Klotho: A Major Shareholder in Vascular Aging Enterprises


Abstract
1. Introduction
2. The Biology of Klotho
2.1. Structure of Klotho
2.2. Tissue Expression of Klotho
2.3. The Klotho–FGF23 Axis
2.4. Klotho Exerts Pleiotropic Functions Independent of FGF23
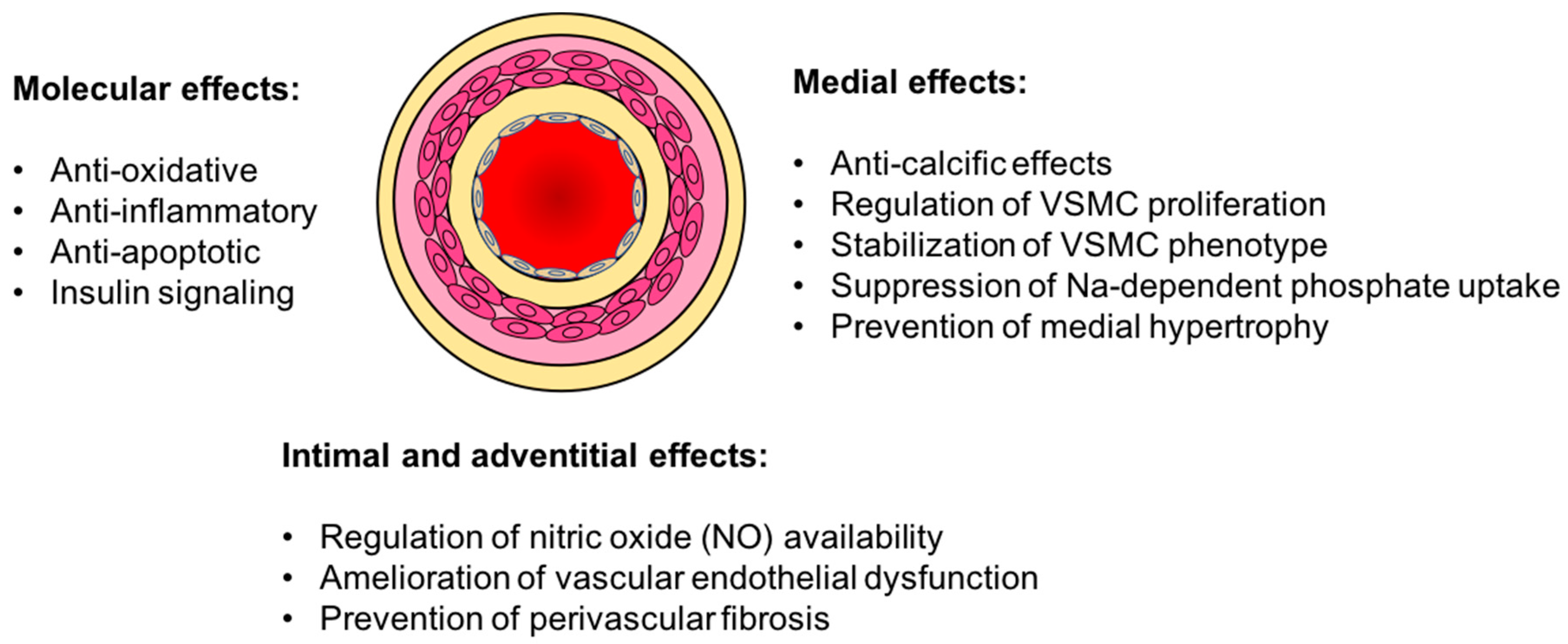
3. Vasculo-Protective Effects of Klotho
3.1. Vascular Calcification
3.2. Endothelial Dysfunction
3.3. Oxidative Stress and Inflammation
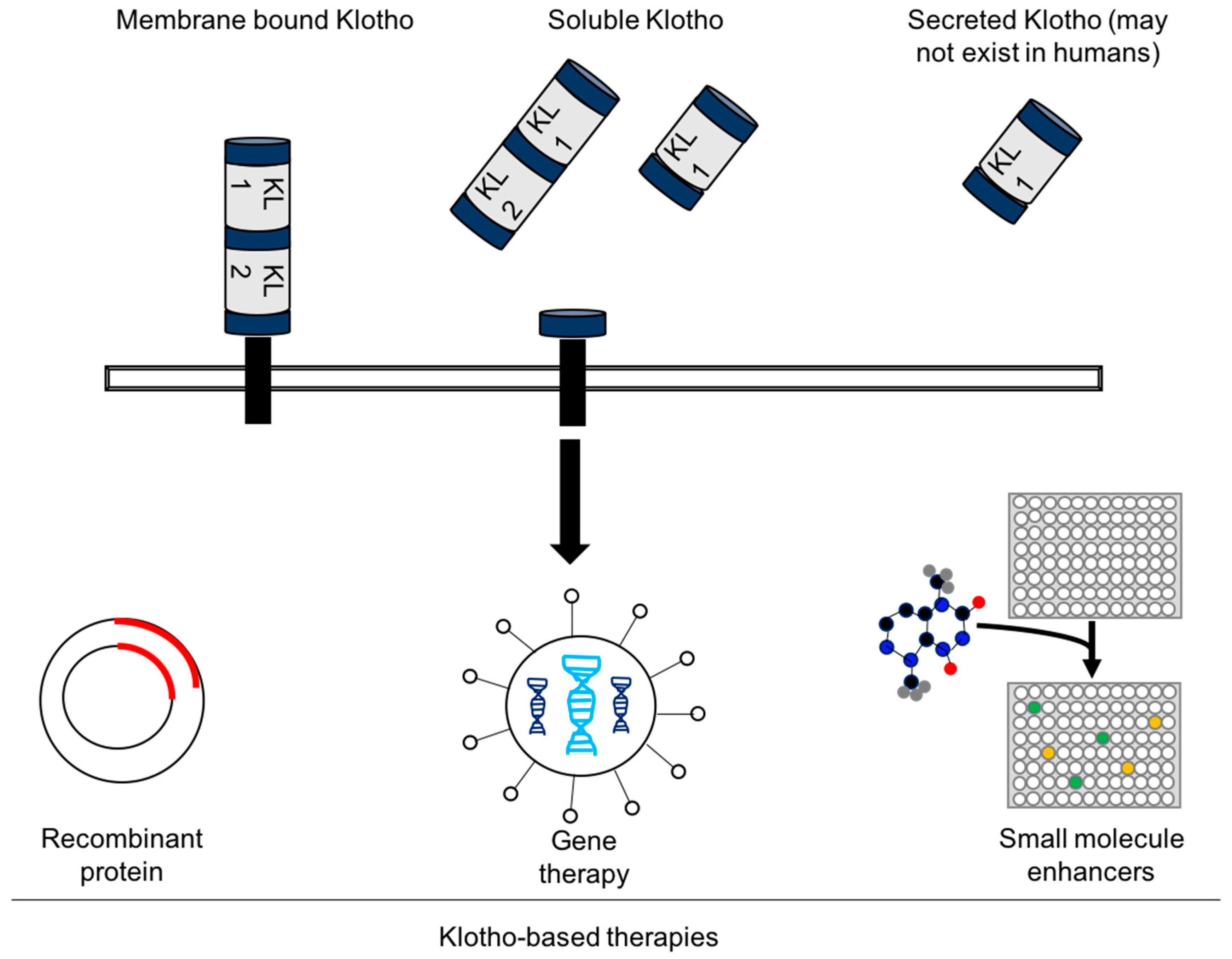
4. Experimental Challenges and the Future of Klotho-Based Therapies
5. Summary
Funding
Conflicts of Interest
Disclosures
References
- Lakatta, E.G.; Levy, D. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part II: The aging heart in health: Links to heart disease. Circulation 2003, 107, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Stenvinkel, P.; Larsson, T.E. Chronic kidney disease: A clinical model of premature aging. Am. J. Kidney Dis. 2013, 62, 339–351. [Google Scholar] [CrossRef]
- Lim, K.; Groen, A.; Molostvov, G.; Lu, T.; Lilley, K.S.; Snead, D.; James, S.; Wilkinson, I.B.; Ting, S.; Hsiao, L.L.; et al. alpha-Klotho Expression in Human Tissues. J. Clin. Endocrinol. Metab. 2015, 100, E1308–E1318. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, M.F. Arterial aging: Pathophysiological principles. Vasc. Med. 2007, 12, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Kwan, G.F.; Benjamin, E.J. Global Health and Cardiovascular Disease. Circulation 2015, 132, 1217. [Google Scholar] [CrossRef]
- Kanasaki, K.; Kitada, M.; Koya, D. Pathophysiology of the aging kidney and therapeutic interventions. Hypertens Res. 2012, 35, 1121–1128. [Google Scholar] [CrossRef]
- Foley, R.N.; Parfrey, P.S.; Sarnak, M.J. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am. J. Kidney Dis. 1998, 32 (Suppl. 3), S112–S119. [Google Scholar] [CrossRef]
- Hodes, R.J. Aging Hearts and Arteries: A Scientific Quest; National Institute of Aging: Bethesda, MA, USA, 1990.
- Roderick, P.J.; Atkins, R.J.; Smeeth, L.; Mylne, A.; Nitsch, D.D.; Hubbard, R.B.; Bulpitt, C.J.; Fletcher, A.E. CKD and mortality risk in older people: A community-based population study in the United Kingdom. Am. J. Kidney Dis. 2009, 53, 950–960. [Google Scholar] [CrossRef]
- Kahn, M.R.; Robbins, M.J.; Kim, M.C.; Fuster, V. Management of cardiovascular disease in patients with kidney disease. Nat. Rev. Cardiol. 2013, 10, 261–273. [Google Scholar] [CrossRef]
- Verbrugge, F.H.; Tang, W.H.; Hazen, S.L. Protein carbamylation and cardiovascular disease. Kidney Int. 2015, 88, 474–478. [Google Scholar] [CrossRef]
- Rhee, E.P.; Gerszten, R.E. Metabolomics and cardiovascular biomarker discovery. Clin. Chem. 2012, 58, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.; Lu, T.S.; Molostvov, G.; Lee, C.; Lam, F.T.; Zehnder, D.; Hsiao, L.L. Vascular Klotho deficiency potentiates the development of human artery calcification and mediates resistance to fibroblast growth factor 23. Circulation 2012, 125, 2243–2255. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Touyz, R.M. Oxidative Stress, Inflammation, and Vascular Aging in Hypertension. Hypertension 2017, 70, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Kobold, S. Inflammation: A common contributor to cancer, aging, and cardiovascular diseases-expanding the concept of cardio-oncology. Cardiovasc. Res. 2019, 115, 824–829. [Google Scholar] [CrossRef] [PubMed]
- Cupit-Link, M.C.; Kirkland, J.L.; Ness, K.K.; Armstrong, G.T.; Tchkonia, T.; LeBrasseur, N.K.; Armenian, S.H.; Ruddy, K.J.; Hashmi, S.K. Biology of premature ageing in survivors of cancer. ESMO Open 2017, 2, e000250. [Google Scholar] [CrossRef] [PubMed]
- Tolle, M.; Reshetnik, A.; Schuchardt, M.; Hohne, M.; van der Giet, M. Arteriosclerosis and vascular calcification: Causes, clinical assessment and therapy. Eur. J. Clin. Investig. 2015, 45, 976–985. [Google Scholar] [CrossRef]
- London, G.; Covic, A.; Goldsmith, D.; Wiecek, A.; Suleymanlar, G.; Ortiz, A.; Massy, Z.; Lindholm, B.; Martinez-Castelao, A.; Fliser, D.; et al. Arterial aging and arterial disease: Interplay between central hemodynamics, cardiac work, and organ flow-implications for CKD and cardiovascular disease. Kidney Int. Suppl. 2011, 1, 10–12. [Google Scholar] [CrossRef]
- Najjar, S.S.; Scuteri, A.; Lakatta, E.G. Arterial aging: Is it an immutable cardiovascular risk factor? Hypertension 2005, 46, 454–462. [Google Scholar] [CrossRef]
- Lakatta, E.G.; Levy, D. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part I: Aging arteries: A “set up” for vascular disease. Circulation 2003, 107, 139–146. [Google Scholar] [CrossRef]
- Kuro-o, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef]
- Kurosu, H.; Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Nandi, A.; Gurnani, P.; McGuinness, O.P.; Chikuda, H.; Yamaguchi, M.; Kawaguchi, H.; et al. Suppression of aging in mice by the hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar] [CrossRef]
- Masuda, H.; Chikuda, H.; Suga, T.; Kawaguchi, H.; Kuro-o, M. Regulation of multiple ageing-like phenotypes by inducible klotho gene expression in klotho mutant mice. Mech. Ageing Dev. 2005, 126, 1274–1283. [Google Scholar] [CrossRef]
- Drew, D.A.; Katz, R.; Kritchevsky, S.; Ix, J.; Shlipak, M.; Gutierrez, O.M.; Newman, A.; Hoofnagle, A.; Fried, L.; Semba, R.D.; et al. Association between Soluble Klotho and Change in Kidney Function: The Health Aging and Body Composition Study. J. Am. Soc. Nephrol. 2017, 28, 1859–1866. [Google Scholar] [CrossRef]
- Wang, Q.; Su, W.; Shen, Z.; Wang, R. Correlation between Soluble alpha-Klotho and Renal Function in Patients with Chronic Kidney Disease: A Review and Meta-Analysis. Biomed. Res. Int. 2018, 2018, 9481475. [Google Scholar]
- Memmos, E.; Sarafidis, P.; Pateinakis, P.; Tsiantoulas, A.; Faitatzidou, D.; Giamalis, P.; Vasilikos, V.; Papagianni, A. Soluble Klotho is associated with mortality and cardiovascular events in hemodialysis. BMC Nephrol. 2019, 20, 217. [Google Scholar] [CrossRef]
- Hu, M.C.; Shi, M.; Zhang, J.; Quinones, H.; Griffith, C.; Kuro-o, M.; Moe, O.W. Klotho deficiency causes vascular calcification in chronic kidney disease. J. Am. Soc. Nephrol. 2011, 22, 124–136. [Google Scholar] [CrossRef]
- Pan, H.C.; Chou, K.M.; Lee, C.C.; Yang, N.I.; Sun, C.Y. Circulating Klotho levels can predict long-term macrovascular outcomes in type 2 diabetic patients. Atherosclerosis 2018, 276, 83–90. [Google Scholar] [CrossRef]
- Matsumura, Y.; Aizawa, H.; Shiraki-Iida, T.; Nagai, R.; Kuro-o, M.; Nabeshima, Y. Identification of the human klotho gene and its two transcripts encoding membrane and secreted klotho protein. Biochem. Biophys. Res. Commun. 1998, 242, 626–630. [Google Scholar] [CrossRef]
- Hu, M.C.; Shi, M.; Cho, H.J.; Adams-Huet, B.; Paek, J.; Hill, K.; Shelton, J.; Amaral, A.P.; Faul, C.; Taniguchi, M.; et al. Klotho and phosphate are modulators of pathologic uremic cardiac remodeling. J. Am. Soc. Nephrol. 2015, 26, 1290–1302. [Google Scholar] [CrossRef]
- Xu, Y.; Sun, Z. Molecular basis of Klotho: From gene to function in aging. Endocr. Rev. 2015, 36, 174–193. [Google Scholar] [CrossRef]
- Chen, C.D.; Podvin, S.; Gillespie, E.; Leeman, S.E.; Abraham, C.R. Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proc. Natl. Acad. Sci. USA 2007, 104, 19796–19801. [Google Scholar] [CrossRef]
- Bloch, L.; Sineshchekova, O.; Reichenbach, D.; Reiss, K.; Saftig, P.; Kuro-o, M.; Kaether, C. Klotho is a substrate for alpha-, beta- and gamma-secretase. FEBS Lett. 2009, 583, 3221–3224. [Google Scholar] [CrossRef]
- Shiraki-Iida, T.; Aizawa, H.; Matsumura, Y.; Sekine, S.; Iida, A.; Anazawa, H.; Nagai, R.; Kuro-o, M.; Nabeshima, Y. Structure of the mouse klotho gene and its two transcripts encoding membrane and secreted protein. FEBS Lett. 1998, 424, 6–10. [Google Scholar] [CrossRef]
- Mencke, R.; Harms, G.; Moser, J.; van Meurs, M.; Diepstra, A.; Leuvenink, H.G.; Hillebrands, J.L. Human alternative Klotho mRNA is a nonsense-mediated mRNA decay target inefficiently spliced in renal disease. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Tohyama, O.; Imura, A.; Iwano, A.; Freund, J.N.; Henrissat, B.; Fujimori, T.; Nabeshima, Y. Klotho is a novel beta-glucuronidase capable of hydrolyzing steroid beta-glucuronides. J. Biol. Chem. 2004, 279, 9777–9784. [Google Scholar] [CrossRef]
- Chen, G.; Liu, Y.; Goetz, R.; Fu, L.; Jayaraman, S.; Hu, M.C.; Moe, O.W.; Liang, G.; Li, X.; Mohammadi, M. alpha-Klotho is a non-enzymatic molecular scaffold for FGF23 hormone signalling. Nature 2018, 553, 461–466. [Google Scholar] [CrossRef]
- Hu, M.C.; Shi, M.; Zhang, J.; Quinones, H.; Kuro-o, M.; Moe, O.W. Klotho deficiency is an early biomarker of renal ischemia-reperfusion injury and its replacement is protective. Kidney Int. 2010, 78, 1240–1251. [Google Scholar] [CrossRef]
- Lindberg, K.; Amin, R.; Moe, O.W.; Hu, M.C.; Erben, R.G.; Ostman Wernerson, A.; Lanske, B.; Olauson, H.; Larsson, T.E. The kidney is the principal organ mediating klotho effects. J. Am. Soc. Nephrol. 2014, 25, 2169–2175. [Google Scholar] [CrossRef]
- Kakareko, K.; Rydzewska-Rosolowska, A.; Brzosko, S.; Gozdzikiewicz-Lapinska, J.; Koc-Zorawska, E.; Samocik, P.; Kozlowski, R.; Mysliwiec, M.; Naumnik, B.; Hryszko, T. The effect of nephrectomy on Klotho, FGF-23 and bone metabolism. Int. Urol. Nephrol. 2017, 49, 681–688. [Google Scholar] [CrossRef]
- Lim, K.; Hamano, T.; Thadhani, R. Vitamin D and Calcimimetics in Cardiovascular Disease. Semin. Nephrol. 2018, 38, 251–266. [Google Scholar] [CrossRef]
- Liu, S.; Zhou, J.; Tang, W.; Jiang, X.; Rowe, D.W.; Quarles, L.D. Pathogenic role of Fgf23 in Hyp mice. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E38–E49. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.Q.; Ward, L.M.; Liu, S.; Lu, Y.; Xie, Y.; Yuan, B.; Yu, X.; Rauch, F.; Davis, S.I.; Zhang, S.; et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat. Genet. 2006, 38, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Kakitani, M.; Yamazaki, Y.; Hasegawa, H.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Tomizuka, K.; Yamashita, T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J. Clin. Investig. 2004, 113, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Tsujikawa, H.; Kurotaki, Y.; Fujimori, T.; Fukuda, K.; Nabeshima, Y. Klotho, a gene related to a syndrome resembling human premature aging, functions in a negative regulatory circuit of vitamin D endocrine system. Mol. Endocrinol. 2003, 17, 2393–2403. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, J.R.; Liu, S.; Tang, W.; Zhou, J.; Wang, Y.; Yao, X.; Quarles, L.D. Role of hyperphosphatemia and 1,25-dihydroxyvitamin D in vascular calcification and mortality in fibroblastic growth factor 23 null mice. J. Am. Soc. Nephrol. 2007, 18, 2116–2124. [Google Scholar] [CrossRef]
- Urakawa, I.; Yamazaki, Y.; Shimada, T.; Iijima, K.; Hasegawa, H.; Okawa, K.; Fujita, T.; Fukumoto, S.; Yamashita, T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 2006, 444, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Goetz, R.; Beenken, A.; Ibrahimi, O.A.; Kalinina, J.; Olsen, S.K.; Eliseenkova, A.V.; Xu, C.; Neubert, T.A.; Zhang, F.; Linhardt, R.J.; et al. Molecular insights into the klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol. Cell Biol. 2007, 27, 3417–3428. [Google Scholar] [CrossRef]
- Goetz, R.; Nakada, Y.; Hu, M.C.; Kurosu, H.; Wang, L.; Nakatani, T.; Shi, M.; Eliseenkova, A.V.; Razzaque, M.S.; Moe, O.W.; et al. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc. Natl. Acad. Sci. USA 2010, 107, 407–412. [Google Scholar] [CrossRef]
- Jimbo, R.; Kawakami-Mori, F.; Mu, S.; Hirohama, D.; Majtan, B.; Shimizu, Y.; Yatomi, Y.; Fukumoto, S.; Fujita, T.; Shimosawa, T. Fibroblast growth factor 23 accelerates phosphate-induced vascular calcification in the absence of Klotho deficiency. Kidney Int. 2014, 85, 1103–1111. [Google Scholar] [CrossRef]
- Murer, H.; Forster, I.; Biber, J. The sodium phosphate cotransporter family SLC34. Pflugers Arch. 2004, 447, 763–767. [Google Scholar] [CrossRef]
- Meyer, M.B.; Benkusky, N.A.; Kaufmann, M.; Lee, S.M.; Onal, M.; Jones, G.; Pike, J.W. A kidney-specific genetic control module in mice governs endocrine regulation of the cytochrome P450 gene Cyp27b1 essential for vitamin D3 activation. J. Biol. Chem. 2017, 292, 17541–17558. [Google Scholar] [CrossRef]
- Di Marco, G.S.; Hausberg, M.; Hillebrand, U.; Rustemeyer, P.; Wittkowski, W.; Lang, D.; Pavenstadt, H. Increased inorganic phosphate induces human endothelial cell apoptosis in vitro. Am. J. Physiol. Renal Physiol. 2008, 294, F1381–F1387. [Google Scholar] [CrossRef]
- Jono, S.; McKee, M.D.; Murry, C.E.; Shioi, A.; Nishizawa, Y.; Mori, K.; Morii, H.; Giachelli, C.M. Phosphate regulation of vascular smooth muscle cell calcification. Circ. Res. 2000, 87, E10–E17. [Google Scholar] [CrossRef]
- Kuro, O.M. Molecular Mechanisms Underlying Accelerated Aging by Defects in the FGF23-Klotho System. Int. J. Nephrol. 2018, 2018, 9679841. [Google Scholar] [CrossRef]
- Kuro, O.M. The Klotho proteins in health and disease. Nat. Rev. Nephrol. 2019, 15, 27–44. [Google Scholar] [CrossRef]
- Reiser, J.; Polu, K.R.; Moller, C.C.; Kenlan, P.; Altintas, M.M.; Wei, C.; Faul, C.; Herbert, S.; Villegas, I.; Avila-Casado, C.; et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat. Genet. 2005, 37, 739–744. [Google Scholar] [CrossRef]
- Haruna, Y.; Kashihara, N.; Satoh, M.; Tomita, N.; Namikoshi, T.; Sasaki, T.; Fujimori, T.; Xie, P.; Kanwar, Y.S. Amelioration of progressive renal injury by genetic manipulation of Klotho gene. Proc. Natl. Acad. Sci. USA 2007, 104, 2331–2336. [Google Scholar] [CrossRef]
- Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Gurnani, P.; Nandi, A.; Kurosu, H.; Miyoshi, M.; Ogawa, Y.; Castrillon, D.H.; Rosenblatt, K.P.; et al. Regulation of oxidative stress by the anti-aging hormone klotho. J. Biol. Chem. 2005, 280, 38029–38034. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, M.M.; Cai, Y.; Zheng, M.F.; Sun, W.L.; Zhang, S.Y.; Kong, W.; Gu, J.; Wang, X.; Xu, M.J. Mammalian target of rapamycin signaling inhibition ameliorates vascular calcification via Klotho upregulation. Kidney Int. 2015, 88, 711–721. [Google Scholar] [CrossRef]
- Rakugi, H.; Matsukawa, N.; Ishikawa, K.; Yang, J.; Imai, M.; Ikushima, M.; Maekawa, Y.; Kida, I.; Miyazaki, J.; Ogihara, T. Anti-oxidative effect of Klotho on endothelial cells through cAMP activation. Endocrine 2007, 31, 82–87. [Google Scholar] [CrossRef]
- Imai, M.; Ishikawa, K.; Matsukawa, N.; Kida, I.; Ohta, J.; Ikushima, M.; Chihara, Y.; Rui, X.; Rakugi, H.; Ogihara, T. Klotho protein activates the PKC pathway in the kidney and testis and suppresses 25-hydroxyvitamin D3 1alpha-hydroxylase gene expression. Endocrine 2004, 25, 229–234. [Google Scholar] [CrossRef]
- Doi, S.; Zou, Y.; Togao, O.; Pastor, J.V.; John, G.B.; Wang, L.; Shiizaki, K.; Gotschall, R.; Schiavi, S.; Yorioka, N.; et al. Klotho inhibits transforming growth factor-beta1 (TGF-beta1) signaling and suppresses renal fibrosis and cancer metastasis in mice. J. Biol. Chem. 2011, 286, 8655–8665. [Google Scholar] [CrossRef] [PubMed]
- Ikushima, M.; Rakugi, H.; Ishikawa, K.; Maekawa, Y.; Yamamoto, K.; Ohta, J.; Chihara, Y.; Kida, I.; Ogihara, T. Anti-apoptotic and anti-senescence effects of Klotho on vascular endothelial cells. Biochem. Biophys. Res. Commun. 2006, 339, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Fergusson, M.M.; Castilho, R.M.; Liu, J.; Cao, L.; Chen, J.; Malide, D.; Rovira, I.I.; Schimel, D.; Kuo, C.J.; et al. Augmented Wnt signaling in a mammalian model of accelerated aging. Science 2007, 317, 803–806. [Google Scholar] [CrossRef] [PubMed]
- Semba, R.D.; Cappola, A.R.; Sun, K.; Bandinelli, S.; Dalal, M.; Crasto, C.; Guralnik, J.M.; Ferrucci, L. Plasma klotho and mortality risk in older community-dwelling adults. J. Gerontol. A Biol. Sci. Med. Sci. 2011, 66, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Arking, D.E.; Atzmon, G.; Arking, A.; Barzilai, N.; Dietz, H.C. Association between a functional variant of the KLOTHO gene and high-density lipoprotein cholesterol, blood pressure, stroke, and longevity. Circ. Res. 2005, 96, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Semba, R.D.; Cappola, A.R.; Sun, K.; Bandinelli, S.; Dalal, M.; Crasto, C.; Guralnik, J.M.; Ferrucci, L. Plasma klotho and cardiovascular disease in adults. J. Am. Geriatr. Soc. 2011, 59, 1596–1601. [Google Scholar] [CrossRef] [PubMed]
- Lau, W.L.; Leaf, E.M.; Hu, M.C.; Takeno, M.M.; Kuro-o, M.; Moe, O.W.; Giachelli, C.M. Vitamin D receptor agonists increase klotho and osteopontin while decreasing aortic calcification in mice with chronic kidney disease fed a high phosphate diet. Kidney Int. 2012, 82, 1261–1270. [Google Scholar] [CrossRef]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial calcification in chronic kidney disease: Key roles for calcium and phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef]
- Ragnauth, C.D.; Warren, D.T.; Liu, Y.; McNair, R.; Tajsic, T.; Figg, N.; Shroff, R.; Skepper, J.; Shanahan, C.M. Prelamin A acts to accelerate smooth muscle cell senescence and is a novel biomarker of human vascular aging. Circulation 2010, 121, 2200–2210. [Google Scholar] [CrossRef]
- Reynolds, J.L.; Joannides, A.J.; Skepper, J.N.; McNair, R.; Schurgers, L.J.; Proudfoot, D.; Jahnen-Dechent, W.; Weissberg, P.L.; Shanahan, C.M. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: A potential mechanism for accelerated vascular calcification in ESRD. J. Am. Soc. Nephrol. 2004, 15, 2857–2867. [Google Scholar] [CrossRef] [PubMed]
- Shaw, L.J.; Raggi, P.; Berman, D.S.; Callister, T.Q. Coronary artery calcium as a measure of biologic age. Atherosclerosis 2006, 188, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Girndt, M.; Seibert, E. Premature cardiovascular disease in chronic renal failure (CRF): A model for an advanced ageing process. Exp. Gerontol. 2010, 45, 797–800. [Google Scholar] [CrossRef] [PubMed]
- Blacher, J.; Safar, M.E.; Guerin, A.P.; Pannier, B.; Marchais, S.J.; London, G.M. Aortic pulse wave velocity index and mortality in end-stage renal disease. Kidney Int. 2003, 63, 1852–1860. [Google Scholar] [CrossRef] [PubMed]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.H.; Kuro, O.M.; Chen, C.H.; Sue, Y.M.; Chen, Y.C.; Wu, H.H.; Cheng, C.Y. The secreted Klotho protein restores phosphate retention and suppresses accelerated aging in Klotho mutant mice. Eur. J. Pharmacol. 2013, 698, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Ginsberg, C.; Sugatani, T.; Monier-Faugere, M.C.; Malluche, H.; Hruska, K.A. Early chronic kidney disease-mineral bone disorder stimulates vascular calcification. Kidney Int. 2014, 85, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, K.; Olauson, H.; Amin, R.; Ponnusamy, A.; Goetz, R.; Taylor, R.F.; Mohammadi, M.; Canfield, A.; Kublickiene, K.; Larsson, T.E. Arterial klotho expression and FGF23 effects on vascular calcification and function. PLoS ONE 2013, 8, e60658. [Google Scholar] [CrossRef]
- Yamazaki, M.; Ozono, K.; Okada, T.; Tachikawa, K.; Kondou, H.; Ohata, Y.; Michigami, T. Both FGF23 and extracellular phosphate activate Raf/MEK/ERK pathway via FGF receptors in HEK293 cells. J. Cell. Biochem. 2010, 111, 1210–1221. [Google Scholar] [CrossRef]
- Donate-Correa, J.; Mora-Fernandez, C.; Martinez-Sanz, R.; Muros-de-Fuentes, M.; Perez, H.; Meneses-Perez, B.; Cazana-Perez, V.; Navarro-Gonzalez, J.F. Expression of FGF23/KLOTHO system in human vascular tissue. Int. J. Cardiol. 2013, 165, 179–183. [Google Scholar] [CrossRef]
- Zhu, D.; Mackenzie, N.C.; Millan, J.L.; Farquharson, C.; MacRae, V.E. A protective role for FGF-23 in local defence against disrupted arterial wall integrity? Mol. Cell. Endocrinol. 2013, 372, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Scialla, J.J.; Lau, W.L.; Reilly, M.P.; Isakova, T.; Yang, H.Y.; Crouthamel, M.H.; Chavkin, N.W.; Rahman, M.; Wahl, P.; Amaral, A.P.; et al. Fibroblast growth factor 23 is not associated with and does not induce arterial calcification. Kidney Int. 2013, 83, 1159–1168. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Gonzalez, J.F.; Donate-Correa, J.; Muros de Fuentes, M.; Perez-Hernandez, H.; Martinez-Sanz, R.; Mora-Fernandez, C. Reduced Klotho is associated with the presence and severity of coronary artery disease. Heart 2014, 100, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A., Jr.; Garcia-Cardena, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Six, I.; Okazaki, H.; Gross, P.; Cagnard, J.; Boudot, C.; Maizel, J.; Drueke, T.B.; Massy, Z.A. Direct, acute effects of Klotho and FGF23 on vascular smooth muscle and endothelium. PLoS ONE 2014, 9, e93423. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Yamagishi, T.; Nakamura, T.; Ohyama, Y.; Aizawa, H.; Suga, T.; Matsumura, Y.; Masuda, H.; Kurabayashi, M.; Kuro-o, M.; et al. Klotho protein protects against endothelial dysfunction. Biochem. Biophys. Res. Commun. 1998, 248, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Nakamura, T.; Ohyama, Y.; Suzuki, T.; Iida, A.; Shiraki-Iida, T.; Kuro-o, M.; Nabeshima, Y.; Kurabayashi, M.; Nagai, R. In vivo klotho gene delivery protects against endothelial dysfunction in multiple risk factor syndrome. Biochem. Biophys. Res. Commun. 2000, 276, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Izbeki, F.; Asuzu, D.T.; Lorincz, A.; Bardsley, M.R.; Popko, L.N.; Choi, K.M.; Young, D.L.; Hayashi, Y.; Linden, D.R.; Kuro-o, M.; et al. Loss of Kitlow progenitors, reduced stem cell factor and high oxidative stress underlie gastric dysfunction in progeric mice. J. Physiol. 2010, 588 Pt 16, 3101–3117. [Google Scholar] [CrossRef]
- Wang, Y.; Kuro-o, M.; Sun, Z. Klotho gene delivery suppresses Nox2 expression and attenuates oxidative stress in rat aortic smooth muscle cells via the cAMP-PKA pathway. Aging Cell 2012, 11, 410–417. [Google Scholar] [CrossRef]
- Emerling, B.M.; Weinberg, F.; Liu, J.L.; Mak, T.W.; Chandel, N.S. PTEN regulates p300-dependent hypoxia-inducible factor 1 transcriptional activity through Forkhead transcription factor 3a (FOXO3a). Proc. Natl. Acad. Sci. USA 2008, 105, 2622–2627. [Google Scholar] [CrossRef]
- Mitobe, M.; Yoshida, T.; Sugiura, H.; Shirota, S.; Tsuchiya, K.; Nihei, H. Oxidative stress decreases klotho expression in a mouse kidney cell line. Nephron Exp. Nephrol. 2005, 101, e67–e74. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wang, X.; Zhao, W.; Wu, J. Klotho inhibits growth and promotes apoptosis in human lung cancer cell line A549. J. Exp. Clin. Cancer Res. 2010, 29, 99. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, Z. Klotho gene delivery prevents the progression of spontaneous hypertension and renal damage. Hypertension 2009, 54, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, Y.; Ishikawa, K.; Yasuda, O.; Oguro, R.; Hanasaki, H.; Kida, I.; Takemura, Y.; Ohishi, M.; Katsuya, T.; Rakugi, H. Klotho suppresses TNF-alpha-induced expression of adhesion molecules in the endothelium and attenuates NF-kappaB activation. Endocrine 2009, 35, 341–346. [Google Scholar] [CrossRef]
- Liu, F.; Wu, S.; Ren, H.; Gu, J. Klotho suppresses RIG-I-mediated senescence-associated inflammation. Nat. Cell Biol. 2011, 13, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Fan, J.; Wang, S.; Sun, Z. Secreted Klotho Attenuates Inflammation-Associated Aortic Valve Fibrosis in Senescence-Accelerated Mice P1. Hypertension 2018, 71, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Abraham, C.R.; Chen, C.; Cuny, G.D.; Glicksman, M.A.; Zeldich, E. Small-molecule Klotho enhancers as novel treatment of neurodegeneration. Future Med. Chem. 2012, 4, 1671–1679. [Google Scholar] [CrossRef]
- Cheikhi, A.; Barchowsky, A.; Sahu, A.; Shinde, S.N.; Pius, A.; Clemens, Z.J.; Li, H.; Kennedy, C.A.; Hoeck, J.D.; Franti, M.; et al. Klotho: An Elephant in Aging Research. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Brownstein, C.A.; Adler, F.; Nelson-Williams, C.; Iijima, J.; Li, P.; Imura, A.; Nabeshima, Y.; Reyes-Mugica, M.; Carpenter, T.O.; Lifton, R.P. A translocation causing increased alpha-klotho level results in hypophosphatemic rickets and hyperparathyroidism. Proc. Natl. Acad. Sci. USA 2008, 105, 3455–3460. [Google Scholar] [CrossRef]
- Smart, A.; Martin, P. The promise of pharmacogenetics: Assessing the prospects for disease and patient stratification. Stud. Hist. Philos. Biol. Biomed. Sci. 2006, 37, 583–601. [Google Scholar] [CrossRef]
- Gat-Viks, I.; Chevrier, N.; Wilentzik, R.; Eisenhaure, T.; Raychowdhury, R.; Steuerman, Y.; Shalek, A.K.; Hacohen, N.; Amit, I.; Regev, A. Deciphering molecular circuits from genetic variation underlying transcriptional responsiveness to stimuli. Nat. Biotechnol. 2013, 31, 342–349. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Barker, S.L.; Pastor, J.; Carranza, D.; Quinones, H.; Griffith, C.; Goetz, R.; Mohammadi, M.; Ye, J.; Zhang, J.; Hu, M.C.; et al. The demonstration of alphaKlotho deficiency in human chronic kidney disease with a novel synthetic antibody. Nephrol. Dial. Transplant. 2015, 30, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Shi, M.; Zhang, J.; Addo, T.; Cho, H.J.; Barker, S.L.; Ravikumar, P.; Gillings, N.; Bian, A.; Sidhu, S.S.; et al. Renal Production, Uptake, and Handling of Circulating alphaKlotho. J. Am. Soc. Nephrol. 2016, 27, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, L.; Pedersen, S.M.; Brasen, C.L.; Rasmussen, L.M. Soluble serum Klotho levels in healthy subjects. Comparison of two different immunoassays. Clin. Biochem. 2013, 46, 1079–1083. [Google Scholar] [CrossRef] [PubMed]
- Neyra, J.A.; Moe, O.W.; Pastor, J.; Gianella, F.; Sidhu, S.S.; Sarnak, M.J.; Ix, J.H.; Drew, D.A. Performance of soluble Klotho assays in clinical samples of kidney disease. Clin. Kidney J. 2019. [Google Scholar] [CrossRef]
- Adema, A.Y.; Vervloet, M.G.; Blankenstein, M.A.; Heijboer, A.C. alpha-Klotho is unstable in human urine. Kidney Int. 2015, 88, 1442–1444. [Google Scholar] [CrossRef] [PubMed]
- Grabner, A.; Faul, C. The role of fibroblast growth factor 23 and Klotho in uremic cardiomyopathy. Curr. Opin. Nephrol. Hypertens 2016, 25, 314–324. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Arterial Klotho Expression | Experimental Observations | Reference |
|---|---|---|
| Decreased mRNA and protein in human CKD | Human aorta Klotho deficiency in CKD can be reversed and calcification is attenuated ex vivo with VDRA | Lim et al. [13] |
| Decreased mRNA and protein in CKD mice | Low aortic Klotho but high circulating Klotho associated with vascular calcification in ldlr -/- CKD mice | Fang et al. [78] |
| mRNA but no protein in mouse aorta | Aortic Klotho has no role in vascular calcification | Lindberg et al. [79] |
| mRNA in human aorta, coronary arteries and thrombus | Klotho mRNA detectable in human arteries and thrombi of occlusive coronary disease | Donate-Correa et al. [81] |
| Increased mRNA and protein in calcified aorta of Enpp1-/- mice | Increased Klotho associated with decreased vascular calcification in CKD mice | Zhu et al. [82] |
| mRNA and protein expression in rat aorta but not in rat vascular smooth muscle cells | No native VSMC Klotho expression, however overexpression worsens calcification | Jimbo et al. [50] |
| No mRNA or protein expression in mouse aorta | VDRA in vivo increases plasma αKlotho an decreases vascular calcification in CKD mice | Lau et al. [69] |
| No mRNA in normal and calcified aortas of CKD mice | No aortic Klotho expression and no Klotho effect in vitro | Scialla et al. [83] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, K.; Halim, A.; Lu, T.-s.; Ashworth, A.; Chong, I. Klotho: A Major Shareholder in Vascular Aging Enterprises. Int. J. Mol. Sci. 2019, 20, 4637. https://doi.org/10.3390/ijms20184637
Lim K, Halim A, Lu T-s, Ashworth A, Chong I. Klotho: A Major Shareholder in Vascular Aging Enterprises. International Journal of Molecular Sciences. 2019; 20(18):4637. https://doi.org/10.3390/ijms20184637
Chicago/Turabian StyleLim, Kenneth, Arvin Halim, Tzong-shi Lu, Alan Ashworth, and Irene Chong. 2019. "Klotho: A Major Shareholder in Vascular Aging Enterprises" International Journal of Molecular Sciences 20, no. 18: 4637. https://doi.org/10.3390/ijms20184637
APA StyleLim, K., Halim, A., Lu, T.-s., Ashworth, A., & Chong, I. (2019). Klotho: A Major Shareholder in Vascular Aging Enterprises. International Journal of Molecular Sciences, 20(18), 4637. https://doi.org/10.3390/ijms20184637