Interstitial Flow Recapitulates Gemcitabine Chemoresistance in A 3D Microfluidic Pancreatic Ductal Adenocarcinoma Model by Induction of Multidrug Resistance Proteins

 , and
, and 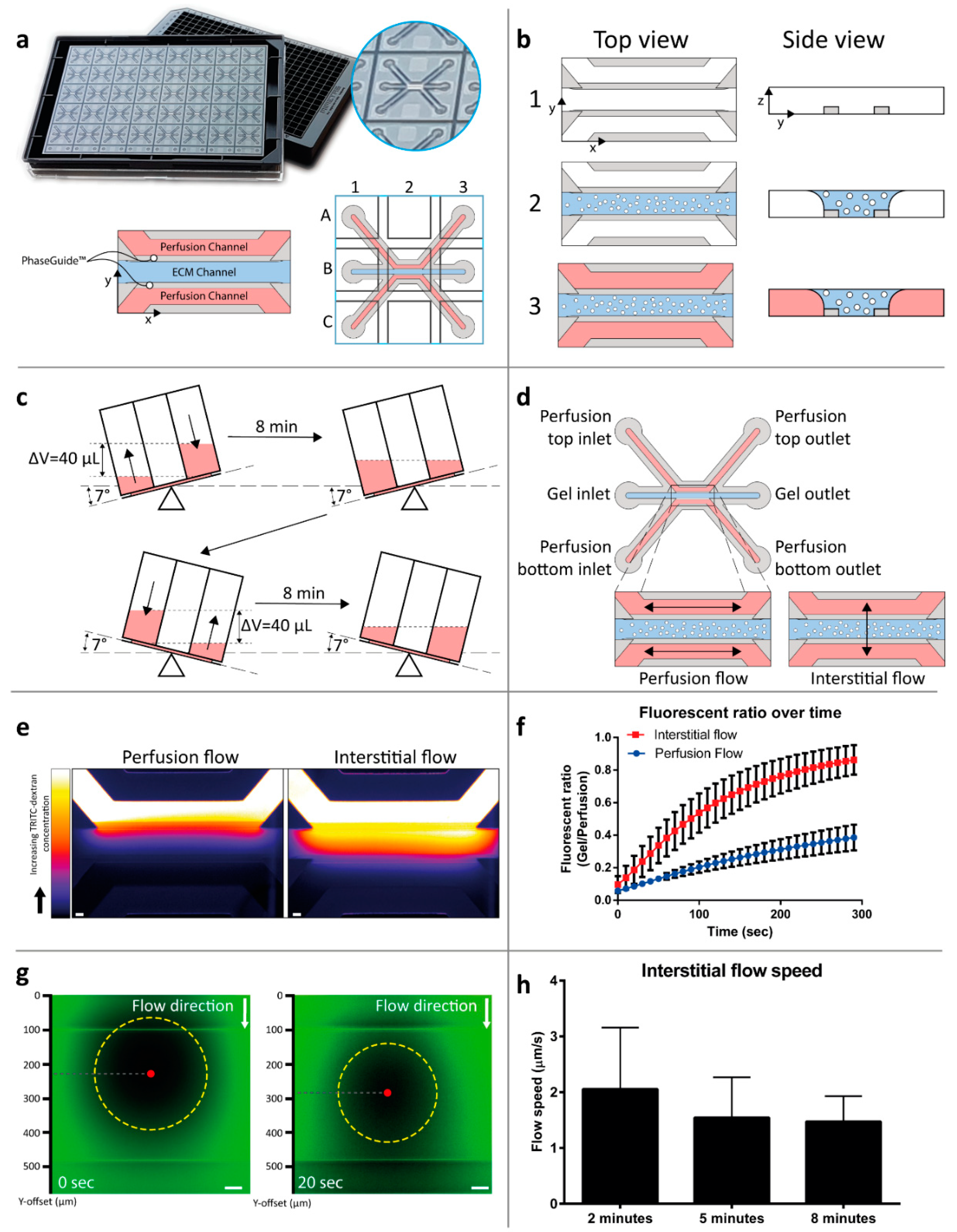{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Interstitial Flow Modeling in a Microfluidic Platform
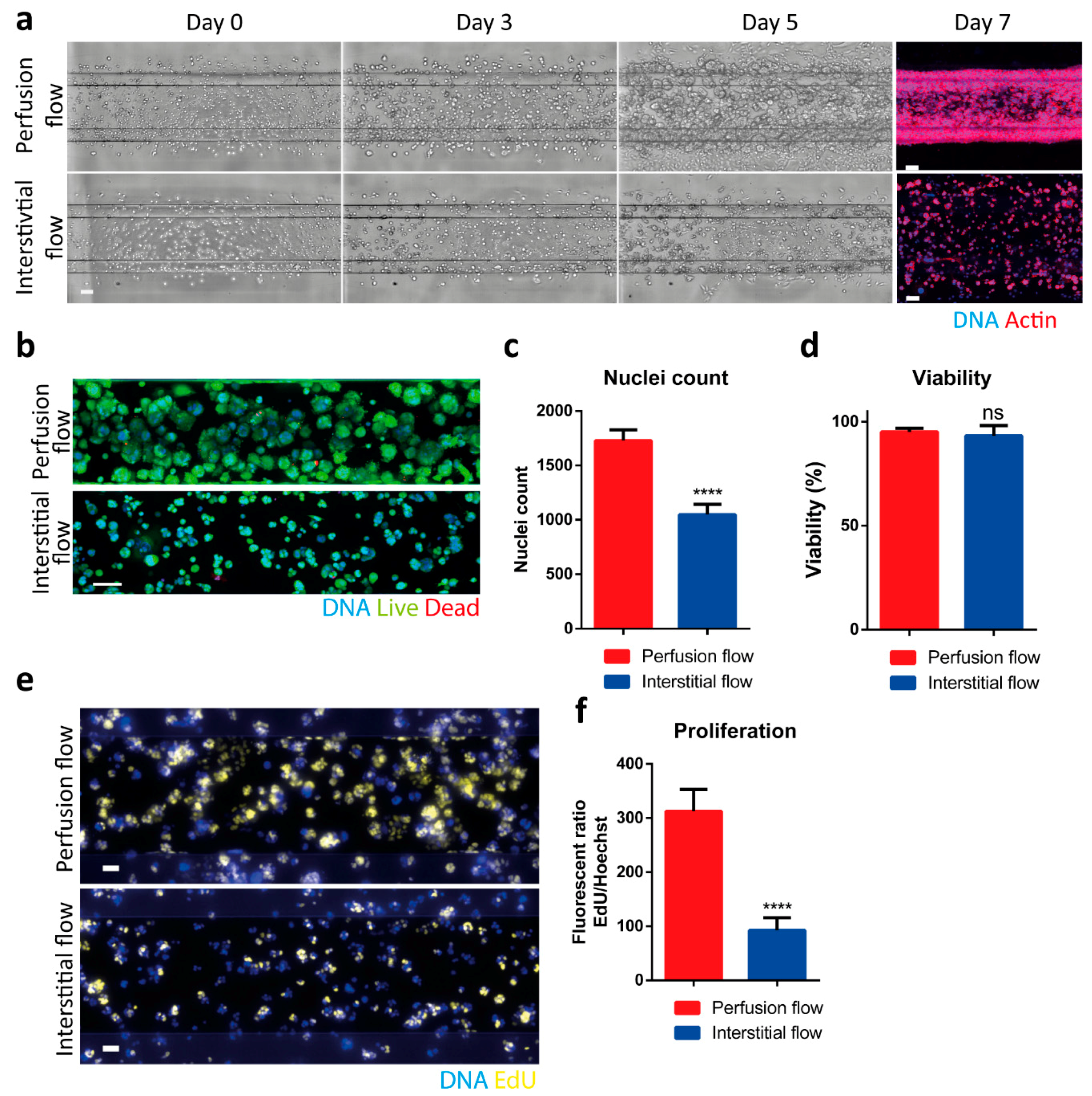
2.2. Interstitial Flow Inhibits the Proliferation of the PDAC Cells
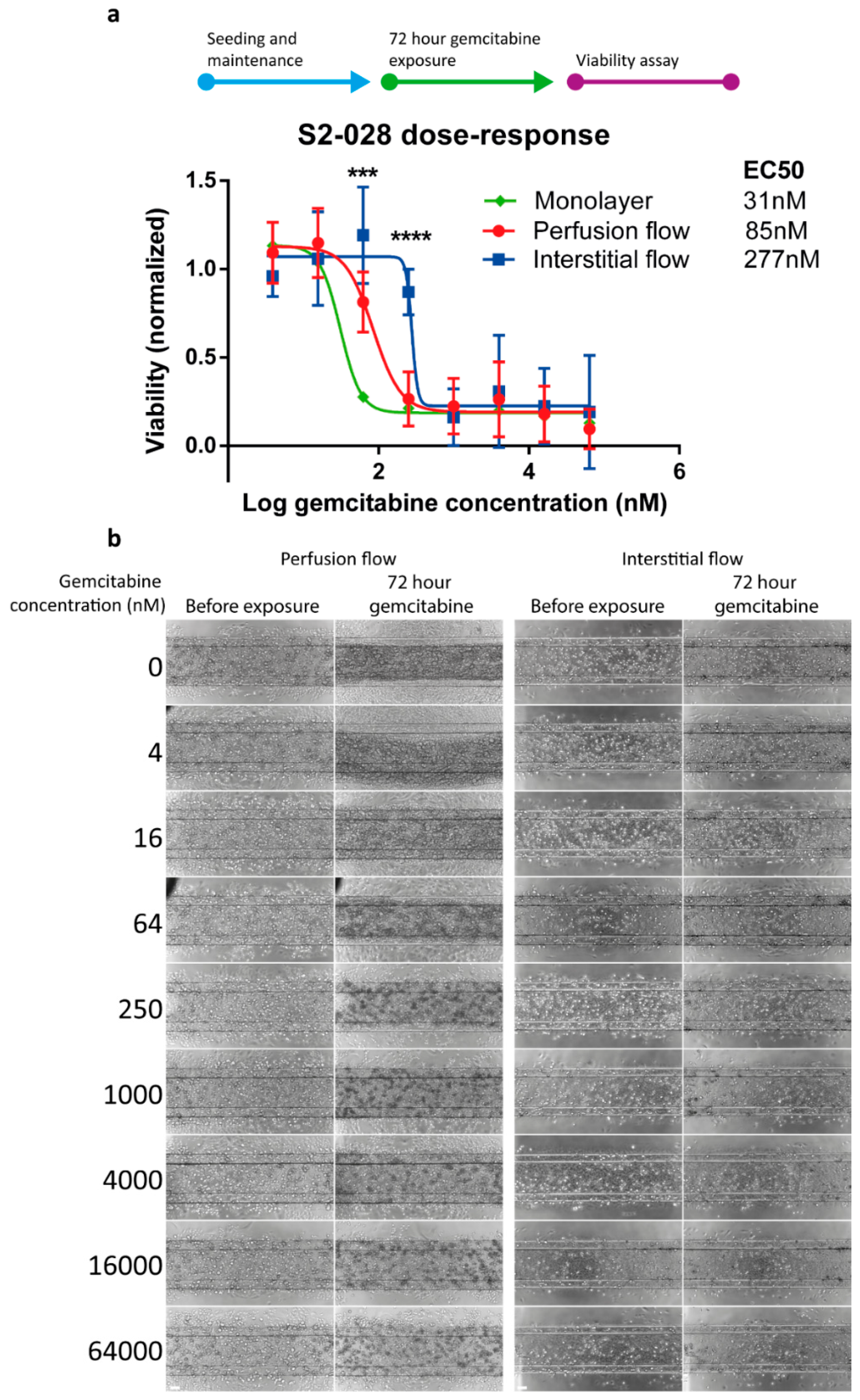
2.3. PDAC Cells are More Resistant to Gemcitabine in the Presence of Interstitial Flow
2.4. MRP Protein Function is Altered under Interstitial Flow
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. OrganoPlate Culture
4.3. Interstitial Flow Simulation
4.4. Live/dead Assay
4.5. EdU Proliferation Assay
4.6. Immunohistochemistry
4.7. Drug Exposure and Viability Assessment
4.8. Enzymatic Activity Assessment
4.9. qPCR
4.10. MRP Efflux Assay
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MRP | multidrug resistance protein |
| PDAC | PancreaticDuctal Adenocarcinoma |
| ABC | ATP-binding cassette |
| ECM | extracellular matrix |
| FRAP | Fluorescence Recovery After Photobleaching |
| CMF | 5-chloromethylfluorescein |
| CMFDA | 5-choloromethylfluorescein diacetate |
| GS-MF | carboxylfluorescein-glutathione |
| TRITC | Tetramethyl Rhodamine Iso-Thiocyanate |
References
- Ghaneh, P.; Costello, E.; Neoptolemos, J.P. Biology and management of pancreatic cancer. Postgrad. Med. J. 2008, 84, 478–497. [Google Scholar] [CrossRef] [PubMed]
- Plunkett, W.; Huang, P.; Xu, Y.Z.; Heinemann, V.; Grunewald, R.; Gandhi, V. Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin. Oncol. 1995, 22, 3–10. [Google Scholar] [PubMed]
- Chandler, N.M.; Canete, J.J.; Callery, M.P. Caspase-3 drives apoptosis in pancreatic cancer cells after treatment with gemcitabine. J. Gastrointest. Surg. 2004, 8, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Habiro, A.; Tanno, S.; Koizumi, K.; Izawa, T.; Nakano, Y.; Osanai, M.; Mizukami, Y.; Okumura, T.; Kohgo, Y. Involvement of p38 mitogen-activated protein kinase in gemcitabine-induced apoptosis in human pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2004, 316, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Amrutkar, M.; Gladhaug, I.P. Pancreatic cancer chemoresistance to gemcitabine. Cancers 2017, 9, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Munson, J.M.; Shieh, A.C. Interstitial fluid flow in cancer: implications for disease progression and treatment. Cancer Manag. Res. 2014, 6, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Dhayat, S.A.; Mardin, W.A.; Seggewiß, J.; Ströse, A.J.; Matuszcak, C.; Hummel, R.; Senninger, N.; Mees, S.T.; Haier, J. MicroRNA profiling implies new markers of gemcitabine chemoresistance in mutant p53 pancreatic ductal adenocarcinoma. PLoS ONE 2015, 10, e0143755. [Google Scholar] [CrossRef] [PubMed]
- Hoarau-Véchot, J.; Rafii, A.; Touboul, C.; Pasquier, J. Halfway between 2D and animal models: Are 3D cultures the ideal tool to study cancer-microenvironment interactions? Int. J. Mol. Sci. 2018, 19, 181. [Google Scholar] [CrossRef]
- Cekanova, M.; Rathore, K. DDDT-49584-animal-models-of-cancer-utility-and-limitations. Dev. Ther. 2014, 8, 1911–1922. [Google Scholar] [CrossRef]
- Zhang, B.; Korolj, A.; Lai, B.F.L.; Radisic, M. Advances in organ-on-a-chip engineering. Nat. Rev. Mater. 2018, 3, 257–278. [Google Scholar] [CrossRef]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef] [PubMed]
- van Duinen, V.; van den heuvel, A.; Trietsch, S.; Lanz, H.; van Gils, J.; van Zonneveld, A.; Vulto, P.; Hankemeier, T. High-throughput permeability assay in vitro on perfused 3D microvessels. Circ. Res. 2017. submitted. [Google Scholar]
- Buchanan, C.F.; Verbridge, S.S.; Vlachos, P.P.; Rylander, M.N. Flow shear stress regulates endothelial barrier function and expression of angiogenic factors in a 3D microfluidic tumor vascular model. Cell Adhes. Migr. 2014, 8, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Wevers, N.R.; van Vught, R.; Wilschut, K.J.; Nicolas, A.; Chiang, C.; Lanz, H.L.; Trietsch, S.J.; Joore, J.; Vulto, P. High-throughput compound evaluation on 3D networks of neurons and glia in a microfluidic platform. Sci. Rep. 2016, 6, 38856. [Google Scholar] [CrossRef] [PubMed]
- Wilmer, M.J.; Ng, C.P.; Lanz, H.L.; Vulto, P.; Suter-Dick, L.; Masereeuw, R. Kidney-on-a-chip technology for drug-induced nephrotoxicity screening. Trends Biotechnol. 2015, 34, 156–170. [Google Scholar] [CrossRef] [PubMed]
- Au, S.H.; Chamberlain, M.D.; Mahesh, S.; Sefton, M.V.; Wheeler, A.R. Hepatic organoids for microfluidic drug screening. Lab Chip 2014, 14, 3290–3299. [Google Scholar] [CrossRef] [PubMed]
- Lanz, H.L.; Saleh, A.; Kramer, B.; Cairns, J.; Ng, C.P.; Yu, J.; Trietsch, S.J.; Hankemeier, T.; Joore, J.; Vulto, P.; et al. Therapy response testing of breast cancer in a 3D high-throughput perfused microfluidic platform. BMC Cancer 2017, 17, 709. [Google Scholar] [CrossRef] [PubMed]
- Iwamura, T.; Katsuki, T.; Ide, K. Establishment and characterization of a human pancreatic cancer cell line (SUIT-2) producing carcinoembryonic antigen and carbohydrate antigen. Jpn. J. Cancer Res. 1987, 78, 54–62. [Google Scholar]
- Vulto, P.; Podszun, S.; Meyer, P.; Hermann, C.; Manz, A.; Urban, G. Phaseguides: A paradigm shift in microfluidic priming and emptying. Lab Chip 2011, 11, 1596–1602. [Google Scholar] [CrossRef] [PubMed]
- Dafni, H.; Israely, T.; Bhujwalla, Z.M.; Benjamin, L.E.; Neeman, M. Overexpression of vascular endothelial growth factor 165 drives peritumor interstitial convection and induces lymphatic drain: Magnetic resonance imaging, confocal microscopy, and histological tracking of triple-labeled albumin. Cancer Res. 2002, 62, 6731–6739. [Google Scholar] [PubMed]
- Miyamoto, H.; Murakami, T.; Tsuchida, K.; Sugino, H.; Miyake, H.; Tashiro, S. Tumor-stroma interaction of human pancreatic cancer: Acquired resistance to anticancer drugs and proliferation regulation is dependent on extracellular matrix proteins. Pancreas 2004, 28, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, H.; Török, M.; Fricker, G.; Huwyler, J.; Beglinger, C.; Drewe, J. Modulation of multidrug resistance protein expression in porcine brain capillary endothelial cells in vitro. Drug Metab. Dispos. 1999, 27, 937–941. [Google Scholar] [PubMed]
- Fang, Y.; Eglen, R.M. Three-dimensional cell cultures in drug discovery and development. SLAS Discov. 2017, 22, 456–472. [Google Scholar] [PubMed]
- Abbott, A. Cell culture: biology’s new dimension. Nature 2003, 424, 870–872. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Galettis, P.; Links, M.; Mitchell, P.L.; McLachlan, A.J. Population pharmacokinetics of gemcitabine and its metabolite in patients with cancer: Effect of oxaliplatin and infusion rate. Br. J. Clin. Pharmacol. 2008, 65, 326–333. [Google Scholar] [CrossRef]
- Jiang, P.H.; Motoo, Y.; Sawabu, N.; Minamoto, T. Effect of gemcitabine on the expression of apoptosis-related genes in human pancreatic cancer cells. World J. Gastroenterol. 2006, 12, 1597–1602. [Google Scholar] [CrossRef]
- Hagmann, W.; Jesnowski, R.; Löhr, J.M. Interdependence of gemcitabine treatment, transporter expression, and resistance in human pancreatic carcinoma cells. Neoplasia 2015, 12, 740–747. [Google Scholar] [CrossRef]
- Rudin, D.; Li, L.; Niu, N.; Kalari, K.R.; Gilbert, J.A.; Ames, M.M.; Wang, L. Gemcitabine cytotoxicity: Interaction of efflux and deamination. J. Drug Metab. Toxicol. 2011, 2, 1–10. [Google Scholar] [CrossRef]
- Falasca, M.; Linton, K.J. Investigational ABC transporter inhibitors. Expert Opin. Investig. Drugs 2012, 21, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-S.; Tiwari, A.K. Multidrug resistance proteins (MRPs/ABCCs) in cancer chemotherapy and genetic diseases. FEBS J. 2011, 278, 3226–3245. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Gu, J.; Mao, Y.; Zhang, X.; Wang, X.; Jin, C.; Fu, D.; Li, J. Dynamic quantitative detection of ABC transporter family promoter methylation by MS-HRM for predicting MDR in pancreatic cancer. Oncol. Lett. 2018, 15, 5602–5610. [Google Scholar] [CrossRef] [PubMed]
- Waldeland, J.O.; Evje, S. Competing tumor cell migration mechanisms caused by interstitial fluid flow. J. Biomech. 2018, 81, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Banyard, J.; Chung, I.; Migliozzi, M.; Phan, D.T.; Wilson, A.M.; Zetter, B.R.; Bielenberg, D.R. Identification of genes regulating migration and invasion using a new model of metastatic prostate cancer. BMC Cancer 2014, 14, 387. [Google Scholar] [CrossRef] [PubMed]
- Trietsch, S.J.; Israëls, G.D.; Joore, J.; Hankemeier, T.; Vulto, P. Microfluidic titer plate for stratified 3D cell culture. Lab Chip 2013, 13, 3548–3554. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Sternberg Biomedical Image Processing. Computer (Long. Beach. Calif). 1983, 16, 22–34.
- Vriend, J.; Nieskens, T.T.G.; Vormann, M.K.; van den Berge, B.T.; van den Heuvel, A.; Russel, F.G.M.; Suter-Dick, L.; Lanz, H.L.; Vulto, P.; Masereeuw, R.; et al. Screening of drug-transporter interactions in a 3D microfluidic renal proximal tubule on a chip. AAPS J. 2018, 20. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kramer, B.; Haan, L.d.; Vermeer, M.; Olivier, T.; Hankemeier, T.; Vulto, P.; Joore, J.; Lanz, H.L. Interstitial Flow Recapitulates Gemcitabine Chemoresistance in A 3D Microfluidic Pancreatic Ductal Adenocarcinoma Model by Induction of Multidrug Resistance Proteins. Int. J. Mol. Sci. 2019, 20, 4647. https://doi.org/10.3390/ijms20184647
Kramer B, Haan Ld, Vermeer M, Olivier T, Hankemeier T, Vulto P, Joore J, Lanz HL. Interstitial Flow Recapitulates Gemcitabine Chemoresistance in A 3D Microfluidic Pancreatic Ductal Adenocarcinoma Model by Induction of Multidrug Resistance Proteins. International Journal of Molecular Sciences. 2019; 20(18):4647. https://doi.org/10.3390/ijms20184647
Chicago/Turabian StyleKramer, Bart, Luuk de Haan, Marjolein Vermeer, Thomas Olivier, Thomas Hankemeier, Paul Vulto, Jos Joore, and Henriëtte L. Lanz. 2019. "Interstitial Flow Recapitulates Gemcitabine Chemoresistance in A 3D Microfluidic Pancreatic Ductal Adenocarcinoma Model by Induction of Multidrug Resistance Proteins" International Journal of Molecular Sciences 20, no. 18: 4647. https://doi.org/10.3390/ijms20184647
APA StyleKramer, B., Haan, L. d., Vermeer, M., Olivier, T., Hankemeier, T., Vulto, P., Joore, J., & Lanz, H. L. (2019). Interstitial Flow Recapitulates Gemcitabine Chemoresistance in A 3D Microfluidic Pancreatic Ductal Adenocarcinoma Model by Induction of Multidrug Resistance Proteins. International Journal of Molecular Sciences, 20(18), 4647. https://doi.org/10.3390/ijms20184647