Polymorphisms of CYP2C8 Alter First-Electron Transfer Kinetics and Increase Catalytic Uncoupling


Abstract
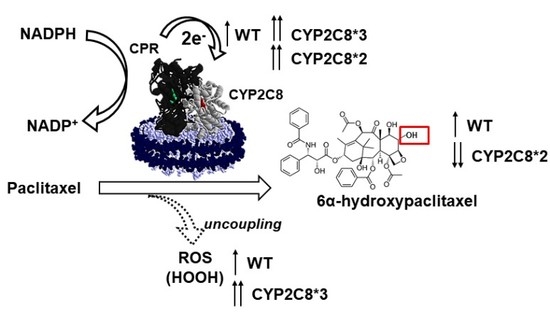
1. Introduction
2. Results and Discussion
2.1. P450 Characterization of CYP2C8*1/*2/*3/R139/K399-ND
2.2. Effect of Polymorphisms on PAC Metabolism
2.3. Polymorphisms in CYP2C8 Lead to Greater HOOH Uncoupling
2.4. Spectral Characterization of Substrate Binding and Reduction Potentials of CYP2C8 Polymorphisms
2.5. Polymorphisms Show Altered First Electron Transfer (FET) Kinetics as Determined by CO Stopped-Flow
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Protein Engineering of CYP2C8*1/*2/*3/R139K/K399R
4.3. Protein Expression and Purification of CYP2C8*1/*2/*3/R139K/K399R
4.4. Expression of Cytochrome P450 Reductase
4.5. Assembly of CYP2C8-Nanodiscs
4.6. Carbon Monoxide Binding Assay
4.7. Paclitaxel Metabolism
4.8. Tandem LC–MS/MS for the Quantification of 6α-Hydroxypaclitaxel
4.9. HOOH Measurements
4.10. NADPH Assay
4.11. Stopped-flow Kinetics of Electron Transfer
4.12. Data Analysis of Stopped-Flow Experiments
4.13. Reduction Potential
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Amp | Ampicillin |
| AA | Arachidonic acid |
| Chlr | Chloramphenicol |
| CYP | Cytochrome P450 |
| CYP2C8 | Cytochrome P450 2C8 |
| CYP2C8*3 | Cytochrome P450 2C8 R139K/K399R |
| CYP2C8*2 | Cytochrome P450 2C8 I269F |
| CPR | Cytochrome P450 reductase |
| δ-ALA | δ-Aminolevulinic acid |
| EETs | Epoxyeicosatrienoic acids |
| FET | First electron transfer |
| HOOH | Hydrogen peroxide |
| IPTG | Isopropyl β-D-1-thiogalactopyranoside |
| ND | Nanodisc |
| POPC | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine |
| PAC | Paclitaxel |
| PAC-OH | 6α-Hydroxypaclitaxel |
| POPS | 1-Palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine |
| ROS | Reactive oxygen species |
References
- VandenBrink, B.M.; Foti, R.S.; Rock, D.A.; Wienkers, L.C.; Wahlstrom, J.L. Evaluation of CYP2C8 inhibition in vitro: Utility of montelukast as a selective CYP2C8 probe substrate. Drug Metab. Dispos. 2011, 39, 1546–1554. [Google Scholar] [CrossRef] [PubMed]
- Bahadur, N.; Leathart, J.B.S.; Mutch, E.; Steimel-Crespi, D.; Dunn, S.A.; Gilissen, R.; Houdt, J.V.; Hendrickx, J.; Mannens, G.; Bohets, H. Daly, CYP2C8 polymorphisms in caucasians and their relationship with paclitaxel 6α-hydroxylase activity in human liver microsomes. Biochem. Pharmacol. 2002, 64, 1579–1589. [Google Scholar] [CrossRef]
- Lundblad, M.S.; Stark, K.; Eliasson, E.; Oliw, E.; Rane, A. Biosynthesis of epoxyeicosatrienoic acids varies between polymorphic CYP2C enzymes. Biochem. Biophys. Res. Commun. 2005, 327, 1052–1057. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Marupudi, N.I.; Han, J.E.; Li, K.W.; Renard, V.M.; Tyler, B.M.; Brem, H. Paclitaxel: a review of adverse toxicities and novel delivery strategies. Expert Opin. Drug Saf. 2007, 6, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Klose, T.S.; Blaisdell, J.A.; Goldstein, J.A. Gene structure of CYP2C8 and extrahepatic distribution of the human CYP2Cs. J. Biochem. Mol. Toxicol. 1999, 13, 289–295. [Google Scholar] [CrossRef]
- Enayetallah, A.E.; French, R.A.; Thibodeau, M.S.; Grant, D.F. Distribution of soluble epoxide hydrolase and of cytochrome P450 2C8, 2C9, and 2J2 in human tissues. J. Histochem. Cytochem. 2004, 52, 447–454. [Google Scholar] [CrossRef] [PubMed]
- DeLozier, T.C.; Kissling, G.E.; Coulter, S.J.; Dai, D.; Foley, J.F.; Bradbury, J.A.; Murphy, E.; Steenbergen, C.; Zeldin, D.C.; Goldstein, J.A. Detection of human CYP2C8, CYP2C9, and CYP2J2 in cardiovascular tissues. Drug Metab. Dispos. 2007, 35, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Node, K.; Huo, Y.; Ruan, X.; Yang, B.; Spiecker, M.; Ley, K.; Zeldin, D.C.; Liao, J.K. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 1999, 285, 1276–1279. [Google Scholar] [CrossRef]
- Michaelis, U.R.; Fisslthaler, B.; Medhora, M.; Harder, D.; Fleming, I.; Busse, R. Cytochrome P450 2C9-derived epoxyeicosatrienoic acids induce angiogenesis via cross-talk with the epidermal growth factor receptor. FASEB J. 2003. [Google Scholar] [CrossRef]
- Sun, J.; Sui, X.; Bradbury, J.A.; Zeldin, D.C.; Conte, M.S.; Liao, J.K. Inhibition of vascular smooth muscle cell migration by cytochrome P450 epoxygenase-derived eicosanoids. Circ. Res. 2002, 90, 1020–1027. [Google Scholar] [CrossRef]
- Zhou, S.F. Polymorphism of human cytochrome P450 2D6 and its clinical significance: Part I. Clin. Pharmacokinet. 2009, 48, 689–723. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F. Polymorphism of human cytochrome P450 2D6 and its clinical significance: part II. Clin. Pharmacokinet. 2009, 48, 761–804. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.; Zeldin, D.C.; Blaisdell, J.A.; Chanas, B.; Coulter, S.J.; Ghanayem, B.I.; Goldstein, J.A. Polymorphisms in human CYP2C8 decrease metabolism of the anticancer drug paclitaxel and arachidonic acid. Pharmacogenet. Genomics 2001, 11, 597–607. [Google Scholar] [CrossRef]
- Yu, L.; Shi, D.; Ma, L.; Zhou, Q.; Zeng, S. Influence of CYP2C8 polymorphisms on the hydroxylation metabolism of paclitaxel, repaglinide and ibuprofen enantiomers in vitro. Biopharm. Drug Dispos. 2013, 34, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Hertz, D.; Roy, S.; Jack, J.; Motsinger-Reif, A.; Drobish, A.; Clark, L.S.; Carey, L.; Dees, E.C.; McLeod, H. Genetic heterogeneity beyond CYP2C8*3 does not explain differential sensitivity to paclitaxel-induced neuropathy. Breast Cancer Res. Treat. 2014, 145, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Hertz, D.L.; Roy, S.; Motsinger-Reif, A.A.; Drobish, A.; Clark, L.S.; McLeod, H.L.; Carey, L.A.; Dees, E.C. CYP2C8*3 increases risk of neuropathy in breast cancer patients treated with paclitaxel. Ann. Oncol. 2013, 24, 1472–1478. [Google Scholar] [CrossRef]
- Marcath, L.A.; Kidwell, K.M.; Robinson, A.C.; Vangipuram, K.; Burness, M.L.; Griggs, J.J.; Poznak, C.V.; Schott, A.F.; Hayes, D.F.; Henry, N.L.; et al. Patients carrying CYP2C8*3 have shorter systemic paclitaxel exposure. Pharmacogenomics 2019, 20, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Soyama, A.; Saito, Y.; Hanioka, N.; Murayama, N.; Nakajima, O.; Katori, N.; Ishida, S.; Sai, K.; Ozawa, S.; Sawada, J.I. Non-synonymous single nucleotide alterations found in the CYP2C8 gene result in reduced in vitro paclitaxel metabolism. Biol. Pharm. Bull. 2001, 24, 1427–1430. [Google Scholar] [CrossRef]
- Kaspera, R.; Naraharisetti, S.B.; Evangelista, E.A.; Marciante, K.D.; Psaty, B.M.; Totah, R.A. Drug metabolism by CYP2C8.3 is determined by substrate dependent interactions with cytochrome P450 reductase and cytochrome b5. Biochem. Pharmacol. 2011, 82, 681–691. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, D.; Wang, H.; Zhu, J.; Chen, C. Functional characterization of five CYP2C8 variants and prediction of CYP2C8 genotype-dependent effects on in vitro and in vivo drug–drug interactions. Xenobiotica 2010, 40, 467–475. [Google Scholar] [CrossRef]
- Soyama, A.; Hanioka, N.; Saito, Y.; Murayama, N.; Ando, M.; Ozawa, S.; Sawada, J. Amiodarone N-deethylation by CYP2C8 and its variants, CYP2C8*3 and CYP2C8 P404A. Pharmacol. Toxicol. 2002, 91, 174–178. [Google Scholar] [CrossRef]
- Meunier, B.; de Visser, S.P.; Shaik, S. Mechanism of oxidation reactions catalyzed by cytochrome p450 enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef] [PubMed]
- Krest, C.M.; Onderko, E.L.; Yosca, T.H.; Calixto, J.C.; Karp, R.F.; Livada, J.; Rittle, J.; Green, M.T. Reactive intermediates in cytochrome p450 catalysis. J. Biol. Chem. 2013, 288, 17074–17081. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Mechanisms of cytochrome P450-catalyzed oxidations. ACS Catal. 2018, 8, 10964–10976. [Google Scholar] [CrossRef] [PubMed]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and chemistry of cytochrome P450. Chem. Rev. 2005, 105. [Google Scholar] [CrossRef] [PubMed]
- Fleming, I.; Michaelis, U.R.; Bredenkötter, D.; Fisslthaler, B.; Dehghani, F.; Brandes, R.P.; Busse, R. Endothelium-derived hyperpolarizing factor synthase (cytochrome P450 2C9) is a functionally significant source of reactive oxygen species in coronary arteries. Circ. Res. 2001, 88, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Edin, M.L.; Wang, Z.; Bradbury, J.A.; Graves, J.P.; Lih, F.B.; DeGraff, L.M.; Foley, J.F.; Torphy, R.; Ronnekleiv, O.K.; Tomer, K.B.; et al. Endothelial expression of human cytochrome P450 epoxygenase CYP2C8 increases susceptibility to ischemia-reperfusion injury in isolated mouse heart. FASEB J. 2011, 25, 3436–3447. [Google Scholar] [CrossRef] [PubMed]
- Nesnow, S.; Grindstaff, R.D.; Lambert, G.; Padgett, W.T.; Bruno, M.; Ge, Y.; Chen, P.J.; Wood, C.E.; Murphy, L. Propiconazole increases reactive oxygen species levels in mouse hepatic cells in culture and in mouse liver by a cytochrome P450 enzyme mediated process. Chem.-Biol. Interact. 2011, 194, 79–89. [Google Scholar] [CrossRef]
- Leung, T.; Rajendran, R.; Singh, S.; Garva, R.; Krstic-Demonacos, M.; Demonacos, C. Cytochrome P450 2E1 (CYP2E1) regulates the response to oxidative stress and migration of breast cancer cells. Breast Cancer Res. 2013, 15, R107. [Google Scholar] [CrossRef]
- Hunter, A.L.; Bai, N.; Laher, I.; Granville, D.J. Cytochrome p450 2C inhibition reduces post-ischemic vascular dysfunction. Vasc. Pharmacol. 2005, 43, 213–219. [Google Scholar] [CrossRef]
- Bayburt, T.H.; Grinkova, Y.V.; Sligar, S.G. Self-assembly of discoidal phospholipid bilayer nanoparticles with membrane scaffold proteins. Nano Lett. 2002, 2, 853–856. [Google Scholar] [CrossRef]
- Orlando, B.J.; McDougle, D.R.; Lucido, M.J.; Eng, E.T.; Graham, L.A.; Schneider, C.; Stokes, D.L.; Das, A.; Malkowski, M.G. Cyclooxygenase-2 catalysis and inhibition in lipid bilayer nanodiscs. Arch. Biochem. Biophys. 2014, 546, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Schoch, G.A.; Yano, J.K.; Wester, M.R.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. Structure of human microsomal cytochrome P450 2C8. Evidence for a peripheral fatty acid binding site. J. Biol. Chem. 2004, 279, 9497–9503. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.A. Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis; John Wiley & Sons: New York, NY, USA, 2004. [Google Scholar]
- Kim, H.K.; Park, S.K.; Zhou, J.L.; Taglialatela, G.; Chung, K.; Coggeshall, R.E.; Chung, J.M. Reactive oxygen species (ROS) play an important role in a rat model of neuropathic pain. Pain 2004, 111, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Scripture, C.D.; Figg, W.D.; Sparreboom, A. Peripheral neuropathy induced by paclitaxel: recent insights and future perspectives. Curr. Neuropharmacol. 2006, 4, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Varma, S.S.; Mularczyk, C.; Meling, D.D. Functional investigations of thromboxane synthase (CYP5A1) in lipid bilayers of nanodiscs. Chembiochem 2014, 15, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Grinkova, Y.V.; Sligar, S.G. Redox potential control by drug binding to cytochrome P450 3A4. J. Am. Chem. Soc. 2007, 129, 13778–13779. [Google Scholar] [CrossRef] [PubMed]
- Arnold, W.R.; Baylon, J.L.; Tajkhorshid, E.; Das, A. Asymmetric binding and metabolism of polyunsaturated fatty acids (PUFAs) by CYP2J2 epoxygenase. Biochemistry 2016, 55, 6969–6980. [Google Scholar] [CrossRef]
- Reed, R.; Hollenberg, P.F. New perspectives on the conformational equilibrium regulating multi-phasic reduction of cytochrome P450 2B4 by cytochrome P450 reductase. J. Inorg. Biochem. 2003, 97, 276–286. [Google Scholar] [CrossRef]
- Meling, D.D.; McDougle, D.R.; Das, A. CYP2J2 epoxygenase membrane anchor plays an important role in facilitating electron transfer from CPR. J. Inorg. Biochem. 2015, 142, 47–53. [Google Scholar] [CrossRef]
- Sokalingam, S.; Raghunathan, G.; Soundrarajan, N.; Lee, S.G. A study on the effect of surface lysine to arginine mutagenesis on protein stability and structure using green fluorescent protein. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Vorobyov, I.; Allen, T.W. The different interactions of lysine and arginine side chains with lipid membranes. J. Phys. Chem. 2013, 117, 11906–11920. [Google Scholar] [CrossRef]
- McDougle, D.R.; Palaria, A.; Magnetta, E.; Meling, D.D.; Das, A. Functional studies of N-terminally modified CYP2J2 epoxygenase in model lipid bilayers. Protein Sci. 2013. [Google Scholar] [CrossRef] [PubMed]
- Zelasko, S.; Palaria, A.; Das, A. Optimizations to achieve high-level expression of cytochrome P450 proteins using Escherichia coli expression systems. Protein Expr. Purif. 2013, 92, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Arnold, W.R.; Baylon, J.L.; Tajkhorshid, E.; Das, A. Arachidonic acid metabolism by human cardiovascular CYP2J2 is modulated by doxorubicin. Biochemistry 2017, 56, 6700–6712. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | Rate (pmol/min/nmolprot) | %WT | kcat/Km (min−1 nM−1) | %WT |
|---|---|---|---|---|
| CYP2C8*1 (WT) | 38.8 ± 0.2 | 100 | 0.381 | 100 |
| CYP2C8*2 | 18.3 ± 3.5 | 47.2 | 0.207 | 54.4 |
| CYP2C8*3 | 34.0 ± 3.0 | 88 | 0.321 | 84.4 |
| CYP2C8-R139K | 103 ± 2 | 265 | 1.02 | 269 |
| CYP2C8-K399R | 47.1 ± 4.3 | 121 | 0.441 | 116 |
| Variant | nmolHOOH/pmolPAC-OH (at 20 min) | %WT |
|---|---|---|
| CYP2C8*1 (WT) | 1.58 | 100 |
| CYP2C8*2 | 2.96 | 187 |
| CYP2C8*3 | 3.25 | 206 |
| CYP2C8-R139K | 0.46 | 29.6 |
| CYP2C8-K399R | 1.24 | 78.4 |
| Variant | Reduction Potential (V) |
|---|---|
| CYP2C8*1 No PAC | −0.283 ± 0.002 |
| CYP2C8*1 | −0.279 ± 0.002 |
| CYP2C8*2 | −0.297 ± 0.021 |
| CYP2C8*3 | −0.281 ± 0.001 |
| Variant | 1:1 CPR:CYP | 3:1 CPR:CYP | |
|---|---|---|---|
| k1 (ms−1) | k2 (ms−1) | k2 (ms−1) | |
| CYP2C8*1 | 468 ± 21 | 20.7 ± 0.4 | 9.70 ± 0.98 |
| CYP2C8*2 | --- | 8.67 ± 0.49 **** | 17.4 ± 1.6 *** |
| CYP2C8*3 | --- | 10.8 ± 1.84 **** | 13.7 ± 0.7 **** |
| CYP2C8-R139K | 406 ± 52 | 18.2 ± 0.65 | 6.02 ± 0.19 ** |
| CYP2C8-K399R | 445 ± 51 | 19.4 ± 0.99 | 3.90 ± 0.10 *** |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
R. Arnold, W.; Zelasko, S.; D. Meling, D.; Sam, K.; Das, A. Polymorphisms of CYP2C8 Alter First-Electron Transfer Kinetics and Increase Catalytic Uncoupling. Int. J. Mol. Sci. 2019, 20, 4626. https://doi.org/10.3390/ijms20184626
R. Arnold W, Zelasko S, D. Meling D, Sam K, Das A. Polymorphisms of CYP2C8 Alter First-Electron Transfer Kinetics and Increase Catalytic Uncoupling. International Journal of Molecular Sciences. 2019; 20(18):4626. https://doi.org/10.3390/ijms20184626
Chicago/Turabian StyleR. Arnold, William, Susan Zelasko, Daryl D. Meling, Kimberly Sam, and Aditi Das. 2019. "Polymorphisms of CYP2C8 Alter First-Electron Transfer Kinetics and Increase Catalytic Uncoupling" International Journal of Molecular Sciences 20, no. 18: 4626. https://doi.org/10.3390/ijms20184626
APA StyleR. Arnold, W., Zelasko, S., D. Meling, D., Sam, K., & Das, A. (2019). Polymorphisms of CYP2C8 Alter First-Electron Transfer Kinetics and Increase Catalytic Uncoupling. International Journal of Molecular Sciences, 20(18), 4626. https://doi.org/10.3390/ijms20184626