. Oncotarget 2017, 8, 22483–22489. [Google Scholar] [CrossRef]
- Qu, X.; Han, L.; Wang, S.; Zhang, Q.; Yang, C.; Xu, S.; Zhang, L. Detection of Chemerin and It’s Clinical Significance in Peripheral Blood of Patients with Lung Cancer. Zhongguo Fei Ai Za Zhi 2009, 12, 1174–1177. [Google Scholar] [CrossRef]
- Kiczmer, P.; Szydło, B.e.; Seńkowska, A.P.; Jopek, J.; Wiewióra, M.; Peicuch, J.; Ostrowska, Z.; Świętochowska, E.b. Serum omentin-1 and chemerin concentrations in pancreatic cancer and chronic panreatitis. Folia Med. Crac. 2018, 58, 77–87. [Google Scholar]
- Siemińska, L.; Borowski, A.; Marek, B.; Nowak, M.; Kajdaniuk, D.; Warakomski, J.; Kos-Kudła, B. Serum concentrations of adipokines in men with prostate cancer and benign prostate hyperplasia. Endokrynol. Pol. 2018, 69, 120–127. [Google Scholar] [PubMed]
- Kang, M.; Byun, S.-S.; Lee, S.E.; Hong, S.K. Clinical significance of serum adipokines according to body mass index in patients with clinically localized prostate cancer undergoing radical porstatectomy. World J. Mens Health 2018, 36, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Adya, R.; Tan, B.K.; Chen, J.; Randeva, H.S. Identification of chemerin receptor (ChemR23) in human endothelial cells: Chemerin-induced endothelial angiogenesis. Biochem. Biophys. Res. Commun. 2010, 391, 1762–1768. [Google Scholar] [CrossRef] [PubMed]
- Bozaoglu, K.; Curran, J.E.; Stocker, C.J.; Zaibi, M.S.; Segal, D.; Konstantopoulos, N.; Morrison, S.; Carless, M.; Dyer, T.D.; Cole, S.A.; et al. Chemerin, a novel adipokine in the regulation of angiogenesis. J. Clin. Endocrinol. Metab. 2010, 95, 2476–2485. [Google Scholar] [CrossRef] [PubMed]
- Somja, J.; Demoulin, S.; Roncarati, P.; Herfs, M.; Bletard, N.; Delvenne, P.; Hubert, P. Dendritic cells in Barrett’s esophagus carcinogenesis: An inadequate microenvironment for antitumor immunity? Am. J. Pathol. 2013, 182, 2168–2179. [Google Scholar] [CrossRef] [PubMed]
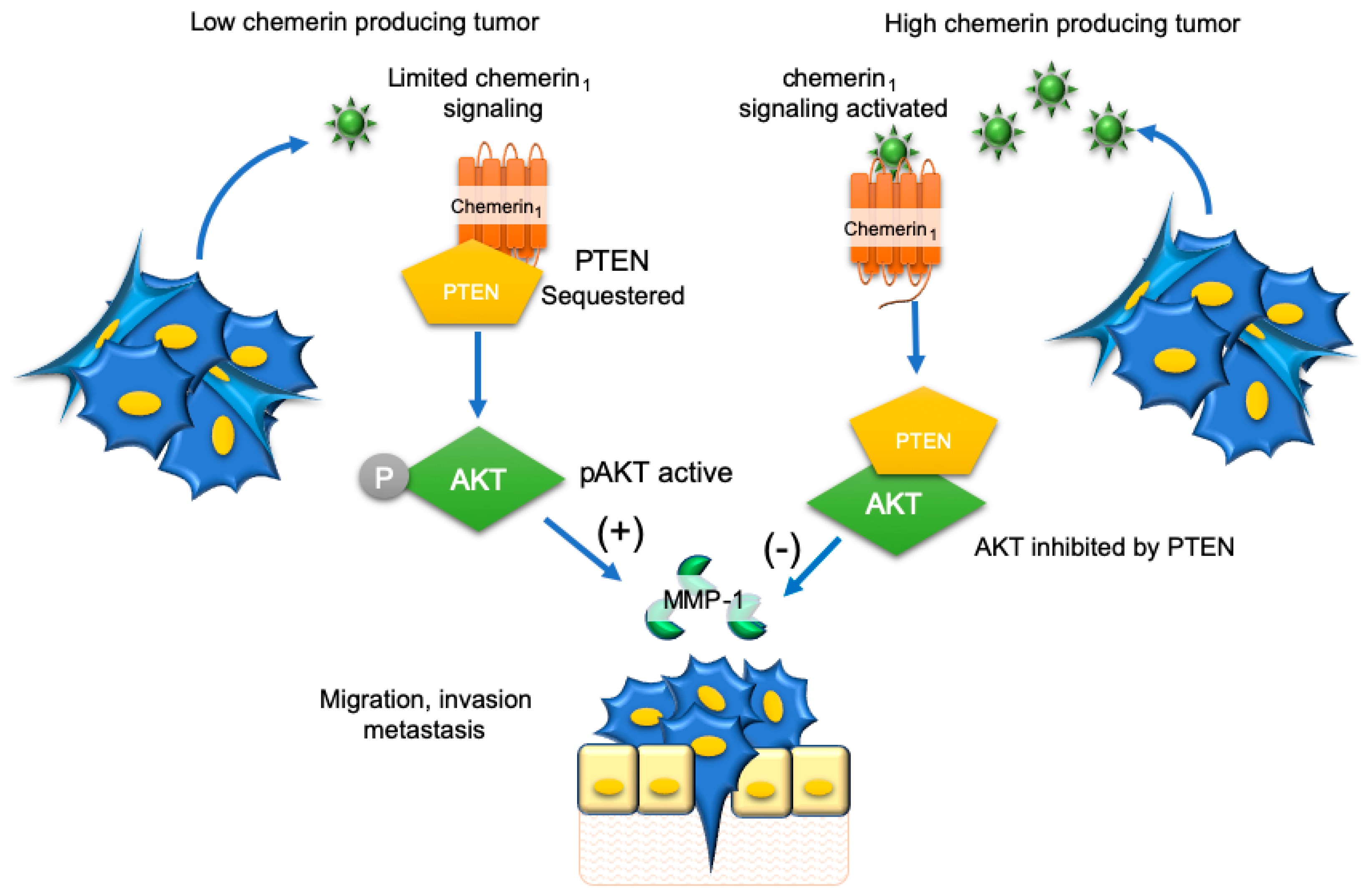
- Li, J.J.; Yin, H.K.; Guan, D.X.; Zhao, J.S.; Feng, Y.X.; Deng, Y.Z.; Wang, X.; Li, N.; Wang, X.F.; Cheng, S.Q.; et al. Chemerin suppresses hepatocellular carcinoma metastasis through CMKLR1-PTEN-Akt axis. Br. J. Cancer 2018, 118, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Chen, Y.L.; Jiang, L.; Chen, J.K. Reduced expression of chemerin is associated with a poor prognosis and a lowed infiltration of both dendritic cells and natural killer cells in human hepatocellular carcinoma. Clin. Lab. 2011, 57, 879–885. [Google Scholar]
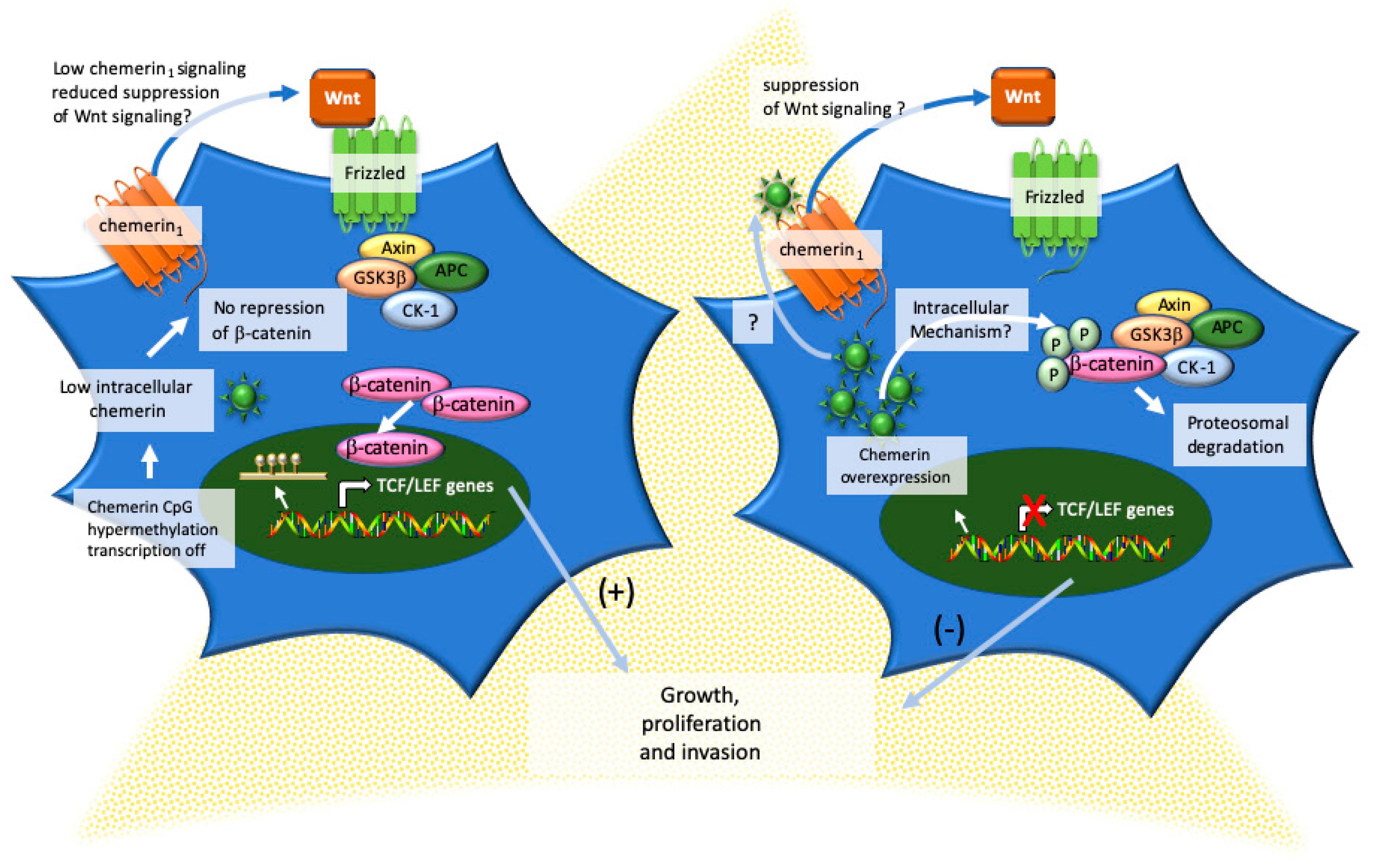
- Liu-Chittenden, Y.; Jain, M.; Gaskins, K.; Wang, S.; Merino, M.J.; Kotian, S.; Kumar Gara, S.; Davis, S.; Zhang, L.; Kebebew, E. RARRES2 functions as a tumor suppressor by promoting beta-catenin phosphorylation/degradation and inhibiting p38 phosphorylation in adrenocortical carcinoma. Oncogene 2017, 36, 3541–3552. [Google Scholar] [CrossRef]
- Adachi, Y.; Yamamoto, H.; Itoh, F.; Hinoda, Y.; Okada, Y.; Imai, K. Contribution of matrilysin (MMP-7) to the cetastatic pathway of human colorectal cancers. Gut 1999, 45, 252–258. [Google Scholar] [CrossRef]
- Ashizawa, T.; Okada, R.; Suzuki, Y.; Takagi, M.; Yamazaki, T.; Sumi, T.; Aoki, T.; Ohnuma, S.; Aoki, T. Clinical significance of interleukin-6 (IL-6) in the spread of gastric cancer: Role of IL-6 as a prognostic factor. Gastric Cancer 2005, 8, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, N.; Naruse, K.; Kobayashi, Y.; Miyade, M.; Saiki, T.; Enomoto, A.; Takahashi, M.; Matsubara, T. Chemerin promotes angiogenesis in vivo. Physiol. Rep. 2018, 6, e13962. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.P.; Fu, L.C.; Guo, H.; Xie, L.X. Expression of vascular endothelial growth factor c correlates with lymphatic vessel density and prognosis in human gastroesophageal junction carcinoma. Onkologie 2012, 35, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Krishna, M.; Narang, H. The complexity of mitogen-activated protein kinases (MAPKs) made simple. Cell. Mol. Life Sci. 2008, 65, 3525–3544. [Google Scholar] [CrossRef] [PubMed]
- Rourke, J.L.; Dranse, H.J.; Sinal, C.J. CMKLR1 and GPR1 mediate chemerin signaling through the RhoA/ROCK pathway. Mol. Cell. Endocrinol. 2015, 417, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.D.; Aolymat, I.; Tiszlavicz, L.; Reisz, Z.; Garalla, H.M.; Beynon, R.; Simpson, D.; Dockray, G.J.; Varro, A. Chemerin acts via CMKLR1 and GPR1 to stimulate migration and invasion of gastric cancer cells: Putative role of decreased TIMP-1 and TIMP-2. Oncotarget 2019, 10, 98–112. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Akram, I.G.; Georges, R.; Hielscher, T.; Adwan, H.; Berger, M.R. The chemokines CCR1 and CCRL2 have a role in colorectal cancer liver metastasis. Tumor Biol. 2016, 37, 2461–2471. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Luo, S.; Wang, G.; Peng, Z.; Zeng, W.; Tan, S.; Xi, Y.; Fan, J. Downregulation of tazarotene induced gene-2 (TIG2) in skin squamous cell carcinoma. Eur. J. Dermatol. 2008, 18, 638–641. [Google Scholar] [CrossRef]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef]
- Di Domizio, J.; Demaria, O.; Gilliet, M. Plasmacytoid dendritic cells in melanoma: Can we revert bad into good? J. Investig. Dermatol. 2014, 134, 1797–1800. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Yang, X.; Liu, W.; Li, B.; Yin, W.; Shi, Y.; He, R. Chemerin has a protective role in hepatocellular carcinoma by inhibiting the expression of IL-6 and GM-CSF and MDSC accumulation. Oncogene 2017, 36, 3599–3608. [Google Scholar] [CrossRef]
- Sun, P.; Wang, S.; Wang, J.; Sun, J.; Peng, M.; Shi, P. The involvement of iron in chemerin induced cell cycle arrest in human hepatic carcinoma SMMC7721 cells. Metallomics 2018, 10, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Rebouissou, S.; Zucman-Rossi, J.; Moreau, R.; Qiu, Z.; Hui, L. Note of caution: Contaminations of hepatocellular cell lines. J. Hepatol. 2017, 67, 896–897. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Chen, C.; Qin, J.; Liu, J.; Zheng, C. Genetic profiling reveals an alarming rate of cross-contamination among human cell lines used in China. FASEB J. 2015, 39, 4268–4272. [Google Scholar] [CrossRef]
- Haberl, E.M.; Pohl, R.; Rein-Fischboeck, L.; Feder, S.; Sinal, C.J.; Buechler, C. Chemerin in a mouse model of non-alcoholic steatohepatitis and hepatocarcinogenesis. Anticancer Res. 2018, 38, 2649–2657. [Google Scholar] [PubMed]
- Ayala-Ramirez, M.; Jasim, S.; Feng, L.; Ejaz, S.; Deniz, F.; Busaidy, N.; Waguespack, S.G.; Naing, A.; Sircar, K.; Wood, C.G.; et al. Adrenocortical carcinoma: Clinical outcomes and prognosis of 330 patients at a tertiary care center. Eur. J. Endocrinol. 2013, 169, 891–899. [Google Scholar] [CrossRef]
- Fernandez-Ranvier, G.G.; Weng, J.; Yeh, R.F.; Khanafshar, E.; Suh, I.; Barker, C.; Duh, Q.Y.; Clark, O.H.; Kebebew, E. Identification of biomarkers of adrenocortical carcinoma using genomewide gene expression profiling. Arch. Surg. 2008, 143, 841–846; discussion 846. [Google Scholar] [CrossRef]
- Velazquez-Fernandez, D.; Laurell, C.; Geli, J.; Hoog, A.; Odeberg, J.; Kjellman, M.; Lundeberg, J.; Hamberger, B.; Nilsson, P.; Backdahl, M. Expression profiling of adrenocortical neoplasms suggests a molecular signature of malignancy. Surgery 2005, 138, 1087–1094. [Google Scholar] [CrossRef]
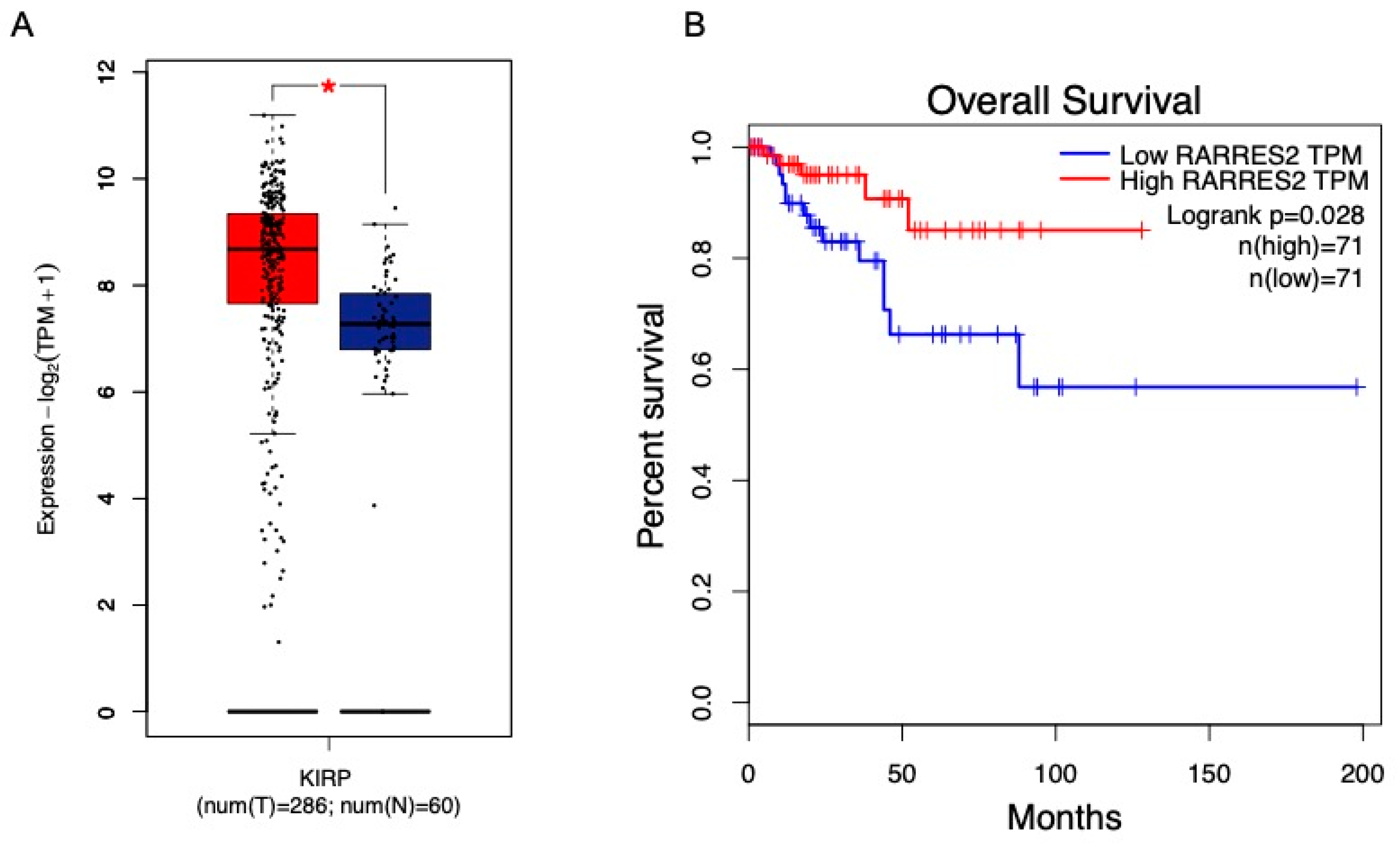
- Liu-Chittenden, Y.; Patel, D.; Gaskins, K.; Giordano, T.J.; Assie, G.; Bertherat, J.; Kebebew, E. Serum RARRES2 Is a Prognostic Marker in Patients With Adrenocortical Carcinoma. J. Clin. Endocrinol. Metab. 2016, 101, 3345–3352. [Google Scholar] [CrossRef]
- Dranse, H.J.; Muruganandan, S.; Fawcett, J.P.; Sinal, C.J. Adipocyte-secreted chemerin is processed to a variety of isoforms and influences MMP3 and chemokine secretion through an NFkB-dependent mechanism. Mol. Cell. Endocrinol. 2016, 436, 114–129. [Google Scholar] [CrossRef] [PubMed]
- Gaujoux, S.; Grabar, S.; Fassnacht, M.; Ragazzon, B.; Launay, P.; Libe, R.; Chokri, I.; Audebourg, A.; Royer, B.; Sbiera, S.; et al. beta-catenin activation is associated with specific clinical and pathologic characteristics and a poor outcome in adrenocortical carcinoma. Clin. Cancer Res. 2011, 17, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Tissier, F.; Cavard, C.; Groussin, L.; Perlemoine, K.; Fumey, G.; Hagnere, A.M.; Rene-Corail, F.; Jullian, E.; Gicquel, C.; Bertagna, X.; et al. Mutations of beta-catenin in adrenocortical tumors: Activation of the Wnt signaling pathway is a frequent event in both benign and malignant adrenocortical tumors. Cancer Res. 2005, 65, 7622–7627. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Cherniack, A.D.; Dewal, N.; Moffitt, R.A.; Danilova, L.; Murray, B.A.; Lerario, A.M.; Else, T.; Knijnenburg, T.A.; Ciriello, G.; et al. Comprehensive Pan-Genomic Characterization of Adrenocortical Carcinoma. Cancer Cell 2016, 29, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Muruganandan, S.; Govindarajan, R.; McMullen, N.M.; Sinal, C.J. Chemokine-Like Receptor 1 Is a Novel Wnt Target Gene that Regulates Mesenchymal Stem Cell Differentiation. Stem Cells 2017, 35, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef]
- Krawczyk, K.M.; Nilsson, H.; Allaoui, R.; Lindgren, D.; Arvidsson, M.; Leandersson, K.; Johansson, M.E. Papillary renal cell carcinoma-derived chemerin, IL-8, and CXCL16 promote monocyte recruitment and differentiation into foam-cell macrophages. Lab. Investig. 2017, 97, 1296–1305. [Google Scholar] [CrossRef]
- Kwon, H.; Han, K.D.; Park, C.Y. Weight change is significantly associated with risk of thyroid cancer: A nationwide population-based cohort study. Sci. Rep. 2019, 9, 1546. [Google Scholar] [CrossRef]
- El-Sagheer, G.; Gayyed, M.; Ahmad, A.; Abd El-Fattah, A.; Mohamed, M. Expression of chemerin correlates with a poor prognosis in female breast cancer patients. Breast Cancer (Dove Med. Press) 2018, 10, 169–176. [Google Scholar] [CrossRef]
- Pachynski, R.K.; Wang, P.; Salazar, N.; Zheng, Y.; Nease, L.; Rosalez, J.; Leong, W.I.; Virdi, G.; Rennier, K.; Shin, W.J.; et al. Chemerin Suppresses Breast Cancer Growth by Recruiting Immune Effector Cells Into the Tumor Microenvironment. Front. Immunol. 2019, 10, 983. [Google Scholar] [CrossRef]
- Boyle, S.T.; Kochetkova, M. Breast cancer stem cells and the immune system: Promotion, evasion and therapy. J. Mammary Gland Biol. Neoplasia 2014, 19, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.J.; Oh, H.K.; Park, S.H.; Bong, J.G. Association between inflammation and cancer stem cell phenotype in breast cancer. Oncol. Lett. 2018, 15, 2380–2386. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Chong, P.Y.; Tan, P.H. Breast cancer stem cells: An update. J. Clin. Pathol. 2013, 66, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Sarmadi, P.; Tunali, G.; Esendagli-Yilmaz, G.; Yilmaz, K.B.; Esendagli, G. CRAM-A indicates IFN-gamma-associated inflammatory response in breast cancer. Mol. Immunol. 2015, 68, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Rak, A.; Ptak, A. Bisphenol A and its derivatives decrease expression of chemerin, which reverses its stimulatory action in ovarian cancer cells. Toxicol. Lett. 2018, 291, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Reverchon, M.; Cornuau, M.; Rame, C.; Guerif, F.; Royere, D.; Dupont, J. Chemerin inhibits IGF-1-induced progesterone and estradiol secretion in human granulosa cells. Hum. Reprod. 2012, 27, 1790–1800. [Google Scholar] [CrossRef] [PubMed]
- Tummler, C.; Snapkov, I.; Wickstrom, M.; Moens, U.; Ljungblad, L.; Maria Elfman, L.H.; Winberg, J.O.; Kogner, P.; Johnsen, J.I.; Sveinbjornsson, B. Inhibition of chemerin/CMKLR1 axis in neuroblastoma cells reduces clonogenicity and cell viability in vitro and impairs tumor growth in vivo. Oncotarget 2017, 8, 95135–95151. [Google Scholar] [CrossRef] [PubMed]
- Ntikoudi, E.; Kiagia, M.; Boura, P.; Syrigos, K.N. Hormones of adipose tissue and their biologic role in lung cancer. Cancer Treat. Rev. 2014, 40, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Lanier, L.L. Natural killer cells and cancer. Adv. Cancer Res. 2003, 90, 127–156. [Google Scholar]
- Zhao, S.; Li, C.; Ye, Y.B.; Peng, F.; Chen, Q. Expression of Chemerin Correlates With a Favorable Prognosis in Patients With Non-Small Cell Lung Cancer. Labmedicine 2011, 42, 553–557. [Google Scholar] [CrossRef]
- Stamey, T.A.; Warrington, J.A.; Caldwell, M.C.; Chen, Z.; Fan, Z.; Mahadevappa, M.; McNeal, J.E.; Nolley, R.; Zhang, Z. Molecular genetic profiling of Gleason grade 4/5 prostate cnacers compared to benign prostatic hyperplasia. J. Urol. 2001, 166, 2171–2177. [Google Scholar] [CrossRef]
- Lin, P.-C.; Giannopoulou, E.G.; Park, K.; Mosquera, J.M.; Sboner, A.; Tewari, A.K.; Garraway, L.A.; Beltran, H.; Rubin, M.A.; Elemento, O. Epigenomic alterations in localized and advanced prostate cancer. Neoplasia 2013, 15, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Reyes, N.; Benedetti, I.; Rebollo, J.; Correa, O.; Geliebter, J. Atypical chemokine receptor CCRL2 is overexpressed in prostate cancer cells. J. Biomed. Res. 2019, 33, 17–23. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Demographics | Serum, Plasma, or Tissue Chemerin in ng/mL | |||
|---|---|---|---|---|---|
| Group, n | Age in Years | Sex | BMI kg/m2 | ||
| OSCC [54] | serum | ||||
| OSCC, 15 | 47.7 ± 14.1 | M6/F9 | 22.8 ± 1.1 | 655 ± 150 † | |
| OPML, 15 | 42.3 ± 11.0 | M5/F10 | 22.4 ± 1.1 | 408 ± 85 * | |
| Controls, 15 | 43.3 ± 11.8 | M7/F8 | 22.7 ± 1.5 | 187 ± 13 | |
| salivary fluid | |||||
| OSCC, 15 | 47.7 ± 14.1 | M6/F9 | 22.8 ± 1.1 | 13.2 ± 3.8 † | |
| OPML, 15 | 42.3 ± 11.0 | M5/F10 | 22.4 ± 1.1 | 9.1 ± 1.9 * | |
| Controls, 15 | 43.3 ± 11.8 | M7/F8 | 22.7 ± 1.5 | 3.1 ± 0.7 | |
| Colorectal [59] | Serum | ||||
| Patients, 41 | 55 (32–75) | M28/F13 | 25.8 (16.2–35.5) | 390 (250–630) | |
| Controls, 27 | 43 (18–64) | M15/F12 | 26.6 (21.5–45.8) | 340 (270–480) | |
| Colorectal [60] | plasma | ||||
| Patients, 221 | 50 ± 9 | 62.1% F | 16.5% > 30 | 148 (50–370) | |
| Gastric [61] | plasma | ||||
| Patients, 196 | 44.4% ≥ 60 | M112/F84 | 23.0 ± 3.1 | 53.1 ± 19.0 * | |
| Controls, 196 | 55.6% < 60 | Matched | 23.4 ± 3.5 | 31.3 ± 11.3 | |
| Colorectal [62] | serum | ||||
| Patients, 32 | 57.6 ± 6.5 | M22/F10 | 25.8 ± 4.2 | 377.0 ± 80 * | |
| Controls, 20 | 58.4 ± 7.2 | M14/F6 | 26.7 ± 5.3 | 87.8 ± 22.0 | |
| Colorectal [63] | serum | ||||
| Survivors, 110 | 56.3 ± 9.3 | M55/F55 | 23.3 ± 3.1 | 105 ± 14 | |
| Gastric [64] | serum | ||||
| Patients, 36 | 47–83 | M19/F17 | 42 * | ||
| Controls, 40 | 31–68 | M27/F13 | non-obese | 28 | |
| HCC [65] | serum | ||||
| Patients, 44 | 71 (50–82) | M29/F15 | 22.5 (15.6–33.5) | 130 (80–312) | |
| Thyroid [66] | serum | ||||
| BMI < 25, 51 | 41.2 ± 11.9 | F51 | 21.8 ± 2.1 | 212 ± 47 | |
| BMI ≥ 25, 126 | 55.4 ± 12.7 | M26/F100 | 30.7 ± 4.1 | 229 ± 50 * | |
| Breast [67] | serum | ||||
| Metastatic, 37 | 52.3 ± 11.8 | F37 | 29.1 ± 5.5 | 250 ± 59 | |
| Non-Met, 80 | 51.7 ± 12.5 | F80 | 28.6 ± 4.9 | 261 ± 73 | |
| All, 117 | 51.9 ± 12.2 | F117 | 28.7 ± 5.1 | 257 ± 69 | |
| CNS [46] | GBM, 12 | N/A | N/A | N/A | CSF |
| chem157S‡—0.2 ± 0.3 | |||||
| chem158K‡—5.1 ± 3.9 | |||||
| chem163S‡—3.0 ± 2.4 | |||||
| ODC, 12 | N/A | N/A | N/A | chem157S—0.7 ± 1.3 | |
| chem158K—3.8 ± 3.8 | |||||
| chem163S—2.9 ± 2.5 | |||||
| NC CNS, 7 | N/A | N/A | N/A | chem157S—1.0 ± 0.8 | |
| chem158K—6.3 ± 4.8 | |||||
| chem163S—5.5 ± 3.8 | |||||
| Controls, 9 | N/A | N/A | N/A | plasma | |
| chem157S—0.7 ± 0.8 | |||||
| chem158K—8.1 ± 2.9 | |||||
| chem163S—40 ± 7.9 | |||||
| NSCLC [68] | serum | ||||
| Patients, 110 | 65.1 | M91/F19 | 26.4 | 245 * | |
| Controls, 110 | 65.0 | M91/F19 | 27.7 | 203 | |
| NSCLC [69] | serum | ||||
| Patients, 189 | 61.8 ± 11.2 | M124/F65 | NA | 1.78 ± 0.57 * | |
| Controls, 120 | 62.6 ± 8.9 | M69/F51 | 1.20 ± 0.23 | ||
| Lung [70] | plasma | ||||
| Patients, 42 | 56 (44–78) | M26/F16 | N/A | 1.97 ± 0.37 * | |
| Controls, 31 | 48 (32–64) | M18/F13 | 1.11 ± 0.25 | ||
| Pancreatic ductal [71] | serum | ||||
| Patients, 25 | 63.0 ± 9.8 | 24.5 (21.7–27.8) | 272 (221–314) * | ||
| Controls, 36 | 37.6 ± 6.4 | M36 | 26.1 (24.2–29.5) | 193 (173–214) | |
| Prostate [72] | serum | ||||
| All patients, 74 | 67.1 ± 8.5 | M74 | 27.9 ± 3.3 | 273 ± 29 | |
| BPH, 66 | 61.5 ± 10.3 | M66 | 27.3 ± 4.0 | 268 ± 83 | |
| WD, 24 | 64.6 ± 8.5 | M24 | 27.2 ± 3.6 | 237 ± 72 + | |
| MD, 28 | 66.7 ± 8.8 | M28 | 28.0 ± 2.8 | 274 ± 60 + | |
| PD, 22 | 70.2 ± 7.5 | M22 | 28.3 ± 3.4 | 313 ± 93 + | |
| Prostate [73] | serum | ||||
| Non-obese, 25 | 68 (64–73) | M25 | 23.0 (21.5–24.3) | 74.0 (59.4–88.1) | |
| Obese, 37 | 64 (60–67) | M37 | 26.7 (25.7–27.6) | 75.0 (65.6–82.3) | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goralski, K.B.; Jackson, A.E.; McKeown, B.T.; Sinal, C.J. More Than an Adipokine: The Complex Roles of Chemerin Signaling in Cancer. Int. J. Mol. Sci. 2019, 20, 4778. https://doi.org/10.3390/ijms20194778
Goralski KB, Jackson AE, McKeown BT, Sinal CJ. More Than an Adipokine: The Complex Roles of Chemerin Signaling in Cancer. International Journal of Molecular Sciences. 2019; 20(19):4778. https://doi.org/10.3390/ijms20194778
Chicago/Turabian StyleGoralski, Kerry B., Ashley E. Jackson, Brendan T. McKeown, and Christopher J. Sinal. 2019. "More Than an Adipokine: The Complex Roles of Chemerin Signaling in Cancer" International Journal of Molecular Sciences 20, no. 19: 4778. https://doi.org/10.3390/ijms20194778
APA StyleGoralski, K. B., Jackson, A. E., McKeown, B. T., & Sinal, C. J. (2019). More Than an Adipokine: The Complex Roles of Chemerin Signaling in Cancer. International Journal of Molecular Sciences, 20(19), 4778. https://doi.org/10.3390/ijms20194778