A Free Web-Based Protocol to Assist Structure-Based Virtual Screening Experiments


 , ,
, , 


Abstract
1. Introduction
2. Results and Discussion
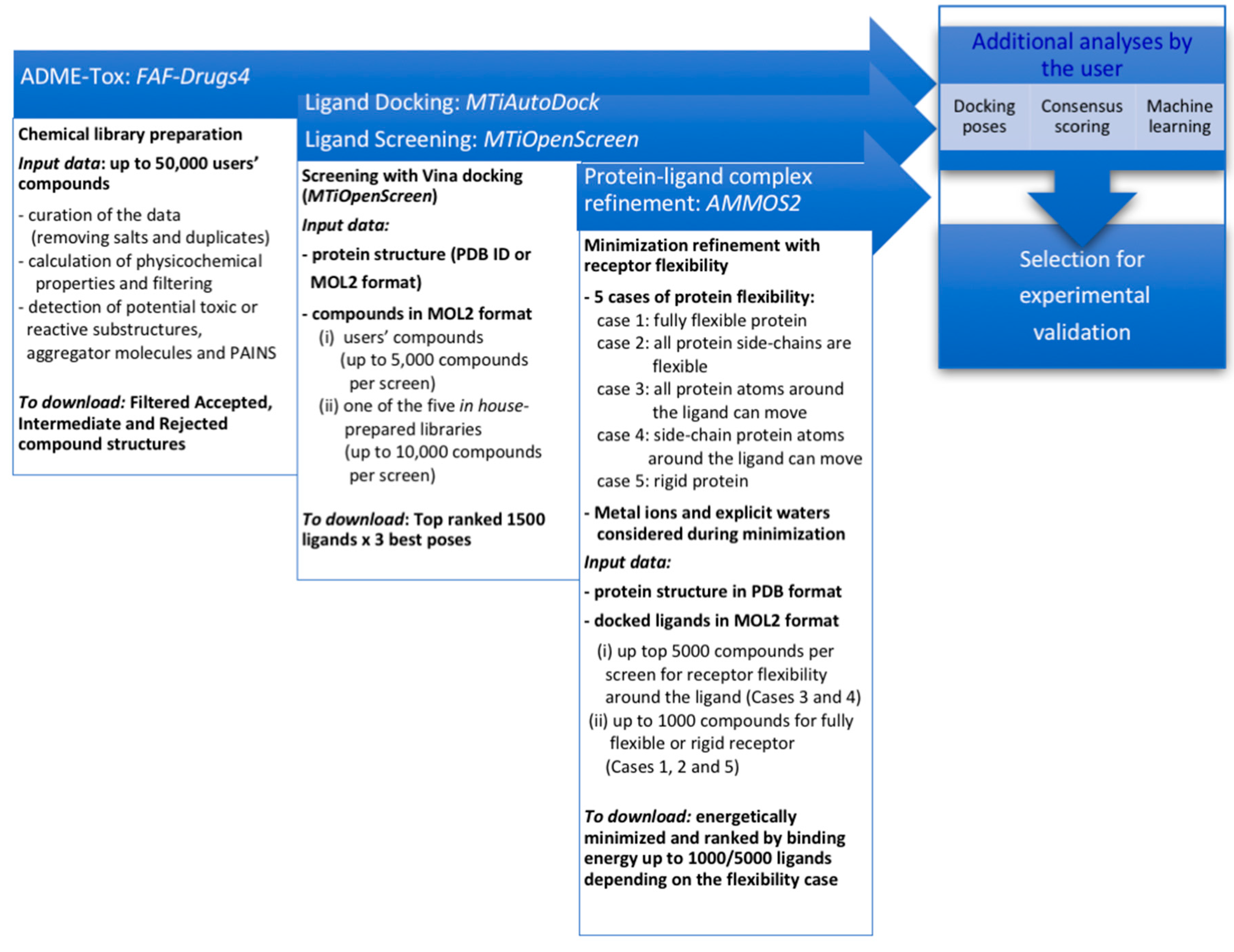
2.1. A Free Web-Based Protocol Dedicated to Small-Molecule Virtual Screening
2.1.1. Chemical Compound Preparation
2.1.2. Docking-Based Virtual Screening
2.1.3. Molecular Mechanics Refinement
2.2. Screening of Approved Drugs Using the Web-Based Protocol for Drug Repositioning
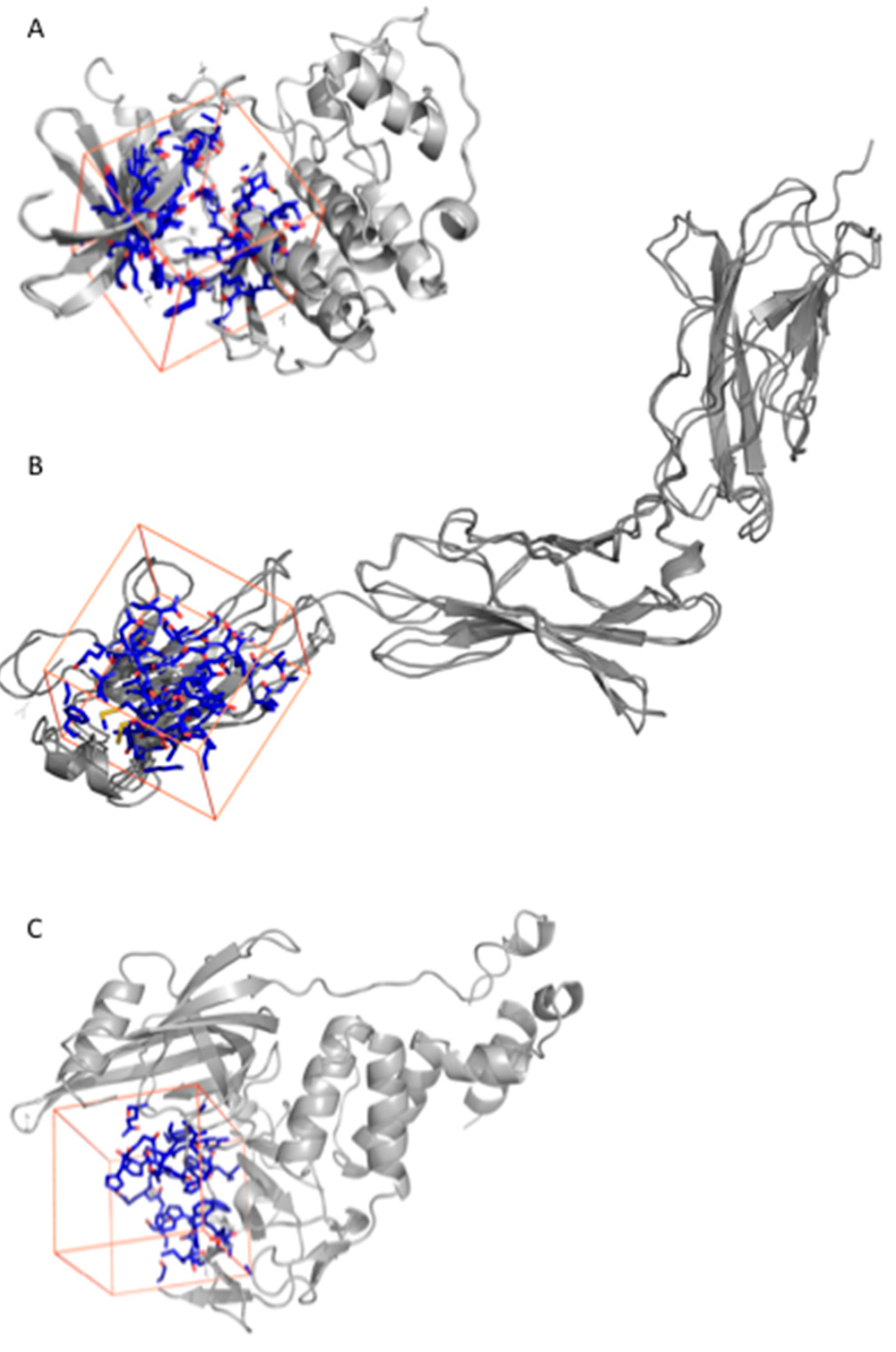
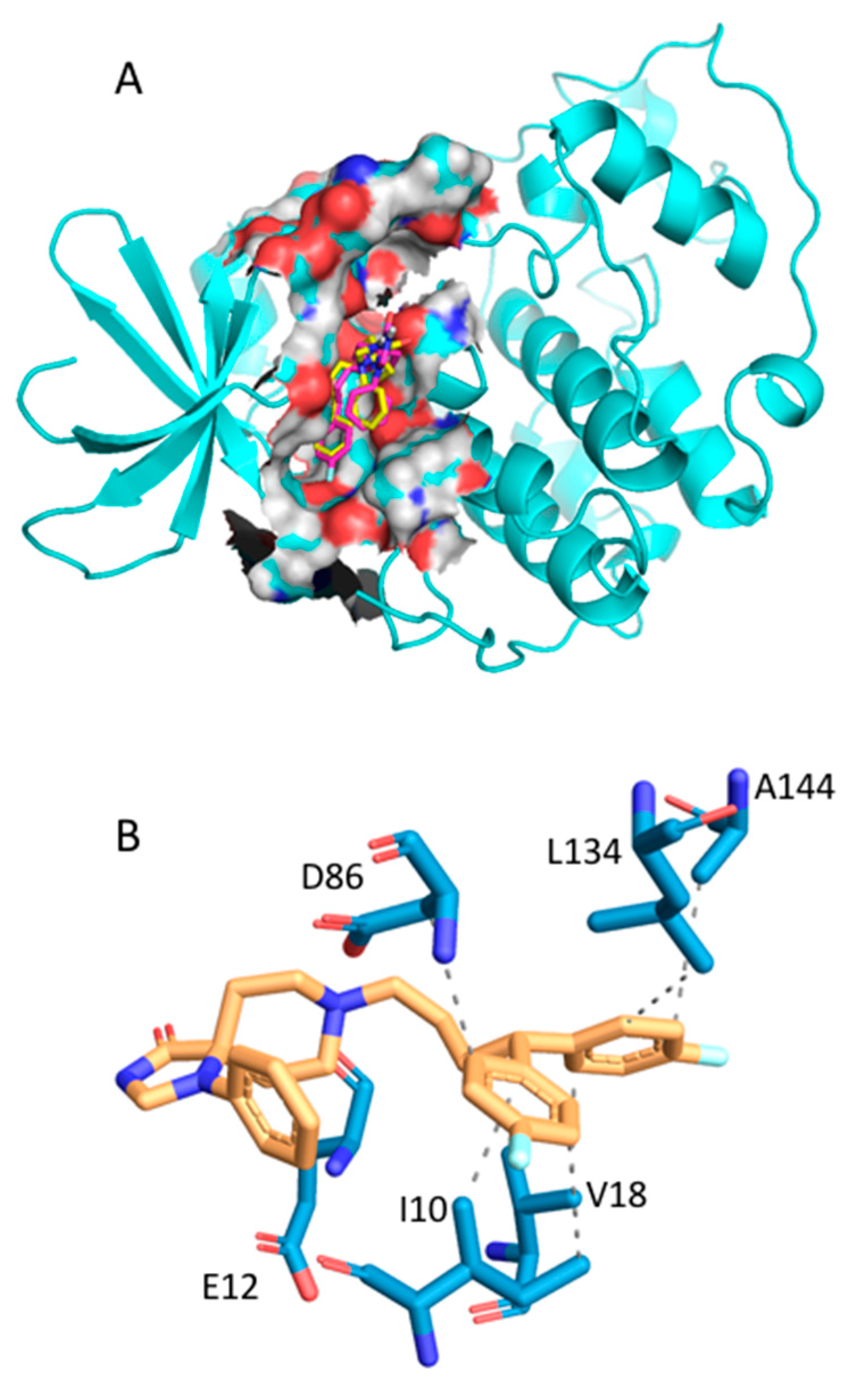
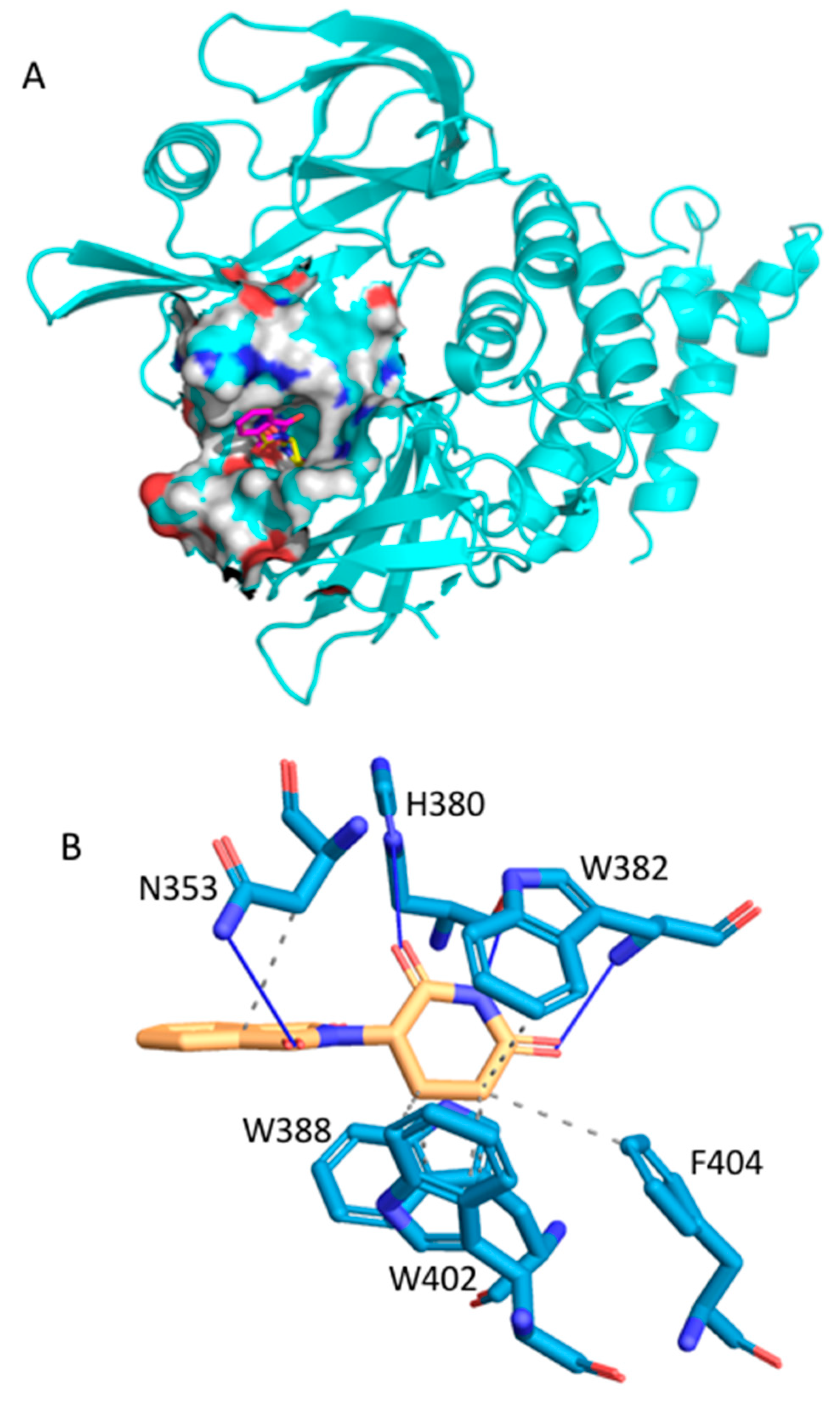
2.2.1. Fluspirilene
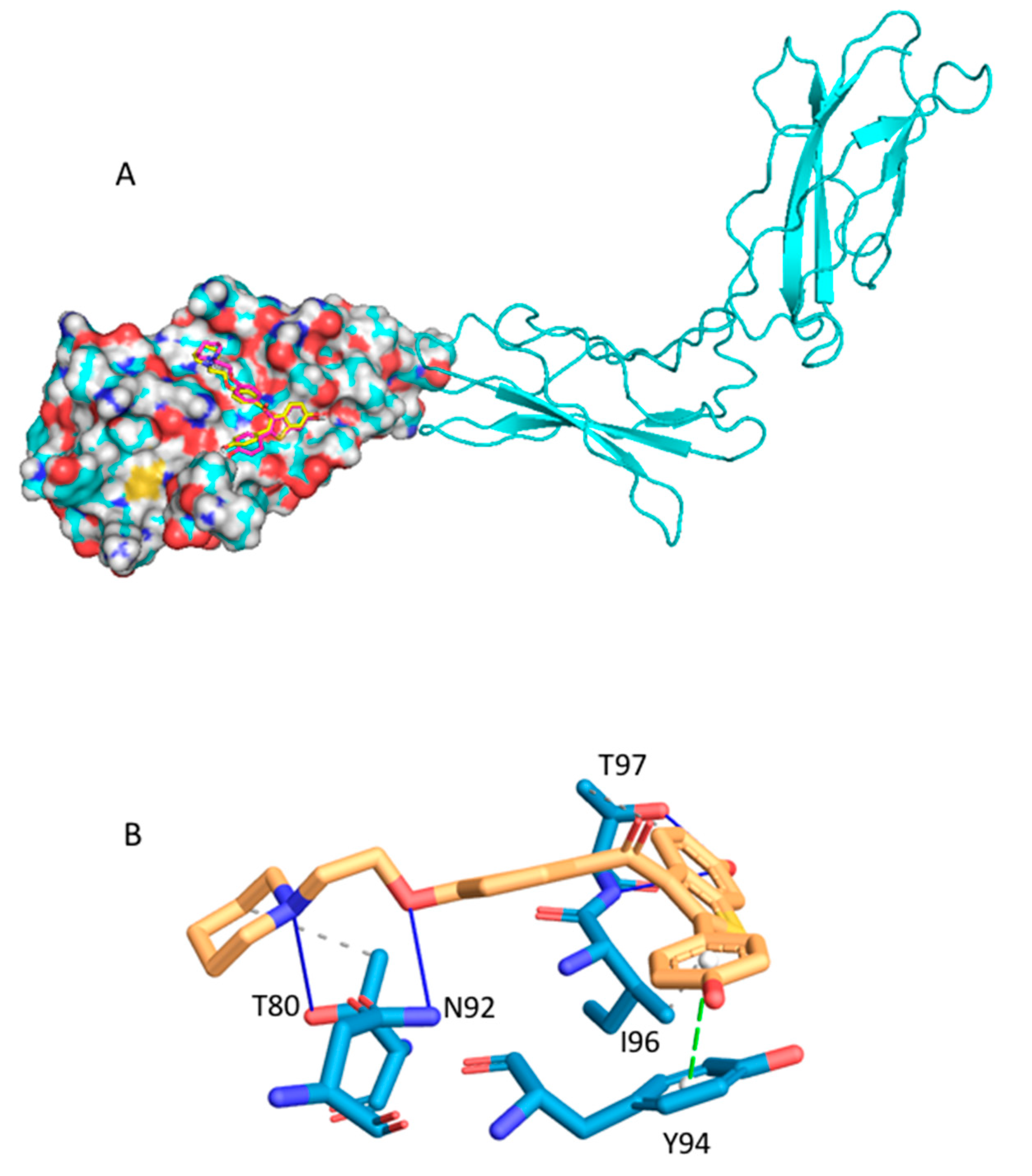
2.2.2. Raloxifene
2.2.3. Thalidomide
3. Materials and Methods
3.1. Web Servers
3.1.1. FAF-Drugs4 Web Server
3.1.2. MTiOpenScreen Web Server
3.1.3. AMMOS2 Web Server
3.2. Preparation of Data for Performance Assessment
3.2.1. Compound Collection Preparation
3.2.2. Protein Structure Preparation and Calculation Parameters
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| RPBS | Ressource Parisienne en Bioinformatique Structurale |
| AMMOS2 | Automatic Molecular Mechanics Optimization for in silico Screening |
| PAINS | pan-assay interference compounds |
| CDK2 | cyclin-dependent kinase 2 |
| ER | estrogen receptor |
| FDA | food drug administration |
| IL-6 | interleukin-6 |
| MW | molecular weight |
| PDB | Protein Data Bank |
References
- Jabeen, A.; Ranganathan, S. Applications of machine learning in GPCR bioactive ligand discovery. Curr. Opin. Struct. Biol. 2019, 55, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Cavasotto, C.N.; Orry, A.J. Ligand Docking and Structure-based Virtual Screening in Drug Discovery. Curr. Top. Med. Chem. 2007, 7, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.L.; Chan, D.S.; Leung, C.H. Drug repositioning by structure-based virtual screening. Chem. Soc. Rev. 2013, 42, 2130–2141. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Roy, K. How far can virtual screening take us in drug discovery? Expert Opin. Drug Discov. 2013, 8, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Gautier, B.; Miteva, M.A.; Goncalves, V.; Huguenot, F.; Coric, P.; Bouaziz, S.; Seijo, B.; Gaucher, J.F.; Broutin, I.; Garbay, C.; et al. Targeting the proangiogenic VEGF-VEGFR protein-protein interface with drug-like compounds by in silico and in vitro screening. Chem. Biol. 2011, 18, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Chevillard, F.; Lagorce, D.; Reynes, C.; Villoutreix, B.O.; Vayer, P.; Miteva, M.A. In silico prediction of aqueous solubility: A multimodel protocol based on chemical similarity. Mol. Pharm. 2012, 9, 3127–3135. [Google Scholar] [CrossRef] [PubMed]
- Moroy, G.; Sperandio, O.; Rielland, S.; Khemka, S.; Druart, K.; Goyal, D.; Perahia, D.; Miteva, M.A. Sampling of conformational ensemble for virtual screening using molecular dynamics simulations and normal mode analysis. Future Med. Chem. 2015, 7, 2317–2331. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Peng, J.; Sidorov, P.; Leung, Y.; Leung, K.S.; Wong, M.H.; Lu, G.; Ballester, P.J. Classical scoring functions for docking are unable to exploit large volumes of structural and interaction data. Bioinformatics 2019. [Google Scholar] [CrossRef]
- Scior, T.; Bender, A.; Tresadern, G.; Medina-Franco, J.L.; Martinez-Mayorga, K.; Langer, T.; Cuanalo-Contreras, K.; Agrafiotis, D.K. Recognizing pitfalls in virtual screening: A critical review. J. Chem. Inf. Modeling 2012, 52, 867–881. [Google Scholar] [CrossRef]
- Ain, Q.U.; Aleksandrova, A.; Roessler, F.D.; Ballester, P.J. Machine-learning scoring functions to improve structure-based binding affinity prediction and virtual screening. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2015, 5, 405–424. [Google Scholar] [CrossRef]
- Yuriev, E.; Holien, J.; Ramsland, P.A. Improvements, trends, and new ideas in molecular docking: 2012–2013 in review. J. Mol. Recognit. 2015, 28, 581–604. [Google Scholar] [CrossRef] [PubMed]
- Douguet, D. e-LEA3D: A computational-aided drug design web server. Nucleic Acids Res. 2010, 38, W615–W621. [Google Scholar] [CrossRef] [PubMed]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Zhou, S.; Ge, Z.; Li, R.; Kwoh, C.K. CovalentDock Cloud: A web server for automated covalent docking. Nucleic Acids Res. 2013, 41, W329–W332. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.; Ascher, D.B. CSM-lig: A web server for assessing and comparing protein-small molecule affinities. Nucleic Acids Res. 2016, 44, W557–W561. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.Y.; Chang, K.W.; Chen, C.Y. iScreen: World’s first cloud-computing web server for virtual screening and de novo drug design based on TCM database@Taiwan. J. Comput.-Aided Mol. Des. 2011, 25, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K.; Mysinger, M.M.; Huang, N.; Colizzi, F.; Wassam, P.; Cao, Y. Automated docking screens: A feasibility study. J. Med. Chem. 2009, 52, 5712–5720. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Leung, K.S.; Wong, M.H.; Ballester, P.J. USR-VS: A web server for large-scale prospective virtual screening using ultrafast shape recognition techniques. Nucleic Acids Res. 2016, 44, W436–W441. [Google Scholar] [CrossRef]
- Lagorce, D.; Bouslama, L.; Becot, J.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs4: Free ADME-tox filtering computations for chemical biology and early stages drug discovery. Bioinform. (Oxf. Engl.) 2017, 33, 3658–3660. [Google Scholar] [CrossRef]
- Labbe, C.M.; Rey, J.; Lagorce, D.; Vavrusa, M.; Becot, J.; Sperandio, O.; Villoutreix, B.O.; Tuffery, P.; Miteva, M.A. MTiOpenScreen: A web server for structure-based virtual screening. Nucleic Acids Res. 2015, 43, W448–W454. [Google Scholar] [CrossRef]
- Labbe, C.M.; Pencheva, T.; Jereva, D.; Desvillechabrol, D.; Becot, J.; Villoutreix, B.O.; Pajeva, I.; Miteva, M.A. AMMOS2: A web server for protein-ligand-water complexes refinement via molecular mechanics. Nucleic Acids Res. 2017, 45, W350–W355. [Google Scholar] [CrossRef] [PubMed]
- Alland, C.; Moreews, F.; Boens, D.; Carpentier, M.; Chiusa, S.; Lonquety, M.; Renault, N.; Wong, Y.; Cantalloube, H.; Chomilier, J.; et al. RPBS: A web resource for structural bioinformatics. Nucleic Acids Res. 2005, 33, W44–W49. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Miteva, M.A.; Violas, S.; Montes, M.; Gomez, D.; Tuffery, P.; Villoutreix, B.O. FAF-Drugs: Free ADME/tox filtering of compound collections. Nucleic Acids Res. 2006, 34, W738–W744. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.D.; Blagg, J.; Price, D.A.; Bailey, S.; Decrescenzo, G.A.; Devraj, R.V.; Ellsworth, E.; Fobian, Y.M.; Gibbs, M.E.; Gilles, R.W.; et al. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, M.P. Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem. 2008, 51, 817–834. [Google Scholar] [CrossRef] [PubMed]
- Przybylak, K.R.; Alzahrani, A.R.; Cronin, M.T. How does the quality of phospholipidosis data influence the predictivity of structural alerts? J. Chem. Inf. Modeling 2014, 54, 2224–2232. [Google Scholar] [CrossRef] [PubMed]
- Bruns, R.F.; Watson, I.A. Rules for identifying potentially reactive or promiscuous compounds. J. Med. Chem. 2012, 55, 9763–9772. [Google Scholar] [CrossRef] [PubMed]
- Miteva, M.A.; Guyon, F.; Tuffery, P. Frog2: Efficient 3D conformation ensemble generator for small compounds. Nucleic Acids Res. 2010, 38, W622–W627. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Lagarde, N.; Rey, J.; Gyulkhandanyan, A.; Tuffery, P.; Miteva, M.A.; Villoutreix, B.O. Online structure-based screening of purchasable approved drugs and natural compounds: Retrospective examples of drug repositioning on cancer targets. Oncotarget 2018, 9, 32346–32361. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. Protein-protein interaction inhibitors get into the groove. Nat. Rev. Drug Discov. 2012, 11, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Betzi, S.; Morelli, X.; Roche, P. Focused chemical libraries--design and enrichment: An example of protein-protein interaction chemical space. Future Med. Chem. 2014, 6, 1291–1307. [Google Scholar] [CrossRef] [PubMed]
- Villoutreix, B.O.; Kuenemann, M.A.; Poyet, J.L.; Bruzzoni-Giovanelli, H.; Labbe, C.; Lagorce, D.; Sperandio, O.; Miteva, M.A. Drug-like protein-protein interaction modulators: Challenges and opportunities for drug discovery and chemical biology. Mol. Inform. 2014, 33, 414–437. [Google Scholar] [CrossRef] [PubMed]
- Weber, I.T.; Harrison, R.W. Molecular mechanics calculations on Rous sarcoma virus protease with peptide substrates. Protein Sci.: A Publ. Protein Soc. 1997, 6, 2365–2374. [Google Scholar] [CrossRef]
- Pencheva, T.; Lagorce, D.; Pajeva, I.; Villoutreix, B.O.; Miteva, M.A. AMMOS: Automated Molecular Mechanics Optimization tool for in silico Screening. BMC Bioinform. 2008, 9, 438. [Google Scholar] [CrossRef]
- Janssen, P.A.; Niemegeers, C.J.; Schellekens, K.H.; Lenaerts, F.M.; Verbruggen, F.J.; Van Nueten, J.M.; Schaper, W.K. The pharmacology of penfluridol (R 16341) a new potent and orally long-acting neuroleptic drug. Eur. J. Pharmacol. 1970, 11, 139–154. [Google Scholar] [CrossRef]
- Hassel, P. Experimental comparison of low doses of 1.5 mg fluspirilene and bromazepam in out-patients with psychovegetative disturbances. Pharmacopsychiatry 1985, 18, 297–302. [Google Scholar] [CrossRef]
- Wang, S.J. Inhibition of glutamate release by fluspirilene in cerebrocortical nerve terminals (synaptosomes). Synapse 2002, 44, 36–41. [Google Scholar] [CrossRef]
- Shi, X.N.; Li, H.; Yao, H.; Liu, X.; Li, L.; Leung, K.S.; Kung, H.F.; Lu, D.; Wong, M.H.; Lin, M.C. In Silico Identification and In Vitro and In Vivo Validation of Anti-Psychotic Drug Fluspirilene as a Potential CDK2 Inhibitor and a Candidate Anti-Cancer Drug. PLoS ONE 2015, 10, e0132072. [Google Scholar] [CrossRef] [PubMed]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Cranney, A.; Adachi, J.D. Benefit-risk assessment of raloxifene in postmenopausal osteoporosis. Drug Saf. 2005, 28, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.U. Mechanism of action and preclinical profile of raloxifene, a selective estrogen receptor modulation. Rev. Endocr. Metab. Disord. 2001, 2, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xiao, H.; Lin, L.; Jou, D.; Kumari, V.; Lin, J.; Li, C. Drug design targeting protein-protein interactions (PPIs) using multiple ligand simultaneous docking (MLSD) and drug repositioning: Discovery of raloxifene and bazedoxifene as novel inhibitors of IL-6/GP130 interface. J. Med. Chem. 2014, 57, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Vargesson, N. Thalidomide-induced teratogenesis: History and mechanisms. Birth Defects Res. Part C Embryo Today Rev. 2015, 105, 140–156. [Google Scholar] [CrossRef]
- Singhal, S.; Mehta, J.; Desikan, R.; Ayers, D.; Roberson, P.; Eddlemon, P.; Munshi, N.; Anaissie, E.; Wilson, C.; Dhodapkar, M.; et al. Antitumor activity of thalidomide in refractory multiple myeloma. New Engl. J. Med. 1999, 341, 1565–1571. [Google Scholar] [CrossRef]
- Stewart, A.K. Medicine. How thalidomide works against cancer. Science 2014, 343, 256–257. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminformatics 2011, 3, 33. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A ‘rule of three’ for fragment-based lead discovery? Drug Discov. Today 2003, 8, 876–877. [Google Scholar] [CrossRef]
- Workman, P.; Collins, I. Probing the probes: Fitness factors for small molecule tools. Chem. Biol. 2010, 17, 561–577. [Google Scholar] [CrossRef] [PubMed]
- Charifson, P.S.; Walters, W.P. Filtering databases and chemical libraries. Mol. Divers. 2002, 5, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. ZINC--a free database of commercially available compounds for virtual screening. J. Chem. Inf. Modeling 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, P.; Summerfield, S. Assessment of the blood-brain barrier in CNS drug discovery. Neurobiol. Dis. 2010, 37, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, T.J.; Luscombe, C.N.; Macdonald, S.J. Analysis of the calculated physicochemical properties of respiratory drugs: Can we design for inhaled drugs yet? J. Chem. Inf. Modeling 2009, 49, 1025–1032. [Google Scholar] [CrossRef]
- Pihan, E.; Colliandre, L.; Guichou, J.F.; Douguet, D. e-Drug3D: 3D structure collections dedicated to drug repurposing and fragment-based drug design. Bioinform. (Oxf. Engl.) 2012, 28, 1540–1541. [Google Scholar] [CrossRef]
- Biasini, M. Zenodo. pv: v1.8.1 (Version V1.8.1). 2015. Available online: http://doi.org/10.5281/zenodo.20980 (accessed on 18 September 2019). [CrossRef]
- Rappé, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard III, W.A.; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Weiner, S.J.; Kollman, P.A.; Nguyen, D.T.; Case, D. An all atom force field for simulations of proteins and nucleic acids. J. Comput. Chem. 1986, 7, 230–252. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Ursu, O.; Holmes, J.; Knockel, J.; Bologa, C.G.; Yang, J.J.; Mathias, S.L.; Nelson, S.J.; Oprea, T.I. DrugCentral: Online drug compendium. Nucleic Acids Res. 2017, 45, D932–D939. [Google Scholar] [CrossRef]
- Siramshetty, V.B.; Eckert, O.A.; Gohlke, B.O.; Goede, A.; Chen, Q.; Devarakonda, P.; Preissner, S.; Preissner, R. SuperDRUG2: A one stop resource for approved/marketed drugs. Nucleic Acids Res. 2018, 46, D1137–D1143. [Google Scholar] [CrossRef]
- Lagorce, D.; Sperandio, O.; Baell, J.B.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs3: A web server for compound property calculation and chemical library design. Nucleic Acids Res. 2015, 43, W200–W207. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15--Ligand Discovery for Everyone. J. Chem. Inf. Modeling 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- ChemAxon. Marvin Calculator Plugins version 17.23. Available online: www.chemaxon.com (accessed on 18 September 2019).
- 3D Structure Generator CORINA Classic, Molecular Networks GmbH, Nuremberg, Germany. Available online: www.mn-am.com (accessed on 18 September 2019).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug, PDB ID | AutoDock Vina | AMMOS2 Case 1 | AMMOS2 Case 3 | AMMOS2 Case 4 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| score | rank | Energy before min | Energy after min | rank | Energy before min | Energy after min | rank | Energy before min | Energy after min | rank | |
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| Fluspirilene 1PXI | −8.7 | 805 | 207.75 | 3.98 | 520 | 195.32 | 8.96 | 665 | >1000.0 | 15.97 | 654 |
| Fluspirilene 1VYZ | −9.2 | 443 | 111.96 | −29.37 | 342 | 657.01 | −13.31 | 683 | >1000.0 | −8.51 | 619 |
| Raloxifene 3L5H | −7.5 | 126 | >1000.0 | −72.11 | 480 | >1000.0 | −59.01 | 609 | >1000.0 | −25.50 | 850 |
| Raloxifene 1P9M | −6.5 | 737 | −49.74 | −124.65 | 292 | −49.73 | −119.48 | 455 | −49.74 | −114.09 | 466 |
| Thalidomide 4CI1 | −9.8 | 196 | −38.11 | −40.31 | 341 | −16.34 | −41.49 | 578 | −38.11 | −40.31 | 555 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lagarde, N.; Goldwaser, E.; Pencheva, T.; Jereva, D.; Pajeva, I.; Rey, J.; Tuffery, P.; Villoutreix, B.O.; Miteva, M.A. A Free Web-Based Protocol to Assist Structure-Based Virtual Screening Experiments. Int. J. Mol. Sci. 2019, 20, 4648. https://doi.org/10.3390/ijms20184648
Lagarde N, Goldwaser E, Pencheva T, Jereva D, Pajeva I, Rey J, Tuffery P, Villoutreix BO, Miteva MA. A Free Web-Based Protocol to Assist Structure-Based Virtual Screening Experiments. International Journal of Molecular Sciences. 2019; 20(18):4648. https://doi.org/10.3390/ijms20184648
Chicago/Turabian StyleLagarde, Nathalie, Elodie Goldwaser, Tania Pencheva, Dessislava Jereva, Ilza Pajeva, Julien Rey, Pierre Tuffery, Bruno O. Villoutreix, and Maria A. Miteva. 2019. "A Free Web-Based Protocol to Assist Structure-Based Virtual Screening Experiments" International Journal of Molecular Sciences 20, no. 18: 4648. https://doi.org/10.3390/ijms20184648
APA StyleLagarde, N., Goldwaser, E., Pencheva, T., Jereva, D., Pajeva, I., Rey, J., Tuffery, P., Villoutreix, B. O., & Miteva, M. A. (2019). A Free Web-Based Protocol to Assist Structure-Based Virtual Screening Experiments. International Journal of Molecular Sciences, 20(18), 4648. https://doi.org/10.3390/ijms20184648