Leptin and Associated Mediators of Immunometabolic Signaling: Novel Molecular Outcome Measures for Neurostimulation to Treat Chronic Pain

 , , and
, , and
Abstract
1. Introduction
2. Methods
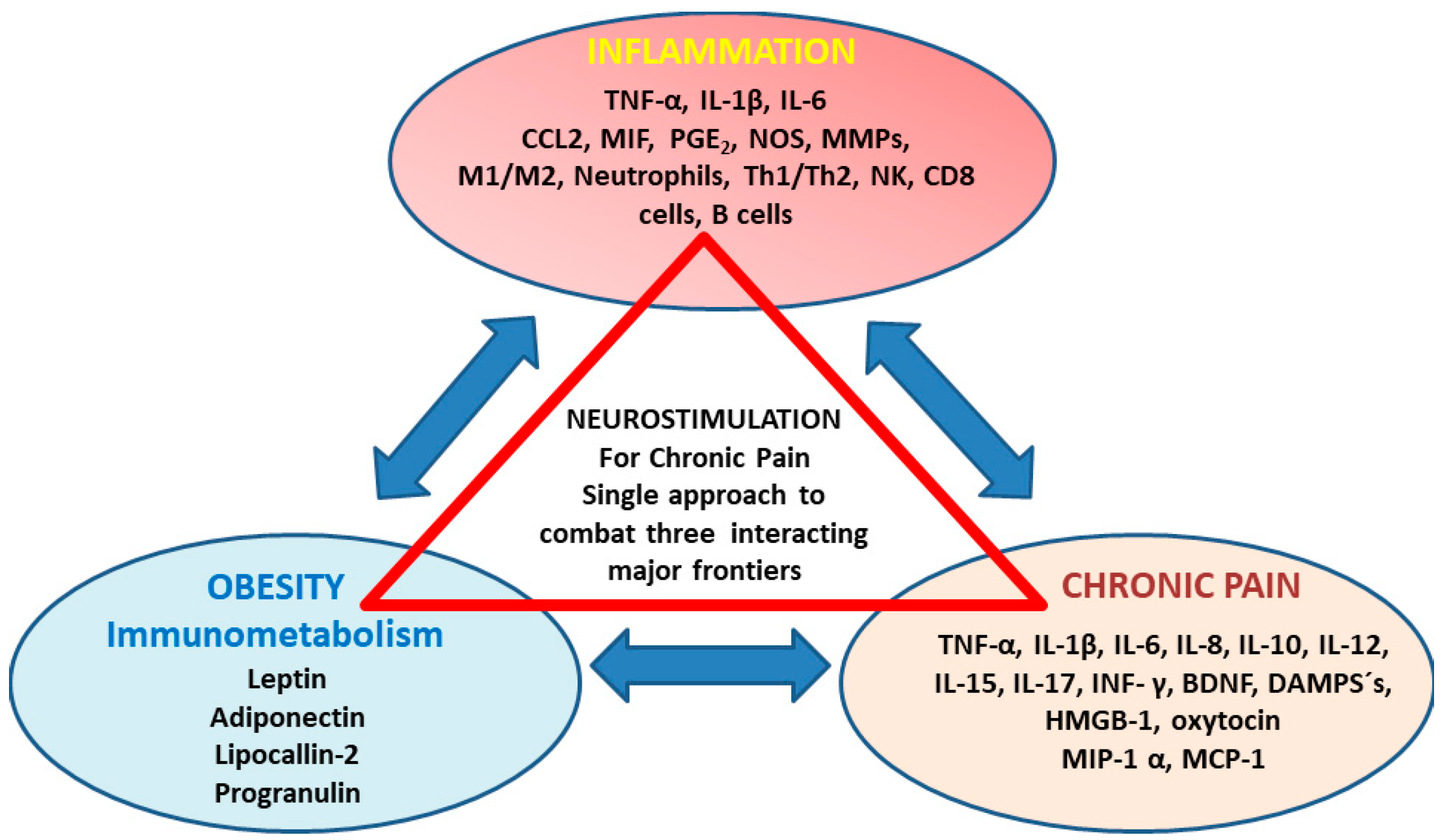
3. The Role of Circulating Inflammatory Mediators in Chronic Pain Development
3.1. Possible Associations between Back Pain (Spino-Nociceptive Traffic) with Immunometabolism
3.2. Possible Associations between Migraine (Trigemino-Nociceptive Signaling) with Immunometabolism
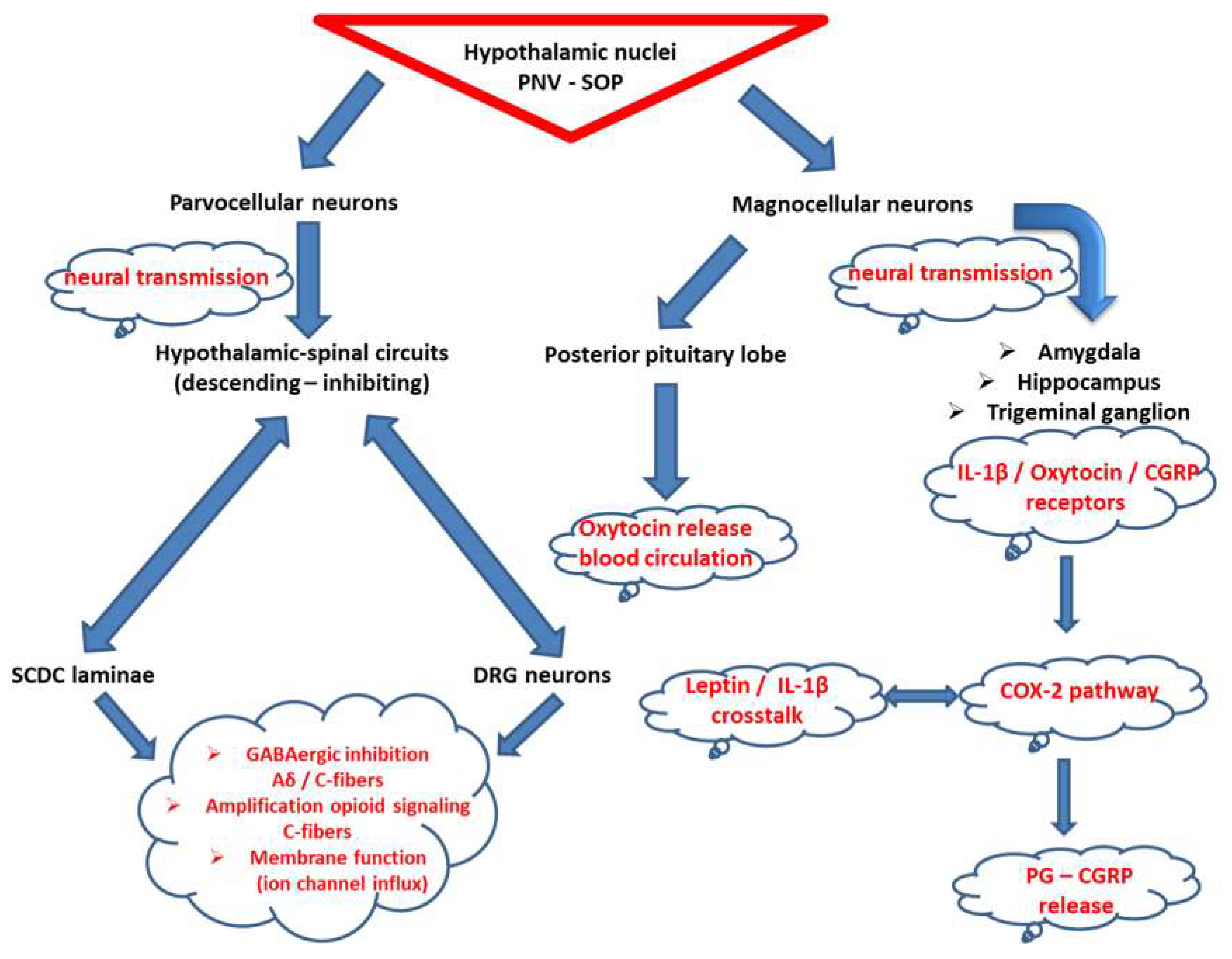
3.3. Oxytocin at the Crossroad of Trigeminal-Spinal Pain Transmission and Immunometabolism
4. Clinical Implications for Neurostimulation Therapies Targeting Chronic Pain Disorders
5. In-Human Chronic Pain-Neurostimulation Studies Addressed to Metabolic Molecular Inflammatory Phenotyping (Cervical Non-Invasive VNS, Tonic and BurstDR SCS, DRG-SCS)
6. Conclusions and Future Targeted Research
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Breivik, H.; Collett, B.; Ventafridda, V.; Cohen, R.; Gallacher, D. Survey of chronic pain in Europe: Prevalence, impact on daily life, and treatment. Eur. J. Pain 2006, 10, 287–333. [Google Scholar] [CrossRef] [PubMed]
- Langley, P.C.; Van Litsenburg, C.; Cappelleri, J.C.; Carroll, D. The burden associated with neuropathic pain in Western Europe. J. Med. Econ. 2013, 16, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Fishman, M.A.; Antony, A.; Esposito, M.; Deer, T.; Levy, R. The Evolution of Neuromodulation in the Treatment of Chronic Pain: Forward-Looking Perspectives. Pain Med. 2019, 20 (Suppl. 1), S58–S68. [Google Scholar] [CrossRef]
- Chaudhry, S.R.; Lendvai, I.S.; Muhammad, S.; Westhofen, P.; Kruppenbacher, J.; Scheef, L.; Boecker, H.; Scheele, D.; Hurlemann, R.; Kinfe, T.M. Inter-ictal assay of peripheral circulating inflammatory mediators in migraine patients under adjunctive cervical non-invasive vagus nerve stimulation (nVNS): A proof-of-concept study. Brain Stimul. 2019, 12, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Gravius, N.; Chaudhry, S.R.; Muhammad, S.; Boström, A.; Gravius, S.; Randau, T.; Scheele, D.; Westhofen, P.; Kruppenbacher, J.; Stoffel-Wagner, B.; et al. Selective L4 Dorsal Root Ganglion Stimulation Evokes Pain Relief and Changes of Inflammatory Markers: Part I Profiling of Saliva and Serum Molecular Patterns. Neuromodul. Technol. Neural Interface 2019, 22, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, S.; Chaudhry, S.R.; Yearwood, T.L.; Krauss, J.K.; Kinfe, T.M. Changes of Metabolic Disorders Associated Peripheral Cytokine/Adipokine Traffic in Non-Obese Chronic Back Patients Responsive to Burst Spinal Cord Stimulation. Neuromodul. Technol. Neural Interface 2018, 21, 31–37. [Google Scholar] [CrossRef]
- Kinfe, T.M.; Muhammad, S.; Link, C.; Roeske, S.; Chaudhry, S.R.; Yearwood, T.L. Burst spinal cord stimulation increases peripheral antineuroinflammatory interleukin 10 levels in failed back surgery syndrome patients with predominant back pain. Neuromodul. Technol. Neural Interface 2017, 20, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.P.; Hakimian, S.; Sherlin, L.H.; Fregni, F. New insights into neuromodulatory approaches for the treatment of pain. J. Pain 2008, 9, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Borowska, M.; Dworacka, M.; Wesolowska, A.; Winiarska, H.; Krzyzagorska, E.; Dworacki, G. The impact of pharmacotherapy of type 2 diabetes mellitus on IL-1beta, IL-6 and IL-10 secretion. Pharmacology 2016, 97, 189–194. [Google Scholar] [CrossRef]
- Barry, J.C.; Shakibakho, S.; Durrer, C.; Simtchouk, S.; Jawanda, K.K.; Cheung, S.T.; Mui, A.L.; Little, J.P. Hyporesponsiveness to the anti-inflammatory action of interleukin-10 in type 2 diabetes. Sci. Rep. 2016, 6, 21244. [Google Scholar] [CrossRef]
- Ma, D.H.; Xu, Q.Y.; Liu, Y.; Zhai, Q.Q.; Guo, M.H. Association between interleukin-10 gene polymorphisms and susceptibility to diabetic nephropathy in a Chinese population. Genet. Mol. Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Dagdeviren, S.; Jung, D.Y.; Friedline, R.H.; Noh, H.L.; Kim, J.H.; Patel, P.R.; Tsitsilianos, N.; Inashima, K.; Tran, D.A.; Hu, X.; et al. IL-10 prevents aging-associated inflammation and insulin resistance in skeletal muscle. FASEB J. 2017, 31, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Apkarian, A.V.; Hashmi, J.A.; Baliki, M.N. Pain and the brain: Specificity and plasticity of the brain in clinical chronic pain. Pain 2011, 152, S49. [Google Scholar] [CrossRef] [PubMed]
- Opatrilova, R.; Caprnda, M.; Kubatka, P.; Valentova, V.; Uramova, S.; Nosal, V.; Gaspar, L.; Zachar, L.; Mozos, I.; Petrovic, D.; et al. Adipokines in neurovascular diseases. Biomed. Pharm. 2018, 98, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Hysing, E.B.; Smith, L.; Thulin, M.; Karlsten, R.; Bothelius, K.; Gordh, T. Detection of systemic inflammation in severely impaired chronic pain patients and effects of a multimodal pain rehabilitation program. Scand. J. Pain 2019, 19, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Wåhlén, K.; Ghafouri, B.; Ghafouri, N.; Gerdle, B. Plasma Protein Pattern Correlates with Pain Intensity and Psychological Distress in Women with Chronic Widespread Pain. Front. Psychol. 2018, 9, 2400. [Google Scholar] [CrossRef] [PubMed]
- Gerdle, B.; Ghafouri, B.; Ghafouri, N.; Bäckryd, E.; Gordh, T. Signs of ongoing inflammation in female patients with chronic widespread pain: A multivariate, explorative, cross-sectional study of blood samples. Medicine (Baltim.) 2017, 96, e6130. [Google Scholar] [CrossRef] [PubMed]
- De La Cruz, P.; Fama, C.; Roth, S.; Haller, J.; Wilock, M.; Lange, S.; Pilitsis, J. Predictors of spinal cord stimulation success. Neuromodulation 2015, 18, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Özaktay, A.C.; Kallakuri, S.; Takebayashi, T.; Cavanaugh, J.M.; Asik, I.; Deleo, J.A.; Weinstein, N.J.N. Effects of interleukin-1 beta, interleukin-6, and tumor necrosis factor on sensitivity of dorsal root ganglion and peripheral receptive fields in rats. Eur. Spine J. 2006, 15, 1529–1537. [Google Scholar] [CrossRef] [PubMed]
- Sorkin, L.; Xiao, W.-H.; Wagner, R.; Myers, R. Tumour necrosis factor-α induces ectopic activity in nociceptive primary afferent fibres. Neuroscience 1997, 81, 255–262. [Google Scholar] [CrossRef]
- Zhang, J.-M.; Li, H.; Liu, B.; Brull, S.J. Acute topical application of tumor necrosis factor α evokes protein kinase A-dependent responses in rat sensory neurons. J. Neurophysiol. 2002, 88, 1387–1392. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.H.; Yang, B.; Donnelly, D.F.; Ma, C.; Lamotte, R.H. MCP-1 enhances excitability of nociceptive neurons in chronically compressed dorsal root ganglia. J. Neurophysiol. 2006, 96, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- White, F.A.; Sun, J.; Waters, S.M.; Ma, C.; Ren, D.; Ripsch, M.; Steflik, J.; Cortright, D.N.; Lamotte, R.H.; Miller, R.J. Excitatory monocyte chemoattractant protein-1 signaling is up-regulated in sensory neurons after chronic compression of the dorsal root ganglion. Proc. Natl. Acad. Sci. USA 2005, 102, 14092–14097. [Google Scholar] [CrossRef] [PubMed]
- Schäfers, M.; Svensson, C.I.; Sommer, C.; Sorkin, L.S. Tumor necrosis factor-α induces mechanical allodynia after spinal nerve ligation by activation of p38 MAPK in primary sensory neurons. J. Neurosci. 2003, 23, 2517–2521. [Google Scholar] [CrossRef] [PubMed]
- Ramer, M.S.; Murphy, P.G.; Richardson, P.M.; Bisby, M.A. Spinal nerve lesion-induced mechanoallodynia and adrenergic sprouting in sensory ganglia are attenuated in interleukin-6 knockout mice. Pain 1998, 78, 115–121. [Google Scholar] [CrossRef]
- Xie, W.R.; Deng, H.; Li, H.; Bowen, T.; Strong, J.; Zhang, J.M. Robust increase of cutaneous sensitivity, cytokine production and sympathetic sprouting in rats with localized inflammatory irritation of the spinal ganglia. Neuroscience 2006, 142, 809–822. [Google Scholar] [CrossRef]
- Zingg, R.W.; Kendall, R. Obesity, vascular disease, and lumbar disk degeneration: Associations of comorbidities in low back pain. PM R 2017, 9, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Samartzis, D.; Karppinen, J.; Cheung, J.P.; Lotz, J. Disk degeneration and low back pain: Are they fat-related conditions? Glob. Spine J. 2013, 3, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Dario, A.; Ferreira, M.; Refshauge, K.; Harmer, A.; Sanchez-Romera, J.; Perez-Riquelme, F.; Cisneros, L.; Ordonana, J.; Ferreira, P. Mapping the association between back pain and type 2 diabetes: A cross-sectional and longitudinal study of adult Spanish twins. PLoS ONE 2017, 12, e0174757. [Google Scholar] [CrossRef] [PubMed]
- Samartzis, D.; Karppinen, J.; Chan, D.; Luk, K.D.; Cheung, K.M. The association of lumbar intervertebral disc degeneration on magnetic resonance imaging with body mass index in overweight and obese adults: A population-based study. Arthritis Rheumatol. 2012, 64, 1488–1496. [Google Scholar] [CrossRef]
- Gupta, N.; White, K.T.; Sandford, P.R. Body mass index in spinal cord injury—A retrospective study. Spinal Cord 2006, 44, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Podichetty, V.K. The aging spine: The role of inflammatory mediators in intervertebral disc degeneration. Cell. Mol. Biol. 2007, 53, 4–18. [Google Scholar] [PubMed]
- Vezzani, A.; Viviani, B. Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology 2015, 96, 70–82. [Google Scholar] [CrossRef]
- Srikanthan, K.; Feyh, A.; Visweshwar, H.; Shapiro, J.I.; Sodhi, K. Systematic review of metabolic syndrome biomarkers: A panel for early detection, management, and risk stratification in the west virginian population. Int. J. Med. Sci. 2016, 13, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Bomfim, G.F.; Rodrigues, F.L.; Carneiro, F.S. Are the innate and adaptive immune systems setting hypertension on fire? Pharm. Res. 2017, 117, 377–393. [Google Scholar] [CrossRef] [PubMed]
- McMaster, W.G.; Kirabo, A.; Madhur, M.S.; Harrison, D.G. Inflammation, immunity, and hypertensive end-organ damage. Circ. Res. 2015, 116, 1022–1033. [Google Scholar] [CrossRef] [PubMed]
- Crowley, S.D. The cooperative roles of inflammation and oxidative stress in the pathogenesis of hypertension. Antioxid. Redox Signal. 2014, 20, 102–120. [Google Scholar] [CrossRef] [PubMed]
- De Ciuceis, C.; Rossini, C.; La Boria, E.; Porteri, E.; Petroboni, B.; Gavazzi, A.; Sarkar, A.; Rosei, E.A.; Rizzoni, D. Immune mechanisms in hypertension. High Blood Press. Cardiovasc. Prev. 2014, 21, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Golia, E.; Limongelli, G.; Natale, F.; Fimiani, F.; Maddaloni, V.; Pariggiano, I.; Bianchi, R.; Crisci, M.; D’Acierno, L.; Giordano, R.; et al. Inflammation and cardiovascular disease: From pathogenesis to therapeutic target. Curr. Atheroscler. Rep. 2014, 16, 435. [Google Scholar] [CrossRef]
- Fernández-Sánchez, A.; Madrigal-Santillán, E.; Bautista, M.; Esquivel-Soto, J.; Morales-González, Á.; Esquivel-Chirino, C.; Durante-Montiel, I.; Sánchez-Rivera, G.; Valadez-Vega, C.; Morales-González, J.A. Inflammation, oxidative stress, and obesity. Int. J. Mol. Sci. 2011, 12, 3117–3132. [Google Scholar] [CrossRef]
- Kotani, K.; Sakane, N. Leptin: Adiponectin ratio and metabolic syndrome in the general Japanese population. Korean J. Lab. Med. 2011, 31, 162–166. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Finucane, F.M.; Luan, J.; Wareham, N.J.; Sharp, S.J.; O’Rahilly, S.; Balkau, B.; Flyvbjerg, A.; Walker, M.; Hojlund, K.; Nolan, J.J.; et al. Correlation of the leptin: Adiponectin ratio with measures of insulin resistance in non-diabetic individuals. Diabetologia 2009, 52, 2345–2349. [Google Scholar] [CrossRef] [PubMed]
- Francisco, V.; Pino, J.; Gonzalez-Gay, M.A.; Mera, A.; Lago, F.; Gómez, R.; Mobasheri, A.; Gualillo, O. Adipokines and inflammation: Is it a question of weight? Br. J. Pharmacol. 2018, 175, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Cong, W.N.; Golden, E.; Pantaleo, N.; White, C.M.; Maudsley, S.; Martin, B. Ghrelin receptor signaling: A promising therapeutic target for metabolic syndrome and cognitive dysfunction. CNS Neurol. Disord. Drug Targets 2010, 9, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Varela, L.; Vazquez, M.J.; Cordido, F.; Nogueiras, R.; Vidal-Puig, A.; Dieguez, C.; Lopez, M. Ghrelin and lipid metabolism: Key partners in energy balance. J. Mol. Endocrinol. 2011, 46, R43–R63. [Google Scholar] [CrossRef] [PubMed]
- Ukkola, O. Ghrelin and metabolic disorders. Curr. Protein Pept. Sci. 2009, 10, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Ukkola, O. Ghrelin and the metabolic balance. J. Endocrinol. Investig. 2005, 28, 849–852. [Google Scholar] [CrossRef]
- Bacha, F.; Arslanian, S.A. Ghrelin suppression in overweight children: A manifestation of insulin resistance? J. Clin. Endocrinol. Metab. 2005, 90, 2725–2730. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, B.; Hulten, L.M.; Hulthe, J. Plasma ghrelin, body fat, insulin resistance, and smoking in clinically healthy men: The atherosclerosis and insulin resistance study. Metabolism 2003, 52, 1460–1463. [Google Scholar] [CrossRef]
- Farajdokht, F.; Babri, S.; Karimi, P.; Alipour, M.R.; Bughchechi, R.; Mohaddes, G. Chronic ghrelin treatment reduced photophobia and anxiety-like behaviors in nitroglycerin-induced migraine: Role of pituitary adenylate cyclase—Activating polypeptide. Eur. J. Neurosci. 2017, 45, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Inoue, W.; Poole, S.; Bristow, A.F.; Luheshi, G.N. Leptin induces cyclo-oxygenase 2 via interaction with interleukin 1-beta in the rat brain. Eur. J. Neurosci. 2006, 24, 2233–2245. [Google Scholar] [CrossRef]
- Boström, A.; Scheele, D.; Stoffel-Wagner, B.; Hönig, F.; Chaudhry, S.R.; Muhammad, S.; Hurlemann, R.; Krauss, J.K.; Lendvai, I.S.; Chakravarthy, K.V.; et al. Saliva molecular inflammatory profiling in female migraine patients responsive to adjunctive cervical non-invasive vagus nerve stimulation: The MOXY Study. J. Transl. Med. 2019, 17, 53. [Google Scholar] [CrossRef]
- Ding, C.; Leow, M.K.; Magkos, F. Oxytocin in metabolic homeostasis: Implication for obesity and diabetes management. Obes. Rev. 2019, 20, 22–40. [Google Scholar] [CrossRef]
- Eliava, M.A. New Population of Parvocellular Oxytocin Neurons Controlling Magnocellular Neuron Activity and Inflammatory Pain Processing. Neuron 2016, 89, 1291–1304. [Google Scholar] [CrossRef]
- Boll, S.; Almeida de Minas, A.C.; Raftogianni, A.; Herpetz, S.C.; Grinevich, V. Oxytocin and pain perception: From animal models to human research. Neuroscience 2018, 387, 149–161. [Google Scholar] [CrossRef]
- Poisbeau, P.; Grinevich, V.; Charlet, A. Oxytocin Signaling in Pain: Cellular, Circuit, System, and Behavioral Levels. Curr. Top. Behav. Neurosci. 2018, 35, 193–211. [Google Scholar] [CrossRef]
- Tzabazis, A.; Mechanic, J.; Miller, J.; Klukinov, M.; Pascual, C.; Manering, N.; Carson, D.S.; Jacobs, A.; Qiao, Y.; Cuellar, J.; et al. Oxytocin receptor: Expression in the trigeminal nociceptive system and potential role in the treatment of headache disorders. Cephalalgia 2016, 36, 943–950. [Google Scholar] [CrossRef]
- Tzabazis, A.; Kori, S.; Mechanic, J.; Miller, J.; Pascual, C.; Manering, N.; Carson, D.; Klukinov, M.; Spierings, E.; Jacobs, D.; et al. Oxytocin and Migraine Headache. Headache 2017, 57 (Suppl. 2), 64–75. [Google Scholar] [CrossRef]
- Hoshiyamata, E.; Tatsumoto, M.; Iwanami, H.; Saisu, A.; Watanabe, H.; Inaba, N.; Hirata, K. Postpartum migraines: A long-term prospective study. Intern. Med. 2012, 51, 3119–3123. [Google Scholar] [CrossRef]
- Phillips, W.J.; Ostrovsky, O.; Galli, R.L.; Dickey, S. Relief of acute migraine headache with intravenous oxytocin: Report of two cases. J. Pain Palliat. Care Pharmacother. 2006, 20, 25–28. [Google Scholar] [CrossRef]
- Noseda, R.; Lee, A.J.; Rony-Reuven, N.; Bernstein, C.A.; Kainz, V.M.; Bertisch, S.M.; Buettner, C.; Borsook, D.; Burstein, R. Neural mechanism for hypothalamic-mediated autonomic responses to light during migraine. Proc. Natl. Acad. Sci. USA 2017, 114, E5683–E5692. [Google Scholar] [CrossRef]
- Strother, L.C.; Srikiatkhachorn, A.; Supronsinchai, W. Targeted orexin and hypothalamic neuropeptides for migraine. Neurotherapeutics 2018, 15, 377–390. [Google Scholar] [CrossRef]
- You, D.S.; Honey, R.; Albu, S.; Meagher, M.W. Generalized pain sensitization and endogenous oxytocin in individuals with symptoms of migraine: A cross-sectional study. Headache 2018, 58, 62–77. [Google Scholar] [CrossRef]
- Martínez-Lorenzana, G.; Espinosa-López, L.; Carranza, M.; Aramburo, C.; Paz-Tres, C.; Rojas-Piloni, G.; Condés-Lara, M. PVN electrical stimulation prolongs withdrawal latencies and releases oxytocin in cerebrospinal fluid, plasma, and spinal cord tissue in intact and neuropathic rats. Pain 2008, 140, 265–273. [Google Scholar]
- Condés-Lara, M.; Maie, I.A.; Dickenson, A.H. Oxytocin actions on afferent evoked spinal cord neuronal activities in neuropathic but not in normal rats. Brain Res. 2005, 1045, 124–133. [Google Scholar] [CrossRef]
- Breton, J.D.; Veinante, P.; Uhl-Bronner, S.; Vergnano, A.M.; Freund-Mercier, M.J.; Schlichter, R.; Poisbeau, P. Oxytocin-induced antinociception in the spinal cord ismediated by a subpopulation of glutamatergic neurons in lamina I-II which amplify GABAergic inhibition. Mol. Pain 2008, 4, 19. [Google Scholar] [CrossRef]
- Condés-Lara, M.; Rojas-Piloni, G.; Martínez-Lorenzana, G.; López-Hidalgo, M.; Rodríguez-Jiménez, J. Hypothalamospinal oxytocinergic antinociception ismediated by GABAergic and opiate neurons that reduce Adelta and C fiber primary afferent excitation of spinal cord cells. Brain Res. 2009, 1247, 38–49. [Google Scholar] [CrossRef]
- Moreno-López, Y.; Martinez-Lorenzana, G.; Condés-Lara, M.; Rojas-Piloni, G. Identification of oxytocin receptor in the dorsal dorn and nociceptive dorsal root ganglion neurons. Neuropeptides 2013, 47, 117–123. [Google Scholar] [CrossRef]
- Tracey, K.J. The inflammatory reflex. Nature 2002, 420, 853–859. [Google Scholar] [CrossRef]
- Pavlov, V.A.; Tracey, K.J. The vagus nerve and the inflammatory reflex—Linking immunity and metabolism. Nat. Rev. Endocrinol. 2012, 8, 743–754. [Google Scholar] [CrossRef]
- Stock, S.; Uvnas-Moberg, K. Increased plasma levels of oxytocin in response to electrical stimulation of the sciatic and vagal nerves and in response to touch and pinch in anaesthetized rats. Acta Physiol. Scand. 1988, 132, 29–34. [Google Scholar] [CrossRef]
- Berthoud, H.R.; Neuhuber, W.L. Vagal mechanisms as neuromodulatory targets for the treatment of metabolic disease. Ann. N. Y. Acad. Sci. 2019. [Google Scholar] [CrossRef]
- Corcoran, C.; Connor, T.J.; O´Keane, V.; Garland, M.R. The effects of vagus nerve stimulation on pro- and anti-inflammatory cytokines in Humans: A preliminary report. Neuroimmunomodulation 2005, 12, 307–309. [Google Scholar] [CrossRef]
- Chakravarthy, K.V.; Xing, F.; Bruno, K.; Kent, A.R.; Raza, A.; Hurlemann, R.; Kinfe, T.M. A Review of Spinal and Peripheral Neuromodulation and Neuroinflammation: Lessons Learned Thus Far and Future Prospects of Biotype Development. Neuromodul. Technol. Neural Interface 2019, 22, 235–243. [Google Scholar] [CrossRef]
- Lind, A.-L.; Khoonsari, E.; Sjodin, M.; Katila, L.; Wetterhall, M.; Gordh, T.; Kultima, K. Spinal cord stimulation alters protein levels in the cerebrospinal fluid of neuropathic pain patients: A proteomic mass spectrometric analysis. Neuromodul. Technol. Neural Interface 2016, 19, 549–562. [Google Scholar] [CrossRef]
- McCarthy, K.F.; Connor, T.; McCrory, C. Cerebrospinal fluid levels of vascular growth factor correlate with reported pain and are reduced by spinal cord stimulation in patients with failed back surgery syndrome. Neuromodul. Technol. Neural Interface 2013, 16, 519–522. [Google Scholar] [CrossRef]
- McCarthy, K.F.; McCrory, C. Cerebrospinal fluid levels of glial cell-derived neurotrophic factor correlate with spinal cord stimulation frequency in patients with neuropathic pain: A preliminary report. Spinal Cord 2014, 52, S8–S10. [Google Scholar] [CrossRef]
- Kriek, N.; Schreurs, M.W.J.; Groeneweg, J.G.; Dik, W.A.; Tjiang, G.C.H.; Gültuna, I.; Stronks, D.L.; Huygen, F.J.P.M. Spinal Cord Stimulation in Patients with Complex Regional Pain Syndrome: A Possible Target for Immunomodulation? Neuromodul. Technol. Neural Interface 2018, 21, 77–86. [Google Scholar] [CrossRef]
- Kinfe, T.M.; Asif, M.; Chakravarthy, K.V.; Deer, T.R.; Kramer, J.M.; Yearwood, T.L.; Hurlemann, R.; Hussain, M.S.; Motameny, S.; Wagle, P.; et al. Unilateral L4-dorsal root ganglion stimulation evokes pain relief in chronic neuropathic postsurgical knee pain and changes of inflammatory markers: Part II whole transcriptome profiling. J. Transl. Med. 2019, 17, 205. [Google Scholar] [CrossRef]
- Lerman, I.; Hauger, R.; Sorkin, L.; Proudfoot, J.; Davis, B.; Huang, A.; Lam, K.; Simon, B.; Baker, D.G. Noninvasive Transcutaneous Vagus Nerve Stimulation Decreases Whole Blood Culture-Derived Cytokines and Chemokines: A Randomized, Blinded, Healthy Control Pilot Trial. Neuromodul. Technol. Neural Interface 2016, 19, 283–290. [Google Scholar] [CrossRef]
- Russo, M.A.; Fiore, N.T.; van Vreden, C.; Bailey, D.; Danielle, M.S.; Helen, M.M.; de St Groth, B.F.; Austin, P.J. Expansion and activation of distinct central memory T lymphocyte subsets in complex regional pain syndrome. Austin J. Neuroinflamm. 2019, 16, 63. [Google Scholar] [CrossRef]
- Dahlem, M.A.; Rode, S.; May, A.; Fujiwara, N.; Hirata, Y.; Kazuyuki, A.; Kurths, J. Towards dynamical network biomarkers in neuromodulation of episodic migraine. Transl. Neurosci. 2013, 4, 282–294. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Year/Study Design | Pain Disorder | Stimulation Target | Patients | Score-Based Parameter | Stimulation Paradigm | Immunometabolic Parameter | Treatment Duration |
|---|---|---|---|---|---|---|---|
| 2019/RCT [4] | Migraine | Cervical branch vagus nerve (nVNS) | [48] | Head pain Severity-Frequency, BMI, PSQI, MIDAS, BDI | 120 sec dose bilateral vagus nerve applied 2 times/day (1 ms bursts of 5 kHz sine waves every 40 ms (25 Hz)) | ELISA serum (leptin, ghrelin, adiponectin, IL-1β, IL-6, IL-10, TNF-α, HMGB-1) | 2 months |
| 2019/pPS [52] | Migraine | Cervical branch vagus nerve (nVNS) | [24] | Head pain Severity -Frequency, BMI, PSQI, MIDAS, BDI, EQ-5D-5L | 120 sec dose bilateral vagus nerve applied 2 times/day (1 ms bursts of 5 kHz sine waves every 40 ms (25 Hz)) | ELISA saliva (IL-1β – oxytocin) | 10 weeks |
| 2019/pPS [5] | CRPS | DRG (L4-DRGSTIM) | [24] | Neuropathic pain Severity, BMI, PSQI, BDI | Chronic stimulation, bipolar, 20 Hz, 200-300 µsec, 300–1600 µA | ELISA serum-saliva (leptin, ghrelin, adiponectin, IL-1β, IL-6, IL-10, TNF-α, HMGB-1, BDNF, oxytocin) | 3 months |
| 2019/pPS [79] | CRPS | DRG (L4-DRGSTIM) | [12] | Neuropathic pain Severity, BMI, PSQI, BDI | Chronic stimulation, bipolar, 20 Hz, 200–300 µsec, 300–1600 µA | Gene expression blood cells (FFAR2, ILRN, IL-17F, PLA2G2, NOX1 (metabolic function)) | 3 months |
| 2018/pPS [6] | FBSS CLBP | Spinal Cord dorsal column level Th10-11 | [24] | FBSS-CLBP Severity, BMI, PSQI, BDI | BurstDR chronic stimulation, bipolar, 40 Hz burst rate, 500 Hz intraburst rate, 1 msec, 2.05–2.45 mA | ELISA-serum (leptin, adiponectin, ghrelin) | 3 months |
| 2017/pPS [7] | FBSS CLBP | Spinal Cord dorsal column level Th10-11 | [24] | FBSS-CLBP Severity, BMI, PSQI, BDI | BurstDR SCS chronic stimulation, bipolar, 40 Hz burst rate, 500 Hz intraburst rate, 1 msec, 2.05–2.45 mA | ELISA-serum (IL-1β, IL-10, TNF-α, HMGB-1) | 3 months |
| 2017/RCT [78] | CRPS | Spinal Cord dorsal column level Th10-11 | [24] | CRPS Severity – Phenotype FBSS – CLBP Severity, BMI, PSQI, BDI | SCS chronic stimulation, bipolar, BurstDR vs 40 Hz vs 500 Hz vs 1200 Hz vs sham | ELISA-artificial skin blister (IL-1b, IL-1ra, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12p70, IL-13, IL-15, IL-17, basic FGF, Eotaxin, G-CSF, GM-CSF, IFN-g, IP-10, MCP-1, MIP-1a, MIP-1b, PDGF-BB, TNF-a, VEGF, T cells) | 3 months |
| 2016/pPS [75] | NP Phantom pain | Spinal Cord dorsal column level Th10-11 | [14] | Pain Severity BMI | Chronic stimulation, bipolar, tonic SCS mode with 50 Hz SCS on vs off | Proteomic Mass Spectrometry-CSF (clusterin, gelsolin, mimecan, angiotensinogen, secretogranin-1, amyloid beta A4 protein, gelsolin, apolipoprotein C1, apolipoprotein E, contactin-1, neural cell adhesion molecule L1-like protein, VGF and dickkopf-related protein 3) | 12 months |
| 2014/pPS [77] | FBSS | Spinal Cord dorsal column level Th10-11 | [9] | Pain Severity, SF-36 | Chronic stimulation, bipolar, tonic SCS mode with 40–100 Hz, 210–360 µsec, 3–7.4 Volt | ELISA-CSF (GDNF) | |
| 2013/pPS [76] | NP Phantom pain | Spinal Cord dorsal column level Th10-11 | [14] | Pain Severity, SF-36 | Chronic stimulation, bipolar, tonic SCS mode with 40–100 Hz, 210–360 µsec, 3–7.4 Volt | ELISA-CSF (VEGF, BDNF, MCP-1, chemokines) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kinfe, T.M.; Buchfelder, M.; Chaudhry, S.R.; Chakravarthy, K.V.; Deer, T.R.; Russo, M.; Georgius, P.; Hurlemann, R.; Rasheed, M.; Muhammad, S.; et al. Leptin and Associated Mediators of Immunometabolic Signaling: Novel Molecular Outcome Measures for Neurostimulation to Treat Chronic Pain. Int. J. Mol. Sci. 2019, 20, 4737. https://doi.org/10.3390/ijms20194737
Kinfe TM, Buchfelder M, Chaudhry SR, Chakravarthy KV, Deer TR, Russo M, Georgius P, Hurlemann R, Rasheed M, Muhammad S, et al. Leptin and Associated Mediators of Immunometabolic Signaling: Novel Molecular Outcome Measures for Neurostimulation to Treat Chronic Pain. International Journal of Molecular Sciences. 2019; 20(19):4737. https://doi.org/10.3390/ijms20194737
Chicago/Turabian StyleKinfe, Thomas M., Michael Buchfelder, Shafqat R. Chaudhry, Krishnan V. Chakravarthy, Timothy R. Deer, Marc Russo, Peter Georgius, Rene Hurlemann, Muhammad Rasheed, Sajjad Muhammad, and et al. 2019. "Leptin and Associated Mediators of Immunometabolic Signaling: Novel Molecular Outcome Measures for Neurostimulation to Treat Chronic Pain" International Journal of Molecular Sciences 20, no. 19: 4737. https://doi.org/10.3390/ijms20194737
APA StyleKinfe, T. M., Buchfelder, M., Chaudhry, S. R., Chakravarthy, K. V., Deer, T. R., Russo, M., Georgius, P., Hurlemann, R., Rasheed, M., Muhammad, S., & Yearwood, T. L. (2019). Leptin and Associated Mediators of Immunometabolic Signaling: Novel Molecular Outcome Measures for Neurostimulation to Treat Chronic Pain. International Journal of Molecular Sciences, 20(19), 4737. https://doi.org/10.3390/ijms20194737