The Roles of Sex Hormones in the Course of Atopic Dermatitis



Abstract
1. Introduction
2. The Effects of Sex Hormones on Immune Responses
2.1. General Tendency (Table 1)
2.2. Female Hormones
2.2.1. Estrogens (Table 2)
2.2.2. Progesterone (Table 3)
2.3. Androgens (Table 4)
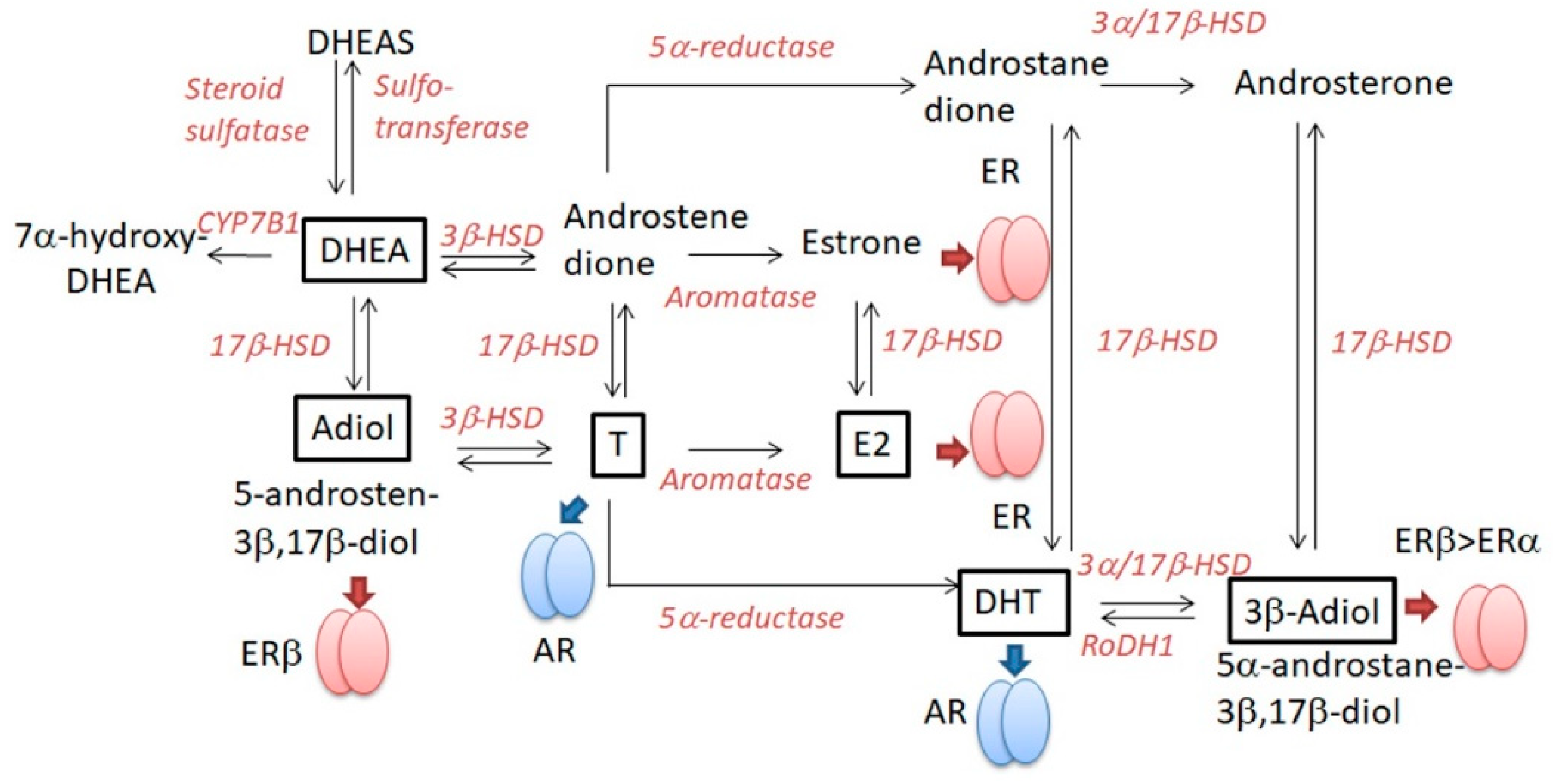
2.4. DHEA (Table 5)
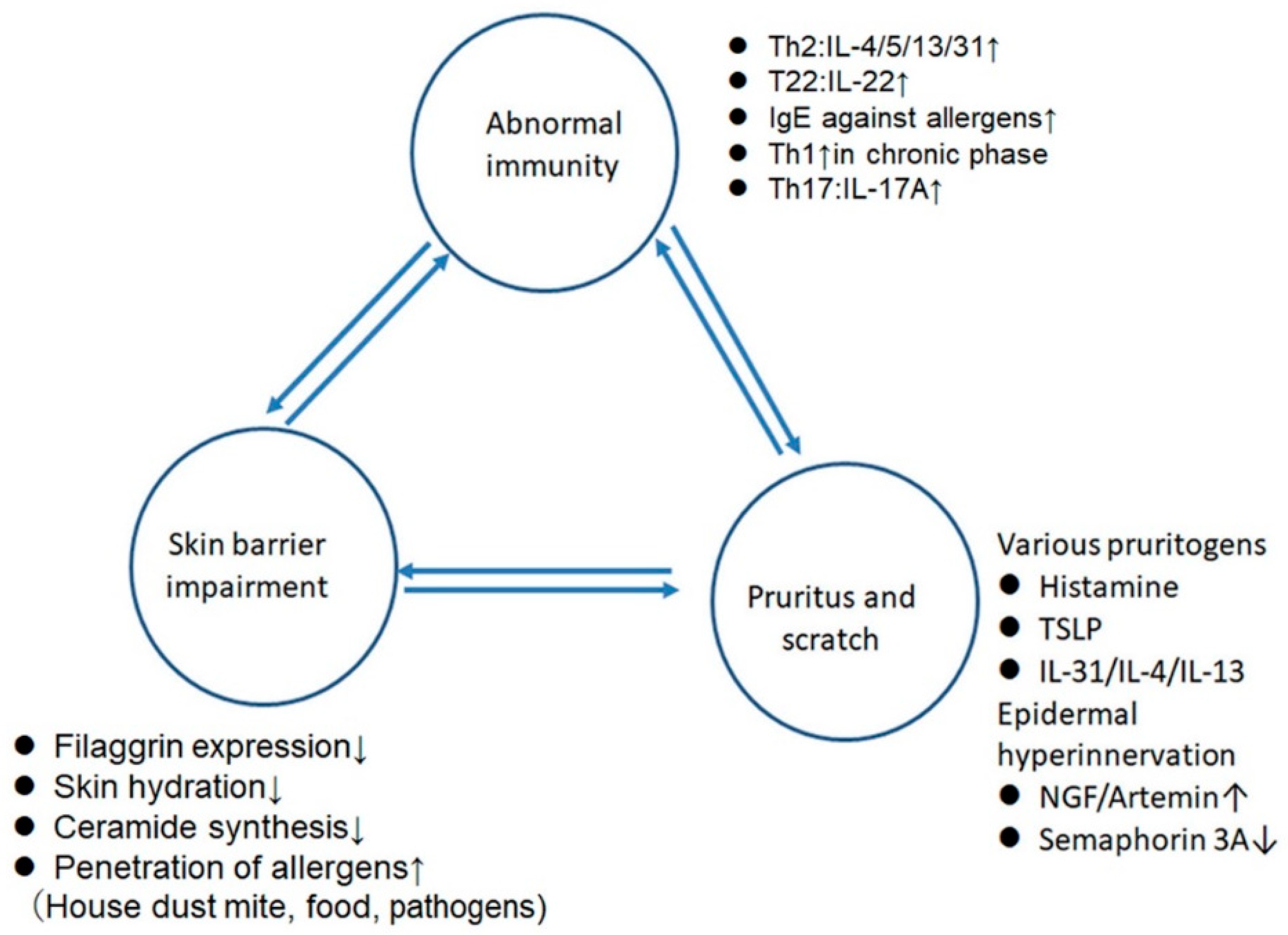
3. The Effects of Sex Hormones on the Skin Barrier (Table 1)
4. The Effects of Sex Hormones on the Pruritus
5. Intrinsic AD
6. Possible Hypotheses on the Generation-Dependent Sexual Difference in the Prevalence of Extrinsic AD (Table 6)
7. Possible Hypotheses on Female Preponderance of Intrinsic AD (Table 7)
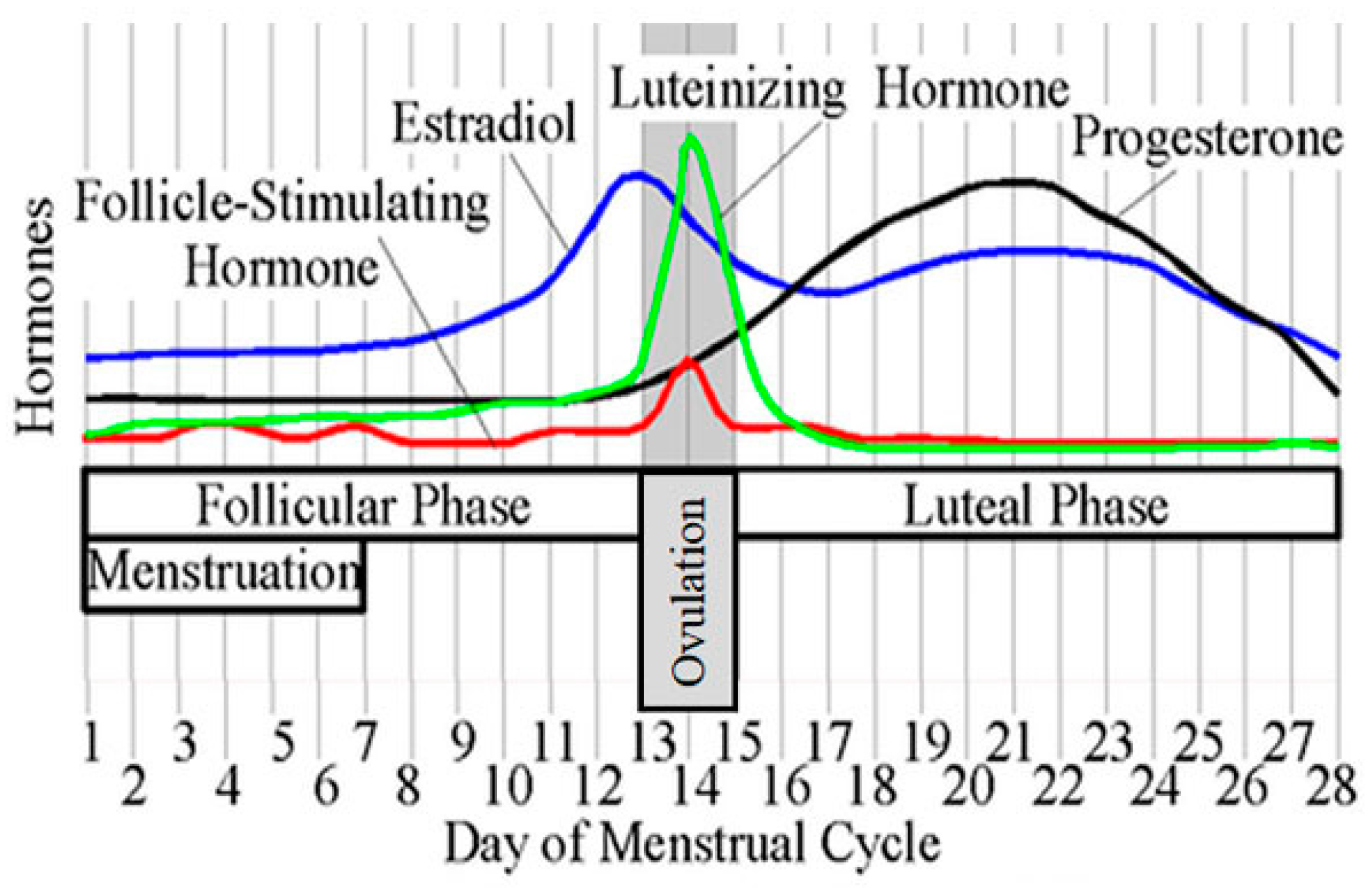
8. Possible Hypotheses on the Premenstrual Deterioration of AD in Females
9. Serum Hormone Concentrations in Patients with Allergic Diseases
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Furue, M.; Chiba, T.; Tsuji, G.; Ulzii, D.; Kido-Nakahara, M.; Nakahara, T.; Kadono, T. Atopic dermatitis: Immune deviation, barrier dysfunction, IgE autoreactivity and new therapies. Allergol. Int. 2017, 66, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Furue, M.; Ulzii, D.; Vu, Y.H.; Tsuji, G.; Kido-Nakahara, M.; Nakahara, T. Pathogenesis of Atopic Dermatitis: Current Paradigm. Iran. J. Immunol. 2019, 16, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Egawa, G.; Kabashima, K. Barrier dysfunction in the skin allergy. Allergol. Int. 2018, 67, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Yuki, T.; Tobiishi, M.; Kusaka-Kikushima, A.; Ota, Y.; Tokura, Y. Impaired tight junctions in atopic dermatitis skin and in a skin-equivalent model treated with interleukin-17. PLoS ONE 2016, 11, e0161759. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Honda, T.; Kabashima, K. Multipolarity of cytokine axes in the pathogenesis of atopic dermatitis in terms of age, race, species, disease stage and biomarkers. Int. Immunol. 2018, 30, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Dirven-Meijer, P.C.; Glazenburg, E.J.; Mulder, P.G.; Oranje, A.P. Prevalence of atopic dermatitis in children younger than 4 years in a demarcated area in central Netherlands: The West Veluwe Study Group. Br. J. Dermatol. 2008, 158, 846–847. [Google Scholar] [CrossRef] [PubMed]
- Saeki, H.; Tsunemi, Y.; Fujita, H.; Kagami, S.; Sasaki, K.; Ohmatsu, H.; Watanabe, A.; Tamaki, K. Prevalence of atopic dermatitis determined by clinical examination in Japanese adults. J. Dermatol. 2006, 33, 817–819. [Google Scholar] [CrossRef]
- Harrop, J.; Chinn, S.; Verlato, G.; Olivieri, M.; Norback, D.; Wjst, M.; Janson, C.; Zock, J.P.; Leynaert, B.; Gislason, D.; et al. Eczema, atopy and allergen exposure in adults: A population-based study. Clin. Exp. Allergy 2007, 37, 526–535. [Google Scholar] [CrossRef]
- Chen, W.; Mempel, M.; Schober, W.; Behrendt, H.; Ring, J. Gender difference, sex hormones, and immediate type hypersensitivity reactions. Allergy 2008, 63, 1418–1427. [Google Scholar] [CrossRef]
- Schatz, M.; Camargo, C.A., Jr. The relationship of sex to asthma prevalence, health care utilization, and medications in a large managed care organization. Ann. Allergy Asthma Immunol. 2003, 91, 553–558. [Google Scholar] [CrossRef]
- Schatz, M.; Clark, S.; Camargo, C.A., Jr. Sex differences in the presentation and course of asthma hospitalizations. Chest 2006, 129, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Tai, P.; Wang, J.; Jin, H.; Song, X.; Yan, J.; Kang, Y.; Zhao, L.; An, X.; Du, X.; Chen, X.; et al. Induction of regulatory T cells by physiological level estrogen. J. Cell Physiol. 2008, 214, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Kuramoto, T.; Yokoe, M.; Tanaka, D.; Yuri, A.; Nishitani, A.; Higuchi, Y.; Yoshimi, K.; Tanaka, M.; Kuwamura, M.; Hiai, H.; et al. Atopic dermatitis-like skin lesions with IgE hyperproduction and pruritus in KFRS4/Kyo rats. J. Dermatol. Sci. 2015, 80, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, P. The laboratory rat: Relating its age with human’s. Int. J. Prev. Med. 2013, 4, 624–630. [Google Scholar] [PubMed]
- Tokura, Y. Extrinsic and intrinsic types of atopic dermatitis. J. Dermatol. Sci. 2010, 58, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Farage, M.A.; Neill, S.; MacLean, A.B. Physiological changes associated with the menstrual cycle: A review. Obstet. Gynecol. Surv. 2009, 64, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Hughes, G.C. Progesterone and autoimmune disease. Autoimmun. Rev. 2012, 11, A502–514. [Google Scholar] [CrossRef]
- Roved, J.; Westerdahl, H.; Hasselquist, D. Sex differences in immune responses: Hormonal effects, antagonistic selection, and evolutionary consequences. Horm. Behav. 2017, 88, 95–105. [Google Scholar] [CrossRef]
- Solano, M.E.; Sander, V.A.; Ho, H.; Motta, A.B.; Arck, P.C. Systemic inflammation, cellular influx and up-regulation of ovarian VCAM-1 expression in a mouse model of polycystic ovary syndrome (PCOS). J. Reprod. Immunol. 2011, 92, 33–44. [Google Scholar] [CrossRef]
- Namazi, M.R. The Th1-promoting effects of dehydroepiandrosterone can provide an explanation for the stronger Th1-immune response of women. Iran. J. Allergy Asthma Immunol. 2009, 8, 65–69. [Google Scholar]
- Xiang, Y.; Jin, Q.; Li, L.; Yang, Y.; Zhang, H.; Liu, M.; Fan, C.; Li, J.; Shan, Z.; Teng, W. Physiological low-dose oestrogen promotes the development of experimental autoimmune thyroiditis through the up-regulation of Th1/Th17 responses. J. Reprod. Immunol. 2018, 126, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Dayoub, O.; Le Lay, S.; Soleti, R.; Clere, N.; Hilairet, G.; Dubois, S.; Gagnadoux, F.; Boursier, J.; Martinez, M.C.; Andriantsitohaina, R. Estrogen receptor alpha/HDAC/NFAT axis for delphinidin effects on proliferation and differentiation of T lymphocytes from patients with cardiovascular risks. Sci. Rep. 2017, 7, 9378. [Google Scholar] [CrossRef] [PubMed]
- Haghmorad, D.; Salehipour, Z.; Nosratabadi, R.; Rastin, M.; Kokhaei, P.; Mahmoudi, M.B.; Amini, A.A.; Mahmoudi, M. Medium-dose estrogen ameliorates experimental autoimmune encephalomyelitis in ovariectomized mice. J. Immunotoxicol. 2016, 13, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Garnier, L.; Laffont, S.; Lelu, K.; Yogev, N.; Waisman, A.; Guery, J.C. Estrogen Signaling in Bystander Foxp3(neg) CD4(+) T Cells Suppresses Cognate Th17 Differentiation in Trans and Protects from Central Nervous System Autoimmunity. J. Immunol. 2018, 201, 3218–3228. [Google Scholar] [CrossRef] [PubMed]
- Lasarte, S.; Elsner, D.; Guia-Gonzalez, M.; Ramos-Medina, R.; Sanchez-Ramon, S.; Esponda, P.; Munoz-Fernandez, M.A.; Relloso, M. Female sex hormones regulate the Th17 immune response to sperm and Candida albicans. Hum. Reprod. 2013, 28, 3283–3291. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lambert, K.C.; Curran, E.M.; Judy, B.M.; Milligan, G.N.; Lubahn, D.B.; Estes, D.M. Estrogen receptor alpha (ERalpha) deficiency in macrophages results in increased stimulation of CD4+ T cells while 17beta-estradiol acts through ERalpha to increase IL-4 and GATA-3 expression in CD4+ T cells independent of antigen presentation. J. Immunol. 2005, 175, 5716–5723. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Denison, M.S.; Tachibana, H.; Yamada, K. Effects of estrogenic compounds on immunoglobulin production by mouse splenocytes. Biol. Pharm. Bull. 2002, 25, 1263–1267. [Google Scholar] [CrossRef] [PubMed]
- Iannello, A.; Rolla, S.; Maglione, A.; Ferrero, G.; Bardina, V.; Inaudi, I.; De Mercanti, S.; Novelli, F.; D’Antuono, L.; Cardaropoli, S.; et al. Pregnancy epigenetic signature in T helper 17 and T regulatory cells in multiple sclerosis. Front. Immunol. 2018, 9, 3075. [Google Scholar] [CrossRef]
- Qin, J.; Li, L.; Jin, Q.; Guo, D.; Liu, M.; Fan, C.; Li, J.; Shan, Z.; Teng, W. Estrogen receptor beta activation stimulates the development of experimental autoimmune thyroiditis through up-regulation of Th17-type responses. Clin. Immunol. 2018, 190, 41–52. [Google Scholar] [CrossRef]
- Adurthi, S.; Kumar, M.M.; Vinodkumar, H.S.; Mukherjee, G.; Krishnamurthy, H.; Acharya, K.K.; Bafna, U.D.; Uma, D.K.; Abhishekh, B.; Krishna, S.; et al. Oestrogen Receptor-alpha binds the FOXP3 promoter and modulates regulatory T-cell function in human cervical cancer. Sci. Rep. 2017, 7, 17289. [Google Scholar] [CrossRef]
- Polanczyk, M.J.; Carson, B.D.; Subramanian, S.; Afentoulis, M.; Vandenbark, A.A.; Ziegler, S.F.; Offner, H. Cutting edge: Estrogen drives expansion of the CD4+CD25+ regulatory T cell compartment. J. Immunol. 2004, 173, 2227–2230. [Google Scholar] [CrossRef] [PubMed]
- Polari, L.; Wiklund, A.; Sousa, S.; Kangas, L.; Linnanen, T.; Harkonen, P.; Maatta, J. SERMs Promote Anti-Inflammatory Signaling and Phenotype of CD14+ Cells. Inflammation 2018, 41, 1157–1171. [Google Scholar] [CrossRef] [PubMed]
- Zaitsu, M.; Narita, S.; Lambert, K.C.; Grady, J.J.; Estes, D.M.; Curran, E.M.; Brooks, E.G.; Watson, C.S.; Goldblum, R.M.; Midoro-Horiuti, T. Estradiol activates mast cells via a non-genomic estrogen receptor-alpha and calcium influx. Mol. Immunol. 2007, 44, 1977–1985. [Google Scholar] [CrossRef] [PubMed]
- Matalka, K.Z. The effect of estradiol, but not progesterone, on the production of cytokines in stimulated whole blood, is concentration-dependent. Neuro Endocrinol. Lett. 2003, 24, 185–191. [Google Scholar] [PubMed]
- Polese, B.; Gridelet, V.; Araklioti, E.; Martens, H.; Perrier d’Hauterive, S.; Geenen, V. The Endocrine Milieu and CD4 T-Lymphocyte Polarization during Pregnancy. Front. Endocrinol. (Lausanne) 2014, 5, 106. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.Y.; Fan, Y.M.; Zhang, Q.; Liu, S.; Li, Q.; Ke, G.L.; Li, C.; You, Z. Estradiol inhibits Th17 cell differentiation through inhibition of RORgammaT transcription by recruiting the ERalpha/REA complex to estrogen response elements of the RORgammaT promoter. J. Immunol. 2015, 194, 4019–4028. [Google Scholar] [CrossRef] [PubMed]
- Narita, S.; Goldblum, R.M.; Watson, C.S.; Brooks, E.G.; Estes, D.M.; Curran, E.M.; Midoro-Horiuti, T. Environmental estrogens induce mast cell degranulation and enhance IgE-mediated release of allergic mediators. Environ. Health Perspect. 2007, 115, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Tajiki-Nishino, R.; Tajima, H.; Fukuyama, T. Role of estrogen receptors alpha and beta in the development of allergic airway inflammation in mice: A possible involvement of interleukin 33 and eosinophils. Toxicology 2019, 411, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Ohtsuka, H.; Tomioka, M.; Oikawa, M. Effect of progesterone on Th1/Th2/Th17 and regulatory T cell-related genes in peripheral blood mononuclear cells during pregnancy in cows. Vet. Res. Commun. 2013, 37, 43–49. [Google Scholar] [CrossRef]
- Kozma, N.; Halasz, M.; Polgar, B.; Poehlmann, T.G.; Markert, U.R.; Palkovics, T.; Keszei, M.; Par, G.; Kiss, K.; Szeberenyi, J.; et al. Progesterone-induced blocking factor activates STAT6 via binding to a novel IL-4 receptor. J. Immunol. 2006, 176, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Dong, B.; Wang, H.; Zeng, Z.; Liu, W.; Chen, N.; Chen, J.; Yang, J.; Li, D.; Duan, Y. Progesterone suppresses Th17 cell responses, and enhances the development of regulatory T cells, through thymic stromal lymphopoietin-dependent mechanisms in experimental gonococcal genital tract infection. Microbes Infect. 2013, 15, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Miyaura, H.; Iwata, M. Direct and indirect inhibition of Th1 development by progesterone and glucocorticoids. J. Immunol. 2002, 168, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Hall, O.J.; Limjunyawong, N.; Vermillion, M.S.; Robinson, D.P.; Wohlgemuth, N.; Pekosz, A.; Mitzner, W.; Klein, S.L. Progesterone-Based Therapy Protects Against Influenza by Promoting Lung Repair and Recovery in Females. PLoS Pathog. 2016, 12, e1005840. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, V.L.; Gershwin, L.J. Progesterone and environmental tobacco smoke act synergistically to exacerbate the development of allergic asthma in a mouse model. Clin. Exp. Allergy 2007, 37, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, T.K.; Heiman, J.R.; Demas, G.E. Sexual activity modulates shifts in TH1/TH2 cytokine profile across the menstrual cycle: An observational study. Fertil. Steril. 2015, 104, 1513–1521. [Google Scholar] [CrossRef]
- Lee, J.H.; Ulrich, B.; Cho, J.; Park, J.; Kim, C.H. Progesterone promotes differentiation of human cord blood fetal T cells into T regulatory cells but suppresses their differentiation into Th17 cells. J. Immunol. 2011, 187, 1778–1787. [Google Scholar] [CrossRef] [PubMed]
- Kissick, H.T.; Sanda, M.G.; Dunn, L.K.; Pellegrini, K.L.; On, S.T.; Noel, J.K.; Arredouani, M.S. Androgens alter T-cell immunity by inhibiting T-helper 1 differentiation. Proc. Natl. Acad. Sci. USA 2014, 111, 9887–9892. [Google Scholar] [CrossRef]
- Zhang, M.A.; Rego, D.; Moshkova, M.; Kebir, H.; Chruscinski, A.; Nguyen, H.; Akkermann, R.; Stanczyk, F.Z.; Prat, A.; Steinman, L.; et al. Peroxisome proliferator-activated receptor (PPAR)alpha and -gamma regulate IFNgamma and IL-17A production by human T cells in a sex-specific way. Proc. Natl. Acad. Sci. USA 2012, 109, 9505–9510. [Google Scholar] [CrossRef]
- Massa, M.G.; David, C.; Jorg, S.; Berg, J.; Gisevius, B.; Hirschberg, S.; Linker, R.A.; Gold, R.; Haghikia, A. Testosterone differentially affects T cells and neurons in murine and human models of neuroinflammation and neurodegeneration. Am. J. Pathol. 2017, 187, 1613–1622. [Google Scholar] [CrossRef]
- Vignozzi, L.; Cellai, I.; Santi, R.; Lombardelli, L.; Morelli, A.; Comeglio, P.; Filippi, S.; Logiodice, F.; Carini, M.; Nesi, G.; et al. Antiinflammatory effect of androgen receptor activation in human benign prostatic hyperplasia cells. J. Endocrinol. 2012, 214, 31–43. [Google Scholar] [CrossRef]
- Trigunaite, A.; Dimo, J.; Jorgensen, T.N. Suppressive effects of androgens on the immune system. Cell Immunol. 2015, 294, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Yamatomo, T.; Okano, M.; Ono, T.; Nakayama, E.; Yoshino, T.; Satoskar, A.R.; Harn, D.A., Jr.; Nishizaki, K. Sex-related differences in the initiation of allergic rhinitis in mice. Allergy 2001, 56, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Fuseini, H.; Yung, J.A.; Cephus, J.Y.; Zhang, J.; Goleniewska, K.; Polosukhin, V.V.; Peebles, R.S., Jr.; Newcomb, D.C. Testosterone Decreases House Dust Mite-Induced Type 2 and IL-17A-Mediated Airway Inflammation. J. Immunol. 2018, 201, 1843–1854. [Google Scholar] [CrossRef] [PubMed]
- Hepworth, M.R.; Hardman, M.J.; Grencis, R.K. The role of sex hormones in the development of Th2 immunity in a gender-biased model of Trichuris muris infection. Eur. J. Immunol. 2010, 40, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Walecki, M.; Eisel, F.; Klug, J.; Baal, N.; Paradowska-Dogan, A.; Wahle, E.; Hackstein, H.; Meinhardt, A.; Fijak, M. Androgen receptor modulates Foxp3 expression in CD4+CD25+Foxp3+ regulatory T-cells. Mol. Biol. Cell 2015, 26, 2845–2857. [Google Scholar] [CrossRef] [PubMed]
- Gubbels Bupp, M.R.; Jorgensen, T.N. Androgen-Induced Immunosuppression. Front. Immunol. 2018, 9, 794. [Google Scholar] [CrossRef]
- Fuseini, H.; Newcomb, D.C. Mechanisms Driving Gender Differences in Asthma. Curr. Allergy Asthma Rep. 2017, 17, 19. [Google Scholar] [CrossRef]
- Khurana, H.; Malhotra, P.; Sachdeva, M.U.; Varma, N.; Bose, P.; Yanamandra, U.; Varma, S.; Khadwal, A.; Lad, D.; Prakash, G. Danazol increases T regulatory cells in patients with aplastic anemia. Hematology 2018, 23, 496–500. [Google Scholar] [CrossRef]
- Liou, C.J.; Huang, W.C. Dehydroepiandrosterone suppresses eosinophil infiltration and airway hyperresponsiveness via modulation of chemokines and Th2 cytokines in ovalbumin-sensitized mice. J. Clin. Immunol. 2011, 31, 656–665. [Google Scholar] [CrossRef]
- Chan, C.C.; Liou, C.J.; Xu, P.Y.; Shen, J.J.; Kuo, M.L.; Len, W.B.; Chang, L.E.; Huang, W.C. Effect of dehydroepiandrosterone on atopic dermatitis-like skin lesions induced by 1-chloro-2,4-dinitrobenzene in mouse. J. Dermatol. Sci. 2013, 72, 149–157. [Google Scholar] [CrossRef]
- Aggelakopoulou, M.; Kourepini, E.; Paschalidis, N.; Simoes, D.C.; Kalavrizioti, D.; Dimisianos, N.; Papathanasopoulos, P.; Mouzaki, A.; Panoutsakopoulou, V. ERbeta-Dependent Direct Suppression of Human and Murine Th17 Cells and Treatment of Established Central Nervous System Autoimmunity by a Neurosteroid. J. Immunol. 2016, 197, 2598–2609. [Google Scholar] [CrossRef] [PubMed]
- Alves, V.B.; Basso, P.J.; Nardini, V.; Silva, A.; Chica, J.E.; Cardoso, C.R. Dehydroepiandrosterone (DHEA) restrains intestinal inflammation by rendering leukocytes hyporesponsive and balancing colitogenic inflammatory responses. Immunobiology 2016, 221, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Yu, L.; Zhao, J.; Ma, H. Effect of dehydroepiandrosterone on the immune function of mice in vivo and in vitro. Mol. Immunol. 2019, 112, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Tabata, N.; Tagami, H.; Terui, T. Dehydroepiandrosterone may be one of the regulators of cytokine production in atopic dermatitis. Arch. Dermatol. Res. 1997, 289, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Sudo, N.; Yu, X.N.; Kubo, C. Dehydroepiandrosterone attenuates the spontaneous elevation of serum IgE level in NC/Nga mice. Immunol. Lett. 2001, 79, 177–179. [Google Scholar] [CrossRef]
- Frantz, M.C.; Prix, N.J.; Wichmann, M.W.; van den Engel, N.K.; Hernandez-Richter, T.; Faist, E.; Chaudry, I.H.; Jauch, K.W.; Angele, M.K. Dehydroepiandrosterone restores depressed peripheral blood mononuclear cell function following major abdominal surgery via the estrogen receptors. Crit. Care Med. 2005, 33, 1779–1786. [Google Scholar] [CrossRef] [PubMed]
- Quiroga, M.F.; Angerami, M.T.; Santucci, N.; Ameri, D.; Francos, J.L.; Wallach, J.; Sued, O.; Cahn, P.; Salomon, H.; Bottasso, O. Dynamics of adrenal steroids are related to variations in Th1 and Treg populations during Mycobacterium tuberculosis infection in HIV positive persons. PLoS ONE 2012, 7, e33061. [Google Scholar] [CrossRef]
- Auci, D.; Kaler, L.; Subramanian, S.; Huang, Y.; Frincke, J.; Reading, C.; Offner, H. A new orally bioavailable synthetic androstene inhibits collagen-induced arthritis in the mouse: Androstene hormones as regulators of regulatory T cells. Ann. NY Acad. Sci. 2007, 1110, 630–640. [Google Scholar] [CrossRef]
- Sulcova, J.; Hill, M.; Hampl, R.; Starka, L. Age and sex related differences in serum levels of unconjugated dehydroepiandrosterone and its sulphate in normal subjects. J. Endocrinol. 1997, 154, 57–62. [Google Scholar] [CrossRef]
- Muller, C.R.; Migl, B.; Traupe, H.; Ropers, H.H. X-linked steroid sulfatase: Evidence for different gene-dosage in males and females. Hum. Genet. 1980, 54, 197–199. [Google Scholar] [CrossRef]
- Carlstrom, K.; Brody, S.; Lunell, N.O.; Lagrelius, A.; Mollerstrom, G.; Pousette, A.; Rannevik, G.; Stege, R.; von Schoultz, B. Dehydroepiandrosterone sulphate and dehydroepiandrosterone in serum: Differences related to age and sex. Maturitas 1988, 10, 297–306. [Google Scholar] [CrossRef]
- Zumoff, B.; Rosenfeld, R.S.; Strain, G.W.; Levin, J.; Fukushima, D.K. Sex differences in the twenty-four-hour mean plasma concentrations of dehydroisoandrosterone (DHA) and dehydroisoandrosterone sulfate (DHAS) and the DHA to DHAS ratio in normal adults. J. Clin. Endocrinol. Metab. 1980, 51, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Prough, R.A.; Clark, B.J.; Klinge, C.M. Novel mechanisms for DHEA action. J. Mol. Endocrinol. 2016, 56, R139–155. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Qiu, X.; Gui, Y.; Xu, Y.; Li, D.; Wang, L. Dehydroepiandrosterone improves the ovarian reserve of women with diminished ovarian reserve and is a potential regulator of the immune response in the ovaries. Biosci. Trends 2015, 9, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Gui, Y.; Xu, Y.; Li, D.; Wang, L. DHEA promotes osteoblast differentiation by regulating the expression of osteoblast-related genes and Foxp3(+) regulatory T cells. Biosci. Trends 2015, 9, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Firooz, A.; Sadr, B.; Babakoohi, S.; Sarraf-Yazdy, M.; Fanian, F.; Kazerouni-Timsar, A.; Nassiri-Kashani, M.; Naghizadeh, M.M.; Dowlati, Y. Variation of biophysical parameters of the skin with age, gender, and body region. Sci. World J. 2012, 2012, 386936. [Google Scholar] [CrossRef] [PubMed]
- Hanley, K.; Rassner, U.; Jiang, Y.; Vansomphone, D.; Crumrine, D.; Komuves, L.; Elias, P.M.; Feingold, K.R.; Williams, M.L. Hormonal basis for the gender difference in epidermal barrier formation in the fetal rat. Acceleration by estrogen and delay by testosterone. J. Clin. Investig. 1996, 97, 2576–2584. [Google Scholar] [CrossRef]
- Tsutsumi, M.; Denda, M. Paradoxical effects of beta-estradiol on epidermal permeability barrier homeostasis. Br. J. Dermatol. 2007, 157, 776–779. [Google Scholar] [CrossRef]
- Harvell, J.; Hussona-Saeed, I.; Maibach, H.I. Changes in transepidermal water loss and cutaneous blood flow during the menstrual cycle. Contact Derm. 1992, 27, 294–301. [Google Scholar] [CrossRef]
- Chen, Y.; Yokozeki, H.; Katagiri, K. Physiological and functional changes in the stratum corneum restored by oestrogen in an ovariectomized mice model of climacterium. Exp. Dermatol. 2017, 26, 394–401. [Google Scholar] [CrossRef]
- Hung, C.F.; Chen, W.Y.; Aljuffali, I.A.; Lin, Y.K.; Shih, H.C.; Fang, J.Y. Skin aging modulates percutaneous drug absorption: The impact of ultraviolet irradiation and ovariectomy. Age (Dordr.) 2015, 37, 21. [Google Scholar] [CrossRef] [PubMed]
- Kao, J.S.; Garg, A.; Mao-Qiang, M.; Crumrine, D.; Ghadially, R.; Feingold, K.R.; Elias, P.M. Testosterone perturbs epidermal permeability barrier homeostasis. J. Investig. Dermatol. 2001, 116, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Hanley, K.; Jiang, Y.; Holleran, W.M.; Elias, P.M.; Williams, M.L.; Feingold, K.R. Glucosylceramide metabolism is regulated during normal and hormonally stimulated epidermal barrier development in the rat. J. Lipid Res. 1997, 38, 576–584. [Google Scholar] [PubMed]
- Komuves, L.G.; Hanley, K.; Jiang, Y.; Elias, P.M.; Williams, M.L.; Feingold, K.R. Ligands and activators of nuclear hormone receptors regulate epidermal differentiation during fetal rat skin development. J. Investig. Dermatol. 1998, 111, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Muizzuddin, N.; Marenus, K.D.; Schnittger, S.F.; Sullivan, M.; Maes, D.H. Effect of systemic hormonal cyclicity on skin. J. Cosmet. Sci. 2005, 56, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Bonamonte, D.; Foti, C.; Antelmi, A.R.; Biscozzi, A.M.; Naro, E.D.; Fanelli, M.; Loverro, G.; Angelini, G. Nickel contact allergy and menstrual cycle. Contact derm. 2005, 52, 309–313. [Google Scholar] [CrossRef]
- Hong, S.H.; Lee, J.E.; Jeong, J.J.; Hwang, S.J.; Bae, S.N.; Choi, J.Y.; Song, H. Small proline-rich protein 2 family is a cluster of genes induced by estrogenic compounds through nuclear estrogen receptors in the mouse uterus. Reprod. Toxicol. 2010, 30, 469–476. [Google Scholar] [CrossRef]
- Hernandez-Monge, J.; Garay, E.; Raya-Sandino, A.; Vargas-Sierra, O.; Diaz-Chavez, J.; Popoca-Cuaya, M.; Lambert, P.F.; Gonzalez-Mariscal, L.; Gariglio, P. Papillomavirus E6 oncoprotein up-regulates occludin and ZO-2 expression in ovariectomized mice epidermis. Exp. Cell Res. 2013, 319, 2588–2603. [Google Scholar] [CrossRef]
- Zhou, Z.; Bian, C.; Luo, Z.; Guille, C.; Ogunrinde, E.; Wu, J.; Zhao, M.; Fitting, S.; Kamen, D.L.; Oates, J.C.; et al. Progesterone decreases gut permeability through upregulating occludin expression in primary human gut tissues and Caco-2 cells. Sci. Rep. 2019, 9, 8367. [Google Scholar] [CrossRef]
- Baulieu, E.E.; Thomas, G.; Legrain, S.; Lahlou, N.; Roger, M.; Debuire, B.; Faucounau, V.; Girard, L.; Hervy, M.P.; Latour, F.; et al. Dehydroepiandrosterone (DHEA), DHEA sulfate, and aging: Contribution of the DHEAge Study to a sociobiomedical issue. Proc. Natl. Acad. Sci. USA 2000, 97, 4279–4284. [Google Scholar] [CrossRef]
- El-Alfy, M.; Deloche, C.; Azzi, L.; Bernard, B.A.; Bernerd, F.; Coutet, J.; Chaussade, V.; Martel, C.; Leclaire, J.; Labrie, F. Skin responses to topical dehydroepiandrosterone: Implications in antiageing treatment? Br. J. Dermatol. 2010, 163, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Calvo, E.; Luu-The, V.; Morissette, J.; Martel, C.; Labrie, C.; Bernard, B.; Bernerd, F.; Deloche, C.; Chaussade, V.; Leclaire, J.; et al. Pangenomic changes induced by DHEA in the skin of postmenopausal women. J. Steroid Biochem. Mol. Biol. 2008, 112, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, D.; Dietze, R.; Shihan, M.; Kirch, U.; Scheiner-Bobis, G. Dehydroepiandrosterone Sulfate Stimulates Expression of Blood-Testis-Barrier Proteins Claudin-3 and -5 and Tight Junction Formation via a Gnalpha11-Coupled Receptor in Sertoli Cells. PLoS ONE 2016, 11, e0150143. [Google Scholar] [CrossRef] [PubMed]
- Yosipovitch, G.; Rosen, J.D.; Hashimoto, T. Itch: From mechanism to (novel) therapeutic approaches. J. Allergy Clin. Immunol. 2018, 142, 1375–1390. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Makino, E.; Tajiki-Nishino, R.; Koyama, A.; Tajima, H.; Ishimota, M.; Fukuyama, T. Involvement of estrogen receptor alpha in pro-pruritic and pro-inflammatory responses in a mouse model of allergic dermatitis. Toxicol. Appl. Pharmacol. 2018, 355, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Ulmann, L.; Rodeau, J.L.; Danoux, L.; Contet-Audonneau, J.L.; Pauly, G.; Schlichter, R. Dehydroepiandrosterone and neurotrophins favor axonal growth in a sensory neuron-keratinocyte coculture model. Neuroscience 2009, 159, 514–525. [Google Scholar] [CrossRef]
- Pediaditakis, I.; Iliopoulos, I.; Theologidis, I.; Delivanoglou, N.; Margioris, A.N.; Charalampopoulos, I.; Gravanis, A. Dehydroepiandrosterone: An ancestral ligand of neurotrophin receptors. Endocrinology 2015, 156, 16–23. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Kabashima-Kubo, R.; Bito, T.; Sakabe, J.; Shimauchi, T.; Ito, T.; Hirakawa, S.; Hirasawa, N.; Ogasawara, K.; Tokura, Y. High frequencies of positive nickel/cobalt patch tests and high sweat nickel concentration in patients with intrinsic atopic dermatitis. J. Dermatol. Sci. 2013, 72, 240–245. [Google Scholar] [CrossRef]
- Hindsen, M.; Christensen, O.B.; Moller, H. Nickel levels in serum and urine in five different groups of eczema patients following oral ingestion of nickel. Acta Derm. Venereol. 1994, 74, 176–178. [Google Scholar] [CrossRef]
- Suarez-Farinas, M.; Dhingra, N.; Gittler, J.; Shemer, A.; Cardinale, I.; de Guzman Strong, C.; Krueger, J.G.; Guttman-Yassky, E. Intrinsic atopic dermatitis shows similar TH2 and higher TH17 immune activation compared with extrinsic atopic dermatitis. J. Allergy Clin. Immunol. 2013, 132, 361–370. [Google Scholar] [CrossRef]
- Minang, J.T.; Troye-Blomberg, M.; Lundeberg, L.; Ahlborg, N. Nickel elicits concomitant and correlated in vitro production of Th1-, Th2-type and regulatory cytokines in subjects with contact allergy to nickel. Scand. J. Immunol. 2005, 62, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Summer, B.; Stander, S.; Thomas, P. Cytokine patterns in vitro, in particular IL-5/IL-8 ratio, to detect patients with nickel contact allergy. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Czarnobilska, E.; Jenner, B.; Kaszuba-Zwoinska, J.; Kapusta, M.; Obtulowicz, K.; Thor, P.; Spiewak, R. Contact allergy to nickel: Patch test score correlates with IL-5, but not with IFN-gamma nickel-specific secretion by peripheral blood lymphocytes. Ann. Agric. Environ. Med. 2009, 16, 37–41. [Google Scholar] [PubMed]
- Ott, H.; Stanzel, S.; Ocklenburg, C.; Merk, H.F.; Baron, J.M.; Lehmann, S. Total serum IgE as a parameter to differentiate between intrinsic and extrinsic atopic dermatitis in children. Acta Derm. Venereol. 2009, 89, 257–261. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kusel, M.M.; Holt, P.G.; de Klerk, N.; Sly, P.D. Support for 2 variants of eczema. J. Allergy Clin. Immunol. 2005, 116, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Zafrir, Y.; Trattner, A.; Hodak, E.; Eldar, O.; Lapidoth, M.; Ben Amitai, D. Patch testing in Israeli children with suspected allergic contact dermatitis: A retrospective study and literature review. Pediatr. Dermatol. 2018, 35, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Vahter, M.; Berglund, M.; Akesson, A.; Liden, C. Metals and women’s health. Environ. Res. 2002, 88, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Cydulka, R.K.; Stahmer, S.A.; Woodruff, P.G.; Camargo, C.A., Jr. Sex differences among adults presenting to the emergency department with acute asthma. Multicenter Asthma Research Collaboration Investigators. Arch. Intern. Med. 1999, 159, 1237–1243. [Google Scholar] [CrossRef]
- Pagtakhan, R.D.; Bjelland, J.C.; Landau, L.I.; Loughlin, G.; Kaltenborn, W.; Seeley, G.; Taussig, L.M. Sex differences in growth patterns of the airways and lung parenchyma in children. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1984, 56, 1204–1210. [Google Scholar] [CrossRef]
- Sears, M.R.; Burrows, B.; Flannery, E.M.; Herbison, G.P.; Holdaway, M.D. Atopy in childhood. I. Gender and allergen related risks for development of hay fever and asthma. Clin. Exp. Allergy 1993, 23, 941–948. [Google Scholar] [CrossRef]
- Mohrenschlager, M.; Schafer, T.; Huss-Marp, J.; Eberlein-Konig, B.; Weidinger, S.; Ring, J.; Behrendt, H.; Kramer, U. The course of eczema in children aged 5–7 years and its relation to atopy: Differences between boys and girls. Br. J. Dermatol. 2006, 154, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Sennhauser, F.H.; Kuhni, C.E. Prevalence of respiratory symptoms in Swiss children: Is bronchial asthma really more prevalent in boys? Pediatr. Pulmonol. 1995, 19, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Biro, F.M.; Huang, B.; Chandler, D.W.; Fassler, C.L.; Pinney, S.M. Impact of pubertal maturation and chronologic age on sex steroids in peripubertal girls. J. Clin. Endocrinol. Metab. 2019, 104, 2971–2977. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, Y.; Aoki, N.; Suzuki, Y.; Ohno, Y.; Imamura, M.; Saika, T.; Sinohara, H.; Yamamoto, T. Sex-steroid-binding plasma protein (SBP), testosterone, oestradiol and dehydroepiandrosterone (DHEA) in prepuberty and puberty. Acta Endocrinol. (Copenh) 1987, 114, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Rouskova, D.; Mittmann, K.; Schumacher, U.; Dietrich, H.; Zimmermann, T. Effectiveness, tolerability and acceptance of a low-dosed estradiol/dienogest formulation (Lafamme 1 mg/2 mg) for the treatment of menopausal complaints: A non-interventional observational study over 6 cycles of 28 days. Gynecol. Endocrinol. 2015, 31, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Yamaura, K.; Tomono, A.; Suwa, E.; Ueno, K. Sex-related differences in SLIGRL-induced pruritus in mice. Life Sci. 2014, 94, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Kitagaki, H.; Ono, N.; Hayakawa, K.; Kitazawa, T.; Watanabe, K.; Shiohara, T. Repeated elicitation of contact hypersensitivity induces a shift in cutaneous cytokine milieu from a T helper cell type 1 to a T helper cell type 2 profile. J. Immunol. 1997, 159, 2484–2491. [Google Scholar] [PubMed]
- Raghunath, R.S.; Venables, Z.C.; Millington, G.W. The menstrual cycle and the skin. Clin. Exp. Dermatol. 2015, 40, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Kemmett, D.; Tidman, M.J. The influence of the menstrual cycle and pregnancy on atopic dermatitis. Br. J. Dermatol. 1991, 125, 59–61. [Google Scholar] [CrossRef]
- Cho, S.; Kim, H.J.; Oh, S.H.; Park, C.O.; Jung, J.Y.; Lee, K.H. The influence of pregnancy and menstruation on the deterioration of atopic dermatitis symptoms. Ann. Dermatol. 2010, 22, 180–185. [Google Scholar] [CrossRef]
- Ebata, T.; Itamura, R.; Aizawa, H.; Niimura, M. Serum sex hormone levels in adult patients with atopic dermatitis. J. Dermatol. 1996, 23, 603–605. [Google Scholar] [CrossRef] [PubMed]
- Kasperska-Zajac, A.; Brzoza, Z.; Rogala, B. Dehydroepiandrosterone and dehydroepiandrosterone sulphate in atopic allergy and chronic urticaria. Inflammation 2008, 31, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Opstad, P.K. The hypothalamo-pituitary regulation of androgen secretion in young men after prolonged physical stress combined with energy and sleep deprivation. Acta Endocrinol. (Copenh) 1992, 127, 231–236. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Hormones | Th1 | Th2 | Th17 | Treg | Skin Barrier Impairment |
|---|---|---|---|---|---|
| Androgen | ↓ | ↓ | ↓ | ↑ | ↑ |
| Estrogen | ↑~⬇ | ⬆ | ↑~⬇ | ⬆ | ⬇ |
| Progesterone | ⬇ | ⬆ | ⬇ | ⬆ | ↑ |
| DHEA | ↑ | ↓ | ? | ? | ? |
| Total activity | F < M | F ≫ M | ? | ? | F < M |
| Effects | Vivo/ Vitro | Species | Th Activities | References |
|---|---|---|---|---|
| Adaptive Immunity | ||||
| T-bet↑ | vivo | mice | Th1↑ | [21] * |
| IFN-γ↑ | vivo | mice | Th1↑ | [21] * |
| IL-12↑ | vivo | mice | Th1↑ | [21] * |
| T-bet↓ | vitro | human | Th1↓ | [22] ** |
| T-bet↓ | vivo | mice | Th1↓ | [23] † |
| IFN-γ↓ | vivo | mice | Th1↓ | [24] † |
| IFN-γ↓ | vitro | mice | Th1↓ | [25] § |
| IFN-γ↓ | vitro | human | Th1↓ | [22] ** |
| IFN-γ↓ | vivo | mice | Th1↓ | [23] † |
| GATA-3↑ | vivo | mice | Th2↑ | [26] |
| GATA-3↑ | vivo | mice | Th2↑ | [23] † |
| IL-4↑ | vivo | mice | Th2↑ | [26] |
| IL-4↑ | vivo | mice | Th2↑ | [23] † |
| B cells IgM, IgE↑ | vitro | mice | Th2↑ | [27] |
| RORc↓ | vivo | mice | Th17↓ | [24] † |
| RORc↓ | vitro | human | Th17↓ | [28] |
| RORc↓ | vitro/vivo | mice | Th17↓ | [25] |
| RORγt↓ | vitro | human | Th17↓ | [22] ** |
| RORγt↓ | vivo | mice | Th17↓ | [23] † |
| IL-17A↓ | vitro | mice | Th17↓ | [25] § |
| IL-17A↓ | vivo | mice | Th17↓ | [24] † |
| IL-17A↓ | vitro | human | Th17↓ | [22] ** |
| IL-17A↓ | vivo | mice | Th17↓ | [23] † |
| IL-22↓ | vitro | mice | Th17↓ | [25] § |
| IL-17A↑ | vivo | mice | Th17↑ | [29] ‡ |
| IL-21↑ | vivo | mice | Th17↑ | [29] ‡ |
| IL-22↑ | vivo | mice | Th17↑ | [21] * |
| RORγt↑ | vivo | mice | Th17↑ | [21] * |
| RORγt↑ | vivo | mice | Th17↑ | [29] ‡ |
| Foxp3↑ | vitro | human | Treg↑ | [28] |
| Foxp3↑ | vitro | human | Treg↑ | [30] |
| Foxp3↑ | vivo | mice | Treg↑ | [23] † |
| Foxp3↑ | vivo/vitro | mice | Treg↑ | [31] |
| IL-10↑ | vivo | mice | Treg↑ | [23] † |
| IL10↑ | vitro | mice | Treg↑ | [12] |
| TGF-↑ | vivo | mice | Treg↑ | [23] † |
| Foxp3↓ | vitro | human | Treg↓ | [22] ** |
| IL-10↓ | vitro | human | Treg↓ | [22] ** |
| Innate immunity | ||||
| Macrophage IL-10↑ | vitro | human | Th2↑ | [32] |
| Macrophage IL-1RA↑ | vitro | human | Th2↑ | [32] |
| Macrophage CD192↑ | vitro | human | Th2↑ | [32] |
| Mast cell degranulation↑ | vitro | mice | Th2↑ | [33] |
| Effects | Vivo/Vitro | Species | Th Activities | References |
|---|---|---|---|---|
| Adaptive Immunity | ||||
| T-bet↓ | vitro | cows | Th1↓ | [39] |
| IFN-γ↓ | vitro | cows | Th1↓ | [39] |
| PIBF- STAT6↑ | vitro | human | Th2↑ | [40] |
| GATA3↑ | vivo/vitro | mice | Th2↑ | [41] |
| IL-4↑ | ex vivo | mice | Th2↑ | [42] |
| IL-4↑ | vitro | cows | Th2↑ | [39] |
| IL-4↑ | vivo | mice | Th2↑ | [42] |
| B cell IgE↑ | vivo | mice | Th2↑ | [42] |
| Vaginal epithelial cell TSLP↑ | vivo/vitro | mice | Th2↑ | [41] |
| STAT3 RORC CCR6 IL-23R IL-6R AHR↓ | vitro | human | Th17↓ | [43] |
| RORγt↓ | vivo/ vitro | mice | Th17↓ | [41] |
| RORC↓ | vitro | cows | Th17↓ | [39] |
| IL-17A↓ | vitro | human | Th17↓ | [43] |
| IL-17A↓ | vitro | cows | Th17↓ | [39] |
| IL-17F↓ | vitro | human | Th17↓ | [43] |
| IL-21↓ | vitro | human | Th17↓ | [43] |
| CD39+ regulatory Th17↑ | vivo | mice | Th17↑※ | [44] |
| IL-17A↑ | vivo | mice | Th17↑※ | [44] |
| IL-22↑ | vivo | mice | Th17↑※ | [44] |
| IL-23↑ | vivo | mice | Th17↑※ | [44] |
| IL-6↑ | vivo | mice | Th17↑※ | [44] |
| TGF-β↑ | vivo | mice | Th17↑※ | [44] |
| Foxp3↑ | vitro | mice | Treg↑ | [43] |
| Foxp3↑ | vivo/vitro | mice | Treg↑ | [41] |
| Innate Immunity | ||||
| Airway epithelial cells Amphiregulin↑ | vivo | mice | [44] | |
| Effects | Vivo/Vitro | Species | Th Activities | References |
|---|---|---|---|---|
| Adaptive Immunity | ||||
| ptpn1↑ STAT4↓ | vivo/ vitro | human and mice | Th1↓ | [47] |
| PPARα↑ | vitro/vivo | mice | Th1↓ | [48] |
| IFN-γ↓ | vitro/vivo | mice | Th1↓ | [48] |
| IFN-γ↓ | vitro | mice | Th1↓ | [49] |
| IFN-γ↓ | vitro | human | Th1↓ | [50] |
| IL-12↓ | vitro | human | Th1↓ | [50] |
| CXCL10↓ | vitro | human | Th1↓ | [50] |
| IL-13↓ | vitro | human | Th2↓ | [50] |
| IL-4↓ | vitro | human | Th2↓ | [50] |
| IL-5↓ | vitro | human | Th2↓ | [50] |
| B cell number↓ | vivo | mice | Th2↓ | [51] |
| B cell Antigen-specific IgE production ↓ | vivo | mice | Th2↓ | [52] |
| PPARγ↓ | vitro/vivo | mice | Th17↑ | [48] |
| IL-17A↑ | vitro/vivo | mice | Th17↑ | [48] |
| IL-23R↓ | vivo | mice | Th17↓ | [53] |
| IL-23R↓ | vitro | Mice | Th17↓ | [54] |
| IL-17A↓ | vitro | mice | Th17↓ | [49] |
| IL-17A↓ | vivo | mice | Th17↓ | [53] |
| IL-17A↓ | vitro | human | Th17↓ | [50] |
| ARE-Foxp3↑ | vitro | human | Treg↑ | [55] |
| IL-10↑ | vitro | human | Treg↑ | [50] |
| Innate Immunity | ||||
| Mast cell IL-4↓ | vivo | mice | Th2↓ | [53] |
| ILC2 IL-13↓ | vivo | mice | Th2↓ | [53] |
| Basophil IL-4↓ | vivo | mice | Th2↓ | [53] |
| Effects | Vivo/Vitro | Species | Th Activities | References |
|---|---|---|---|---|
| Adaptive Immunity | ||||
| IFN-γ↑ | vivo | mice | Th1↑ | [59] |
| IFN-γ↑ | ex vivo | mice | Th1↑ | [60] |
| IFN-γ↑ | vitro | mice | Th1↑ | [19] |
| IL-12↑ | vitro | mice | Th1↑ | [10] |
| IFN-γ↓ | vivo | mice | Th1↓ | [61] * |
| IFN-γ↓ | vivo | mice | Th1↓ | [62] |
| IL-4↓ | vivo | mice | Th2↓ | [59] |
| IL-4↓ | vitro | mice | Th2↓ | [63] |
| IL-4↓ | vitro | human | Th2↓ | [64] |
| IL-4↓ | vivo | mice | Th2↓ | [61] * |
| IL-4↓ | vivo | mice | Th2↓ | [60] |
| IL-4↓ | ex vivo | mice | Th2↓ | [60] |
| IL-5↓ | vivo | mice | Th2↓ | [59] |
| IL-5↓ | vivo | mice | Th2↓ | [62] |
| IL-5↓ | vitro | human | Th2↓ | [64] |
| IL-5↓ | ex vivo | mice | Th2↓ | [60] |
| IL-13↓ | vivo | mice | Th2↓ | [59] |
| CCL11↓ | vivo | mice | Th2↓ | [59] |
| CCL24↓ | vivo | mice | Th2↓ | [59] |
| B cell IgE ↓ | vivo | mice | Th2↓ | [65] |
| B cell IgE ↓ | vivo | mice | Th2↓ | [60] |
| B cell IgG1↓ | vivo | mice | [60] | |
| IL-13↑ | vivo | mice | Th2↑ | [62] |
| RORC↓ | vivo | mice | Th17↓ | [61] * |
| IL-17A↓ | vivo | mice | Th17↓ | [61] * |
| IL-17A↓ | vivo | mice | Th17↓ | [62] |
| TNF-α↓ | vivo | mice | Th17↓ | [60] |
| IL-6↓ | vivo | mice | Th17↓ | [62] |
| TGFβ↓ | vivo | mice | Th17↓ | [62] |
| TNF-α↑ | vitro | human | Th17↑ | [66] † |
| IL-6↑ | vitro | human | Th17↑ | [66] † |
| IL-1β↑ | vitro | human | Th17↑ | [66] † |
| Foxp3↓ | vitro | human | Treg↓ | [67] |
| IL-10↓ | vitro | mice | Treg↓ | [62] |
| IL-10↓ | vitro | mice | Treg↓ | [63] |
| Foxp3↑ | vivo | mice | Treg↑ | [68] ** |
| IL-10↑ | vivo | mice | Treg↑ | [61] * |
| Innate Immunity | ||||
| Mast cell infiltration↓ | vivo | mice | Th2↓ | [60] |
| Eosinophil infiltration↓ | vivo | mice | Th2↓ | [59] |
| Eosinophil infiltration↓ | vivo | mice | Th2↓ | [60] |
| HaCat cells CCL17↓ | vitro | human | Th2↓ | [60] |
| HaCat cells CCL22↓ | vitro | human | Th2↓ | [60] |
| BEAS-2B CCL11↓ | vitro | human | Th2↓ | [59] |
| BEAS-2B CCL24↓ | vitro | human | Th2↓ | [59] |
| Ovary granulosa cell ICAM1/VCAM1↑ | vivo | mice | [19] | |
| Child | Adolescent–Adult | |||||
| Atopic Asthma | Atopic Diathesis | Th2 Regulation by DHEA | Th2 Regulation by Sex Hormones | Th2 Regulation by DHEA | ||
| M | +~++ | ↓ | ↓↓by A | ↓ | ||
| F | + | ↓↓ | ↑↑↑↑by E, P | ↓↓ | ||
| Prevalence | M > F | M ≪ F | ||||
| Extrinsic AD | Atopic Diathesis | Filaggrin Gene Nutation | Th2 Regulation by DHEA | Th2 Regulation by Sex Hormones | Th2 Regulation by DHEA | Skin Barrier Impairment by Sex Hormones |
| M | +~++ | + or − | ↓ | ↓↓by A | ↓ | ↑by A |
| F | + | + or − | ↓↓ | ↑↑↑↑by E, P | ↓↓ | ↓↓by E ↑by P Totally↓ |
| Prevalence | M > F | M < F | ||||
| Intrinsic AD | Child | Adolescent–Adult | |||||
|---|---|---|---|---|---|---|---|
| Atopic Diathesis | Filaggrin Gene Nutation | Exposure to Ni | Stimulation of Th1 Response to Ni by DHEA | Exposure to Ni | Regulation of Th2 Response to Ni by Sex Hormones | Stimulation of Th1 Response to Ni by DHEA | |
| M | − | − | + | ↑ | + | ↓↓by A | ↑ |
| F | − | − | +~++ | ↑↑ | ++ | ↑↑↑↑by E, P | ↑↑ |
| Prevalence | M < F | M ≪ F | |||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanda, N.; Hoashi, T.; Saeki, H. The Roles of Sex Hormones in the Course of Atopic Dermatitis. Int. J. Mol. Sci. 2019, 20, 4660. https://doi.org/10.3390/ijms20194660
Kanda N, Hoashi T, Saeki H. The Roles of Sex Hormones in the Course of Atopic Dermatitis. International Journal of Molecular Sciences. 2019; 20(19):4660. https://doi.org/10.3390/ijms20194660
Chicago/Turabian StyleKanda, Naoko, Toshihiko Hoashi, and Hidehisa Saeki. 2019. "The Roles of Sex Hormones in the Course of Atopic Dermatitis" International Journal of Molecular Sciences 20, no. 19: 4660. https://doi.org/10.3390/ijms20194660
APA StyleKanda, N., Hoashi, T., & Saeki, H. (2019). The Roles of Sex Hormones in the Course of Atopic Dermatitis. International Journal of Molecular Sciences, 20(19), 4660. https://doi.org/10.3390/ijms20194660