Calcium Mechanisms in Limb-Girdle Muscular Dystrophy with CAPN3 Mutations


Abstract
1. Overview of Calcium Homeostasis in the Skeletal Muscle
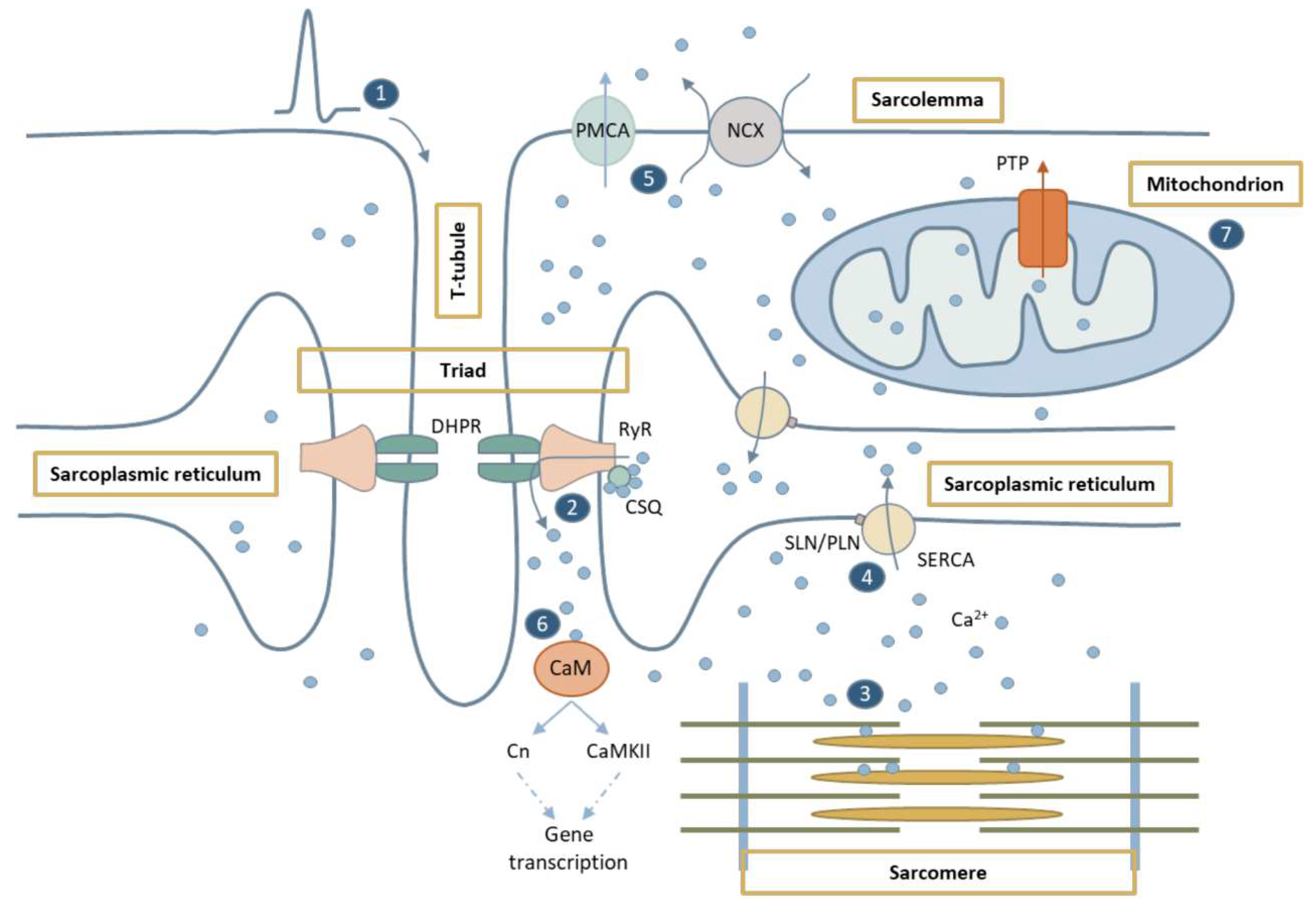
1.1. Ca2+ in Excitation-Contraction Coupling
1.2. Ca2+-Mediated Signaling Pathways
1.3. Ca2+ in Mitochondria
2. Limb-Girdle Muscular Dystrophy-Recessive 1
3. CAPN3 Localization and Function
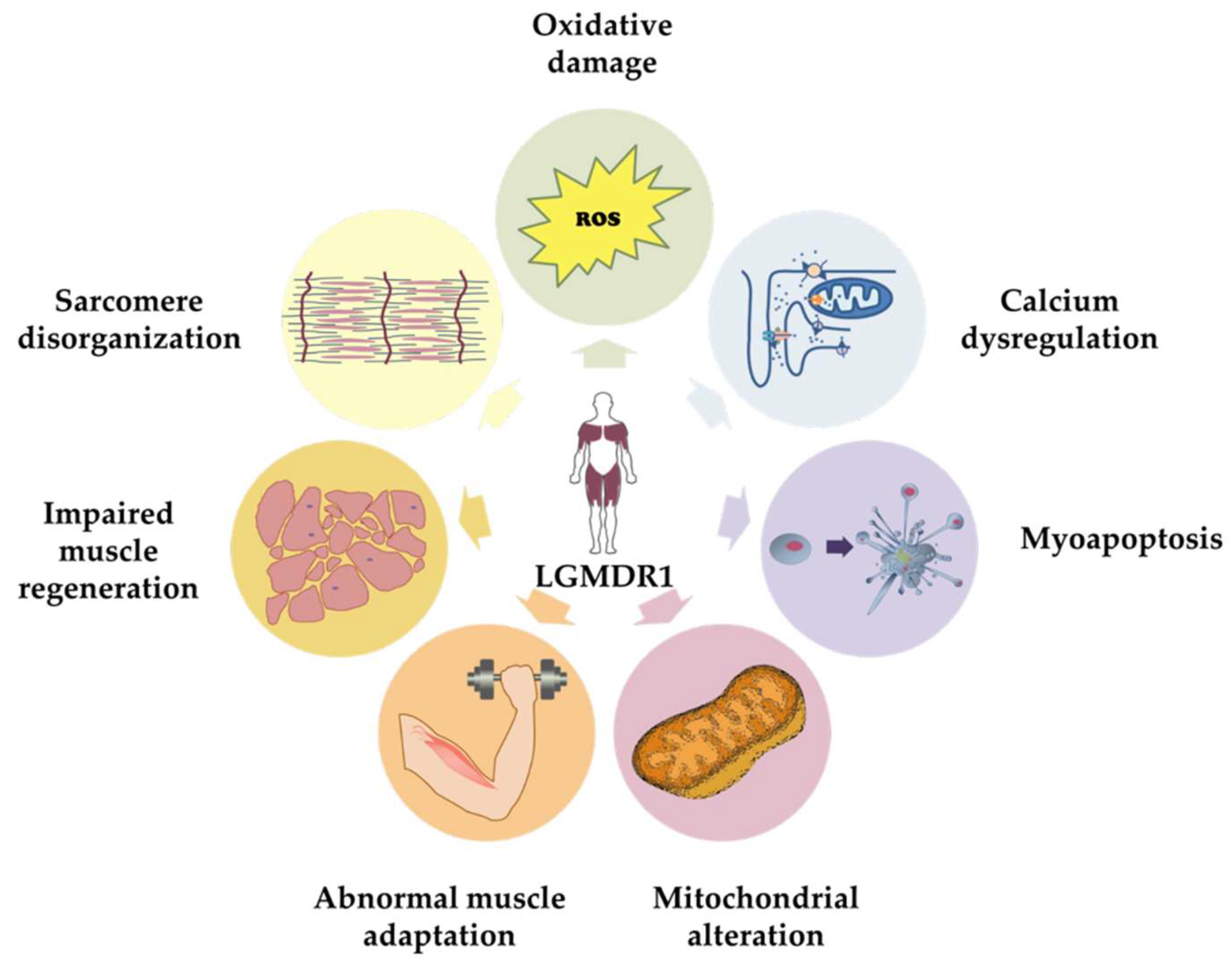
4. Ca2+-Mediated Pathogenic Mechanisms Involved in CAPN3 Deficiency
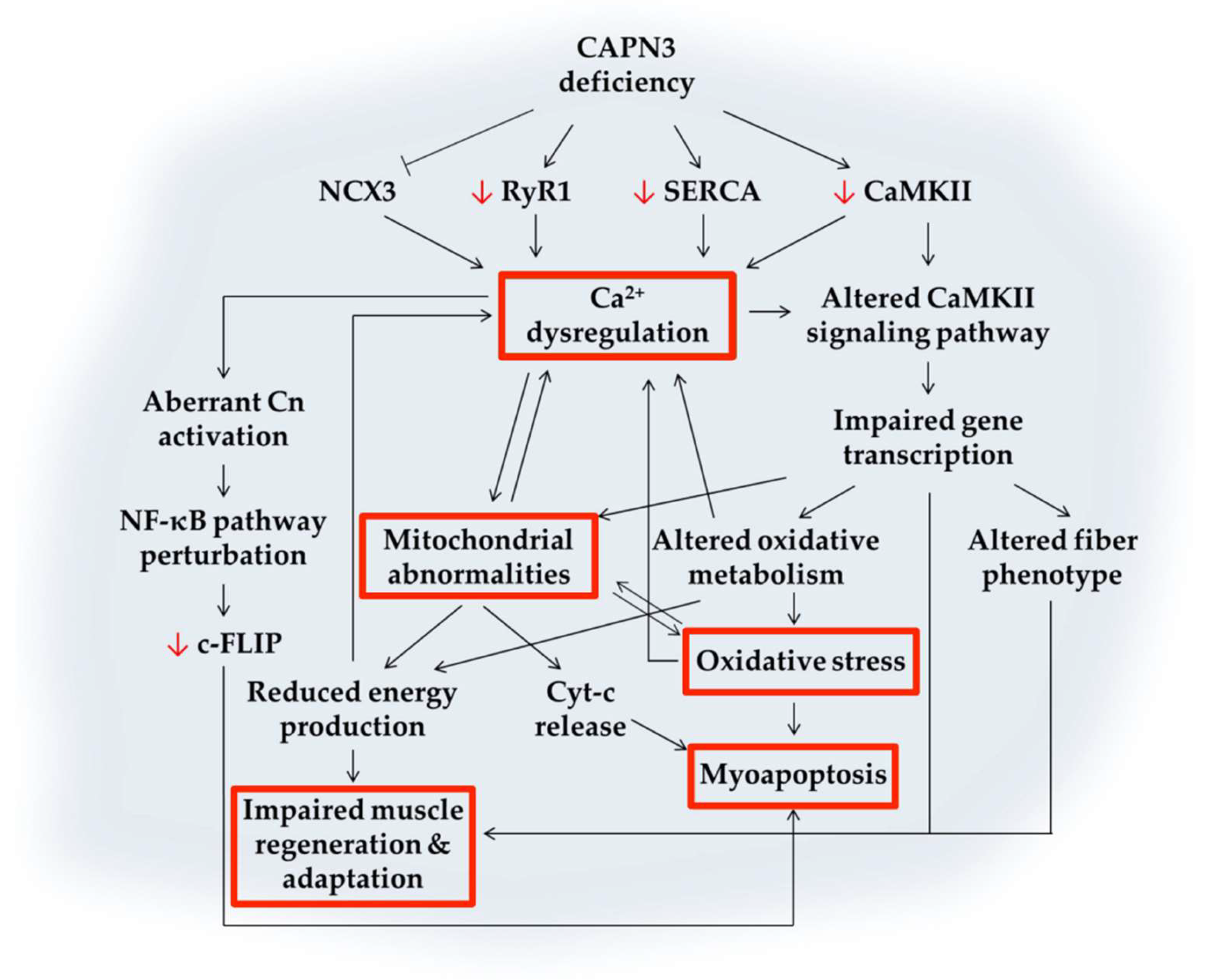
4.1. Calcium Dysregulation
4.2. Abnormal Muscle Adaptation
4.3. Mitochondrial Abnormalities
4.4. Oxidative Stress
4.5. Impaired Muscle Regeneration
4.6. Myoapoptosis
5. Therapeutic Approaches for LGMDR1
6. Future Directions and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAV | Adeno-associated virus |
| Akt | Protein kinase B |
| AldoA | Aldolase isoform A |
| AMP | Adenosine monophosphate |
| AMPK | AMP-activated protein kinase |
| ATP | Adenosine triphosphate |
| ATP5D | ATP synthase subunit delta |
| C3KO | Calpain 3 knockout mouse |
| CaM | Calmodulin |
| CAMK | Ca2+/calmodulin-dependent protein kinase family |
| CAMKII | Ca2+/calmodulin-dependent protein kinase type II |
| CAPN1 | Calpain 1 |
| CAPN2 | Calpain 2 |
| CAPN3 | Human calpain 3 gene |
| CAPN3 | Human calpain 3 protein |
| Capn3−/− | Calpain 3 knockout mouse |
| CBSW | Calpain-type beta-sandwich |
| c-FLIP | Cellular FLICE inhibitory protein |
| CK | Creatine Kinase |
| Cn | Calcineurin |
| CSQ | Calsequestrin |
| CTX | Cardiotoxin |
| Cyt-c | Cytochrome C |
| DHPR | Dihydropyridine receptor |
| ECC | Excitation-contraction coupling |
| FRZB | Frizzled Related Protein |
| HDAC | Class II histone deacetylases |
| IκBα | Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, isoform alpha |
| iMOC | Intermolecular complementation |
| iPSC | Induced pluripotent stem cell |
| IS1 | Insertion sequence 1 |
| IS2 | Insertion sequence 2 |
| LGMD | Limb girdle muscular dystrophy |
| LGMD2A | Limb girdle muscular dystrophy type 2A, renamed LGMDR1 |
| LGMD2B | Limb girdle muscular dystrophy type 2B, renamed LGMDR2 |
| LGMDD4 | Limb girdle muscular dystrophy dominant 4 |
| LGMDR1 | Limb girdle muscular dystrophy recessive 1, caused by mutations in CAPN3 |
| LGMDR2 | Limb girdle muscular dystrophy recessive 2, caused by mutations in dysferlin |
| LGMDR4 | Limb girdle muscular dystrophy recessive 4, caused by mutations in β-sarcoglycan |
| LKB1 | Liver kinase B1 |
| MAM | Mitochondria-associated sarcoplasmic reticulum membrane |
| MARP-2 | Muscle Ankyrin Repeat Protein-2 |
| MEF2 | Myocyte enhancer factor-2 |
| miRNA | Micro ribonucleic acid |
| mRNA | Messenger ribonucleic acid |
| mTOR | Mammalian target of rapamycin |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| MyoD | Myoblast determination protein 1 |
| NCX | Na+/Ca2+ exchanger |
| NCX3 | Na+/Ca2+ exchanger isoform 3 |
| NFAT | Nuclear factor of activated T-cells |
| NF-κB | Nuclear factor kappa-ligjht-chain-enhancer of activated B cells |
| NS | N-terminal addition sequence |
| Pax7 | Paired box protein 7 |
| PC1 | Protease core subdomain 1 |
| PC2 | Protease core subdomain 2 |
| PEF | Penta E-F hand |
| PGC1α | Peroxisome proliferator activated receptor gamma coactivator 1 alpha |
| PKA | Protein kinase A |
| PLN | Phospholamban |
| PMCA | Plasma membrane calcium ATPase |
| PTP | Permeability Transient Pore |
| ROS | Reactive oxygen species |
| RyR | Ryanodine receptor |
| RyR1 | Ryanodine receptor isoform 1 |
| SERCA | Sarco/endoplasmic reticulum Ca2+-ATPase |
| SERCA1 | Sarco/endoplasmic reticulum Ca2+-ATPase isoform 1 |
| SERCA2a | Sarco/endoplasmic reticulum Ca2+-ATPase isoform 2a |
| SLN | Sarcolipin |
| SOD | Superoxide dismutase |
| SR | Sarcoplasmic reticulum |
| T-tubule | Transverse tubule |
| UPR | Unfolded protein rsponse |
| Wnt | Wingless-related integration site |
References
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The Versatility and Universality of Calcium Signalling. Nat. Rev. Mol. Cell. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Zhang, X.; Yu, L.; Xu, H. Calcium Signaling in Membrane Repair. Semin. Cell Dev. Biol. 2015, 45, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Gehlert, S.; Bloch, W.; Suhr, F. Ca2+-Dependent Regulations and Signaling in Skeletal Muscle: From Electro-Mechanical Coupling to Adaptation. Int. J. Mol. Sci. 2015, 16, 1066–1095. [Google Scholar] [CrossRef] [PubMed]
- Al-Qusairi, L.; Laporte, J. T-Tubule Biogenesis and Triad Formation in Skeletal Muscle and Implication in Human Diseases. Skelet. Muscle 2011, 1, 26. [Google Scholar] [CrossRef] [PubMed]
- Vallejo-Illarramendi, A.; Toral-Ojeda, I.; Aldanondo, G.; López de Munain, A. Dysregulation of Calcium Homeostasis in Muscular Dystrophies. Expert Rev. Mol. Med. 2014, 16, e16. [Google Scholar] [CrossRef]
- Protasi, F. Structural Interaction between RyRs and DHPRs in Calcium Release Units of Cardiac and Skeletal Muscle Cells. Front. Biosci. 2002, 7, 650–658. [Google Scholar] [CrossRef]
- Santulli, G.; Lewis, D.R.; Marks, A.R. Physiology and Pathophysiology of Excitation-Contraction Coupling: The Functional Role of Ryanodine Receptor. J. Muscle Res. Cell Motil. 2017, 38, 37–45. [Google Scholar] [CrossRef]
- Lanner, J.T.; Georgiou, D.K.; Joshi, A.D.; Hamilton, S.L. Ryanodine Receptors: Structure, Expression, Molecular Details, and Function in Calcium Release. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef]
- Capes, E.M.; Loaiza, R.; Valdivia, H.H. Ryanodine Receptors. Skelet. Muscle 2011, 1, 18. [Google Scholar] [CrossRef]
- Rossi, A.E.; Dirksen, R.T. Sarcoplasmic Reticulum: The Dynamic Calcium Governor of Muscle. Muscle Nerve 2006, 33, 715–731. [Google Scholar] [CrossRef]
- Rodney, G.G.; Williams, B.Y.; Strasburg, G.M.; Beckingham, K.; Hamilton, S.L. Regulation of RYR1 Activity by Ca2+ and Calmodulin. Biochemistry 2000, 39, 7807–7812. [Google Scholar] [CrossRef] [PubMed]
- Beard, N.A.; Wei, L.; Dulhunty, A.F. Ca2+ Signaling in Striated Muscle: The Elusive Roles of Triadin, Junctin, and Calsequestrin. Eur. Biophys. J. 2009, 39, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Novák, P.; Soukup, T. Calsequestrin Distribution, Structure and Function, Its Role in Normal and Pathological Situations and the Effect of Thyroid Hormones. Physiol. Res. 2011, 60, 439–452. [Google Scholar] [PubMed]
- Calderón, J.C.; Bolaños, P.; Caputo, C. The Excitation-Contraction Coupling Mechanism in Skeletal Muscle. Biophys. Rev. 2014, 6, 133–160. [Google Scholar] [CrossRef] [PubMed]
- Kuo, I.Y.; Ehrlich, B.E. Signaling in Muscle Contraction. Cold Spring Harb. Perspect. Biol. 2015, 7, a006023. [Google Scholar] [CrossRef] [PubMed]
- Periasamy, M.; Kalyanasundaram, A. SERCA Pump Isoforms: Their Role in Calcium Transport and Disease. Muscle Nerve 2007, 35, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.M.; Larkins, N.T.; Mollica, J.P.; Beard, N.A.; Lamb, G.D. Calsequestrin Content and SERCA Determine Normal and Maximal Ca2+ Storage Levels in Sarcoplasmic Reticulum of Fast- and Slow-Twitch Fibres of Rat. J. Physiol. 2009, 587, 443–460. [Google Scholar] [CrossRef]
- Vangheluwe, P.; Raeymaekers, L.; Dode, L.; Wuytack, F. Modulating Sarco (Endo) Plasmic Reticulum Ca2+ ATPase2 (SERCA2) Activity: Cell Biological Implications. Cell Calcium 2005, 38, 291–302. [Google Scholar] [CrossRef]
- Shaikh, S.A.; Sahoo, S.K.; Periasamy, M. Phospholamban and Sarcolipin: Are They Functionally Redundant or Distinct Regulators of the Sarco(Endo)Plasmic Reticulum Calcium ATPase? J. Mol. Cell. Cardiol. 2016, 91, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Tavi, P.; Westerblad, H. The Role of in Vivo Ca2+ Signals Acting on Ca2+-Calmodulin-Dependent Proteins for Skeletal Muscle Plasticity. J. Physiol. 2011, 589, 5021–5031. [Google Scholar] [CrossRef] [PubMed]
- Chin, E.R.; Olson, E.N.; Richardson, J.A.; Yang, Q.; Humphries, C.; Shelton, J.M.; Wu, H.; Zhu, W.; Bassel-Duby, R.; Williams, R.S. A Calcineurin-Dependent Transcriptional Pathway Controls Skeletal Muscle Fiber Type. Genes Dev. 1998, 12, 2499–2509. [Google Scholar] [CrossRef] [PubMed]
- Alzuherri, H.; Chang, K.C. Calcineurin Activates NF-κB in Skeletal Muscle C2C12 Cells. Cell. Signal. 2003, 15, 471–478. [Google Scholar] [CrossRef]
- Valdés, J.A.; Gaggero, E.; Hidalgo, J.; Leal, N.; Jaimovich, E.; Carrasco, M.A. NFAT Activation by Membrane Potential Follows a Calcium Pathway Distinct from Other Activity-Related Transcription Factors in Skeletal Muscle Cells. Am. J. Physiol. Physiol. 2008, 294, C715–C725. [Google Scholar] [CrossRef] [PubMed]
- McCullagh, K.J.A.; Calabria, E.; Pallafacchina, G.; Ciciliot, S.; Serrano, A.L.; Argentini, C.; Kalhovde, J.M.; Lomo, T.; Schiaffino, S. NFAT Is a Nerve Activity Sensor in Skeletal Muscle and Controls Activity-Dependent Myosin Switching. Proc. Natl. Acad. Sci. USA 2004, 101, 10590–10595. [Google Scholar] [CrossRef]
- Peterson, J.M.; Bakkar, N.; Guttridge, D.C. NF-κB Signaling in Skeletal Muscle Health and Disease. In Myogenesis; Pavlath, G.K., Ed.; Elsevier Inc.: Cambridge, MA, USA, 2011. [Google Scholar]
- Kaltschmidt, B.; Kaltschmidt, C.; Hofmann, T.G.; Hehner, S.P.; Dröge, W.; Schmitz, M.L. The Pro- or Anti-Apoptotic Function of NF-κB Is Determined by the Nature of the Apoptotic Stimulus. Eur. J. Biochem. 2000, 267, 3828–3835. [Google Scholar] [CrossRef]
- Eilers, W.; Jaspers, R.T.; De Haan, A.; Ferri, C.; Valdivieso, P.; Flück, M. CaMKII Content Affects Contractile, but Not Mitochondrial, Characteristics in Regenerating Skeletal Muscle. BMC Physiol. 2014, 14. [Google Scholar] [CrossRef]
- Potthoff, M.J.; Wu, H.; Arnold, M.A.; Shelton, J.M.; Backs, J.; Mcanally, J.; Richardson, J.A.; Bassel-duby, R.; Olson, E.N. Histone Deacetylase Degradation and MEF2 Activation Promote the Formation of Slow-Twitch Myofibers. J. Clin. Investig. 2007, 117, 2459–2467. [Google Scholar] [CrossRef]
- Kramerova, I.; Torres, J.A.; Eskin, A.; Nelson, S.F.; Spencer, M.J. Calpain 3 and CaMKIIβ Signaling Are Required to Induce HSP70 Necessary for Adaptive Muscle Growth after Atrophy. Hum. Mol. Genet. 2018, 27, 1642–1653. [Google Scholar] [CrossRef]
- Chin, E.R. Role of Ca2+/Calmodulin-Dependent Kinases in Skeletal Muscle Plasticity. J. Appl. Physiol. 2005, 99, 414–423. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Shaw, R.J. The AMPK Signalling Pathway Coordinates Cell Growth, Autophagy and Metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef]
- Zhou, J.; Dhakal, K.; Yi, J. Mitochondrial Ca2+ Uptake in Skeletal Muscle Health and Disease. Sci. China Life Sci. 2016, 59, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Eisner, V.; Csordas, G.; Hajnoczky, G. Interactions between Sarco-Endoplasmic Reticulum and Mitochondria in Cardiac and Skeletal Muscle—Pivotal Roles in Ca2+ and Reactive Oxygen Species Signaling. J. Cell Sci. 2013, 126, 2965–2978. [Google Scholar] [CrossRef] [PubMed]
- Fraysse, B.; Nagi, S.M.; Boher, B.; Ragot, H.; Lainé, J.; Salmon, A.; Fiszman, M.Y.; Toussaint, M.; Fromes, Y. Ca2+ Overload and Mitochondrial Permeability Transition Pore Activation in Living δ-Sarcoglycan-Deficient Cardiomyocytes. Am. J. Physiol. Physiol. 2010, 299, C706–C713. [Google Scholar] [CrossRef] [PubMed]
- Celsi, F.; Pizzo, P.; Brini, M.; Leo, S.; Fotino, C.; Pinton, P.; Rizzuto, R. Mitochondria, Calcium and Cell Death: A Deadly Triad in Neurodegeneration. Biochim. Biophys. Acta-Bioenerg. 2009, 1787, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Weaver, D.; Hajnóczky, G. Control of Mitochondrial Motility and Distribution by the Calcium Signal: A Homeostatic Circuit. J. Cell Biol. 2004, 167, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.E.; Boncompagni, S.; Dirksen, R.T. Sarcoplasmic Reticulum-Mitochondrial Symbiosis: Bidirectional Signaling in Skeletal Muscle. Exerc. Sport Sci. Rev. 2009, 37, 29–35. [Google Scholar] [CrossRef]
- Giorgi, C.; Baldassari, F.; Bononi, A.; Bonora, M.; De Marchi, E.; Marchi, S.; Missiroli, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; et al. Mitochondrial Ca2+ and Apoptosis. Cell Calcium 2012, 52, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; von Stockum, S. The Permeability Transition Pore as a Ca2+ Release Channel: New Answers to an Old Question. Cell Calcium 2012, 52, 22–27. [Google Scholar] [CrossRef]
- Wright, D.C. Mechanisms of Calcium-Induced Mitochondrial Biogenesis and GLUT4 Synthesis. Appl. Physiol. Nutr. Metab. 2008, 32, 840–845. [Google Scholar] [CrossRef]
- Straub, V.; Murphy, A.; Udd, B. 229th ENMC International Workshop: Limb Girdle Muscular Dystrophies—Nomenclature and Reformed Classification Naarden, the Netherlands, 17–19 March 2017. Neuromuscul. Disord. 2018, 28, 702–710. [Google Scholar] [CrossRef]
- Richard, I.; Broux, O.; Allamand, V.; Fougerousse, F.; Chiannilkulchai, N.; Bourg, N.; Brenguier, L.; Devaud, C.; Pasturaud, P.; Roudaut, C.; et al. Mutations in the Proteolytic Enzyme Calpain 3 Cause Limb-Girdle Muscular Dystrophy Type 2A. Cell 1995, 81, 27–40. [Google Scholar] [CrossRef]
- Vissing, J.; Barresi, R.; Witting, N.; Van Ghelue, M.; Gammelgaard, L.; Bindoff, L.A.; Straub, V.; Lochmüller, H.; Hudson, J.; Wahl, C.M.; et al. A Heterozygous 21-Bp Deletion in CAPN3 Causes Dominantly Inherited Limb Girdle Muscular Dystrophy. Brain 2016, 139, 2154–2163. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Thompson, J.M.; Niu, Z.; Tracy, J.A.; Moore, S.A.; Swenson, A.; Wieben, E.D.; Milone, M. Autosomal Dominant Calpainopathy Due to Heterozygous CAPN3 c.643_663del21. Muscle Nerve 2018, 57, 657–683. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Thompson, J.M.; Moore, S.A.; Liewluck, T. A Novel CAPN3 Mutation in Late-Onset Limb-Girdle Muscular Dystrophy with Early Respiratory Insufficiency. J. Clin. Neurosci. 2018, 53, 229–231. [Google Scholar] [CrossRef] [PubMed]
- Sáenz, A.; López de Munain, A. Dominant LGMD2A: Alternative Diagnosis or Hidden Digenism? Brain 2017, 140, e7. [Google Scholar] [CrossRef] [PubMed]
- Dinçer, P.; Leturcq, F.; Richard, I.; Piccolo, F.; Yalnizoǧlu, D.; De Toma, C.; Akçören, Z.; Broux, O.; Deburgrave, N.; Brenguier, L.; et al. A Biochemical, Genetic, and Clinical Survey of Autosomal Recessive Limb Girdle Muscular Dystrophies in Turkey. Ann. Neurol. 1997, 42, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Topaloğlu, H.; Dincer, P.; Richard, I.; Akçören, Z.; Alehan, D.; Ozme, S.; Caglar, M.; Karaduman, A.; Urtizberea, J.A.; Beckmann, J. Calpain-3 Deficiency Causes a Mild Muscular Dystrophy in Childhood. Neuropediatrics 1997, 28, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Richard, I.; Brenguier, L.; Dincer, P.; Roudaut, C.; Bady, B.; Burgunder, J.; Chemaly, R.; Garcia, C.; Halaby, G.; Jackson, C.; et al. Multiple Independent Molecular Etiology for Limb-Girdle Muscular Dystrophy Type 2A Patients from Various Geographical Origins. Am. J. Hum. Genet. 1997, 60, 1128–1138. [Google Scholar]
- Chae, J.; Minami, N.; Jin, Y.; Nakagawa, M.; Murayama, K.; Igarashi, F.; Nonaka, I. Calpain 3 Gene Mutations: Genetic and Clinico-Pathologic Findings in Limb-Girdle Muscular Dystrophy. Neuromuscul. Disord. 2001, 11, 547–555. [Google Scholar] [CrossRef]
- De Paula, F.; Vainzof, M.; Passos-Bueno, M.R.; Rita de Cássia, M.P.; Matioli, S.R.; Anderson, L.V.B.; Nigro, V.; Zatz, M. Clinical Variability in Calpainopathy: What Makes the Difference? Eur. J. Hum. Genet. 2002, 10, 825–832. [Google Scholar] [CrossRef]
- Zatz, M.; Starling, A. Calpains and Disease. N. Engl. J. Med. 2005, 352, 2413–2423. [Google Scholar] [CrossRef] [PubMed]
- Orphanet. Available online: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=ES&data_id=870&Disease_Disease_Search_diseaseGroup=LGMD2A&Disease_Disease_Search_diseaseType=Pat&Enfermedad(es)/grupo de enfermedades=Distrofia-muscular-de-cinturas-autos-mica-recesiva-tipo-2 (accessed on 2 September 2019).
- Fardeau, M.; Hillaire, D.; Mignard, C.; Feingold, N.; Feingold, J.; Mignard, D.; De Ubeda, B.; Collin, H.; Tomé, F.M.S.; Richard, I.; et al. Juvenile Limb-Girdle Muscular Dystrophy Clinical, Histopathological and Genetic Data from a Small Community Living in the Reunion Island. Brain 1996, 119, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Urtasun, M.; Sáenz, A.; Roudaut, C.; Poza, J.J.; Urtizberea, J.A.; Cobo, A.M.; Richard, I.; García Bragado, F.; Leturcq, F.; Kaplan, J.C.; et al. Limb-Girdle Muscular Dystrophy in Guipúzcoa (Basque Country, Spain). Brain 1998, 121, 1735–1747. [Google Scholar] [CrossRef] [PubMed]
- Allamand, V.; Broux, O.; Bourg, N.; Richard, I.; Tischfield, J.A.; Hodes, M.E.; Conneally, P.M.; Fardeau, M.; Jackson, C.E.; Beckmann, J.S. Genetic Heterogeneity of Autosomal Recessive Limb-Girdle Muscular Dystrophy in a Genetic Isolate (Amish) and Evidence for a New Locus. Hum. Mol. Genet. 1995, 4, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Pantoja-Melendez, C.A.; Miranda-Duarte, A.; Roque-Ramirez, B.; Zenteno, J.C. Epidemiological and Molecular Characterization of a Mexican Population Isolate with High Prevalence of Limb-Girdle Muscular Dystrophy Type 2A Due to a Novel Calpain-3 Mutation. PLoS ONE 2017, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fanin, M.; Benedicenti, F.; Fritegotto, C.; Nascimbeni, A.; Peterle, E.; Stanzial, F.; Cristofoletti, A.; Castellan, C.; Angelini, C. An Intronic Mutation Causes Severe LGMD2A in a Large Inbred Family Belonging to a Genetic Isolate in the Alps. Clin. Genet. 2012, 82, 601–602. [Google Scholar] [CrossRef] [PubMed]
- Leiden Database. Available online: https://databases.lovd.nl/shared/genes/CAPN3 (accessed on 2 September 2019).
- Blázquez, L.; Azpitarte, M.; Sáenz, A.; Goicoechea, M.; Otaegui, D.; Ferrer, X.; Illa, I.; Gutierrez-Rivas, E.; Vilchez, J.J.; López De Munain, A. Characterization of Novel CAPN3 Isoforms in White Blood Cells: An Alternative Approach for Limb-Girdle Muscular Dystrophy 2A Diagnosis. Neurogenetics 2008, 9, 173–182. [Google Scholar] [CrossRef]
- Richard, I.; Hogrel, J.-Y.; Stockholm, D.; Payan, C.A.M.; Fougerousse, F.; Calpainopathy Study Group; Eymard, B.; Mignard, C.; López de Munain, A.; Fardeau, M.; et al. Natural History of LGMD2A for Delineating Outcome Measures in Clinical Trials. Ann. Clin. Transl. Neurol. 2016, 3, 248–265. [Google Scholar] [CrossRef]
- Gallardo, E.; Saenz, A.; Illa, I. Limb-Girdle Muscular Dystrophy 2A. In Muscular Dystrophies; Aminoff, M.J., Boller, F., Swaab, D.F., Eds.; Elsevier, B.V.: Amsterdam, The Netherlands, 2011. [Google Scholar] [CrossRef]
- Sáenz, A.; Ono, Y.; Sorimachi, H.; Goicoechea, M.; Leturcq, F.; Blázquez, L.; García-Bragado, F.; Marina, A.; Poza, J.J.; Azpitarte, M.; et al. Does the Severity of the LGMD2A Phenotype in Compound Heterozygotes Depend on the Combination of Mutations? Muscle Nerve 2011, 44, 710–714. [Google Scholar] [CrossRef]
- Schessl, J.; Walter, M.C.; Schreiber, G.; Schara, U.; Müller, C.R.; Lochmüller, H.; Bönnemann, C.G.; Korinthenberg, R.; Kirschner, J. Phenotypic Variability in Siblings with Calpainopathy (LGMD2A). Acta Myol. 2008, 27, 54–58. [Google Scholar]
- Rajakumar, D.; Alexander, M.; Oommen, A. Oxidative Stress, NF-KB and the Ubiquitin Proteasomal Pathway in the Pathology of Calpainopathy. Neurochem. Res. 2013, 38, 2009–2018. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.I.; Macneil, L.G.; Kitaoka, Y.; Alqarni, F.; Suri, R.; Akhtar, M.; Haikalis, M.E.; Dhaliwal, P.; Saeed, M.; Tarnopolsky, M.A. Redox State and Mitochondrial Respiratory Chain Function in Skeletal Muscle of LGMD2A Patients. PLoS ONE 2014, 9, e102549. [Google Scholar] [CrossRef] [PubMed]
- Toral-Ojeda, I.; Aldanondo, G.; Lasa-Elgarresta, J.; Lasa-Fernández, H.; Fernández-Torrón, R.; López de Munain, A.; Vallejo-Illarramendi, A.; Lasa-Fernandez, H.; Fernandez-Torron, R.; Lopez de Munain, A.; et al. Calpain 3 Deficiency Affects SERCA Expression and Function in the Skeletal Muscle. Expert Rev. Mol. Med. 2016, 18, e7. [Google Scholar] [CrossRef] [PubMed]
- Toral-Ojeda, I.; Aldanondo, G.; Lasa-Elgarresta, J.; Lasa-Fernandez, H.; Vesga-Castro, C.; Mouly, V.; de Munain, A.L.; Vallejo-Illarramendi, A. A novel functional in vitro model that recapitulates human muscle disorders. In Muscle Cell and Tissue-Current Status of Research Field; IntechOpen: London, UK, 2018; pp. 133–153. [Google Scholar] [CrossRef]
- Kramerova, I.; Kudryashova, E.; Tidball, J.G.; Spencer, M.J. Null Mutation of Calpain 3 (P94) in Mice Causes Abnormal Sarcomere Formation in Vivo and in Vitro. Hum. Mol. Genet. 2004, 13, 1373–1388. [Google Scholar] [CrossRef]
- El-Khoury, R.; Traboulsi, S.; Hamad, T.; Lamaa, M.; Sawaya, R.; Ahdab-Barmada, M. Divergent Features of Mitochondrial Deficiencies in LGMD2A Associated With Novel Calpain-3 Mutations. J. Neuropathol. Exp. Neurol. 2019, 78, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Yalvac, M.E.; Amornvit, J.; Braganza, C.; Chen, L.; Hussain, S.R.A.; Shontz, K.M.; Montgomery, C.L.; Flanigan, K.M.; Lewis, S.; Sahenk, Z. Impaired Regeneration in Calpain-3 Null Muscle Is Associated with Perturbations in MTORC1 Signaling and Defective Mitochondrial Biogenesis. Skelet. Muscle 2017, 7, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Kramerova, I.; Kudryashova, E.; Wu, B.; Germain, S.; Vandenborne, K.; Romain, N.; Haller, R.G.; Verity, M.A.; Spencer, M.J. Mitochondrial Abnormalities, Energy Deficit and Oxidative Stress Are Features of Calpain 3 Deficiency in Skeletal Muscle. Hum. Mol. Genet. 2009, 18, 3194–3205. [Google Scholar] [CrossRef] [PubMed]
- Kramerova, I.; Kudryashova, E.; Ermolova, N.; Saenz, A.; Jaka, O.; López de munain, A.; Spencer, M.J. Impaired Calcium Calmodulin Kinase Signaling and Muscle Adaptation Response in the Absence of Calpain 3. Hum. Mol. Genet. 2012, 21, 3193–3204. [Google Scholar] [CrossRef]
- Kramerova, I.; Ermolova, N.; Eskin, A.; Hevener, A.; Quehenberger, O.; Armando, A.M.; Haller, R.; Romain, N.; Nelson, S.F.; Spencer, M.J. Failure to Up-Regulate Transcription of Genes Necessary for Muscle Adaptation Underlies Limb Girdle Muscular Dystrophy 2A (Calpainopathy). Hum. Mol. Genet. 2016, 25, 2194–2207. [Google Scholar] [CrossRef]
- Fanin, M.; Angelini, C. Protein and Genetic Diagnosis of Limb Girdle Muscular Dystrophy Type 2A: The Yield and the Pitfalls. Muscle Nerve 2015, 52, 163–173. [Google Scholar] [CrossRef]
- Krahn, M.; Goicoechea, M.; Hanisch, F.; Groen, E.; Bartoli, M.; Pécheux, C.; Garcia-Bragado, F.; Leturcq, F.; Jeannet, P.Y.; Lobrinus, J.A.; et al. Eosinophilic Infiltration Related to CAPN3 Mutations: A Pathophysiological Component of Primary Calpainopathy? Clin. Genet. 2011, 80, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Carafoli, E.; Molinari, M. Calpain: A Protease in Search of a Function? Biochem. Biophys. Res. Commun. 1998, 247, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Santella, L.; Kyozuka, K.; De Riso, L.; Carafoli, E. Calcium, Protease Action, and the Regulation of the Cell Cycle. Cell Calcium 1998, 23, 123–130. [Google Scholar] [CrossRef]
- Wang, K.K.W. Calpain and Caspase: Can You Tell the Difference? Trends Neurosci. 2000, 23, 20–26. [Google Scholar] [CrossRef]
- Glading, A.; Lauffenburger, D.A.; Wells, A. Cutting to the Chase: Calpain Proteases in Cell Motility. Trends Cell Biol. 2002, 12, 46–54. [Google Scholar] [CrossRef]
- Ono, Y.; Ojima, K.; Shinkai-Ouchi, F.; Hata, S.; Sorimachi, H. An Eccentric Calpain, CAPN3/P94/Calpain-3. Biochimie 2016, 122, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Campbell, R.L.; Davies, P.L. Structures of Human Calpain-3 Protease Core with and without Bound Inhibitor Reveal Mechanisms of Calpain Activation. J. Biol. Chem. 2018, 293, 4056–4070. [Google Scholar] [CrossRef] [PubMed]
- Sorimachi, H.; Toyama-Sorimachi, N.; Saido, T.C.; Kawasaki, H.; Sugita, H.; Miyasaka, M.; Arahata, K.I.; Ishiura, S.; Suzuki, K. Muscle-Specific Calpain, P94, Is Degraded by Autolysis Immediately after Translation, Resulting in Disappearance from Muscle. J. Biol. Chem. 1993, 268, 10593–10605. [Google Scholar]
- Ono, Y.; Shindo, M.; Doi, N.; Kitamura, F.; Gregorio, C.C.; Sorimachi, H. The N- and C-Terminal Autolytic Fragments of CAPN3/P94/Calpain-3 Restore Proteolytic Activity by Intermolecular Complementation. Proc. Natl. Acad. Sci. USA 2014, 111, E5527–E5536. [Google Scholar] [CrossRef]
- Ono, Y.; Ojima, K.; Torii, F.; Takaya, E.; Doi, N.; Nakagawa, K.; Hata, S.; Abe, K.; Sorimachi, H. Skeletal Muscle-Specific Calpain Is an Intracellular Na+- Dependent Protease. J. Biol. Chem. 2010, 285, 22986–22998. [Google Scholar] [CrossRef]
- Ono, Y.; Torii, F.; Ojima, K.; Doi, N.; Yoshioka, K.; Kawabata, Y.; Labeit, D.; Labeit, S.; Suzuki, K.; Abe, K.; et al. Suppressed Disassembly of Autolyzing P94/CAPN3 by N2A Connectin/Titin in a Genetic Reporter System. J. Biol. Chem. 2006, 281, 18519–18531. [Google Scholar] [CrossRef] [PubMed]
- Kramerova, I.; Kudryashova, E.; Venkatraman, G.; Spencer, M.J. Calpain 3 Participates in Sarcomere Remodeling by Acting Upstream of the Ubiquitin-Proteasome Pathway. Hum. Mol. Genet. 2005, 14, 2125–2134. [Google Scholar] [CrossRef] [PubMed]
- Ojima, K.; Ono, Y.; Ottenheijm, C.; Hata, S.; Suzuki, H.; Granzier, H.; Sorimachi, H. Non-Proteolytic Functions of Calpain-3 in Sarcoplasmic Reticulum in Skeletal Muscles. J. Mol. Biol. 2011, 407, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Kramerova, I.; Kudryashova, E.; Wu, B.; Ottenheijm, C.; Granzier, H.; Spencer, M.J. Novel Role of Calpain-3 in the Triad-Associated Protein Complex Regulating Calcium Release in Skeletal Muscle. Hum. Mol. Genet. 2008, 17, 3271–3280. [Google Scholar] [CrossRef] [PubMed]
- Dayanithi, G.; Richard, I.; Viero, C.; Mazuc, E.; Mallie, S.; Valmier, J.; Bourg, N.; Herasse, M.; Marty, I.; Lefranc, G.; et al. Alteration of Sarcoplasmic Reticulum Ca2+ Release in Skeletal Muscle from Calpain 3-Deficient Mice. Int. J. Cell Biol. 2009, 2009, 340346. [Google Scholar] [CrossRef]
- Baghdiguian, S.; Martin, M.; Richard, I.; Pons, F.; Astier, C.; Bourg, N.; Hay, R.T.; Chemaly, R.; Halaby, G.; Loiselet, J.; et al. Calpain 3 Deficiency Is Associated with Myonuclear Apoptosis and Profound Perturbation of the IκBα/NF-κB Pathway in Limb-Girdle Muscular Dystrophy Type 2A. Nat. Med. 1999, 5, 503–511. [Google Scholar] [CrossRef]
- Sorimachi, H.; Kinbara, K.; Kimura, S.; Takahashi, M.; Ishiura, S.; Sasagawa, N.; Sorimachi, N.; Shimada, H.; Tagawa, K.; Maruyama, K.; et al. Muscle-Specific Calpain, P94, Responsible for Limb Girdle Muscular Dystrophy Type 2A, Associates with Connectin through IS2, a P94-Specific Sequence. J. Biol. Chem. 1995, 270, 31158–31162. [Google Scholar] [CrossRef]
- Ojima, K.; Ono, Y.; Doi, N.; Yoshioka, K.; Kawabata, Y.; Labeit, S.; Sorimachi, H. Myogenic Stage, Sarcomere Length, and Protease Activity Modulate Localization of Muscle-Specific Calpain. J. Biol. Chem. 2007, 282, 14493–14504. [Google Scholar] [CrossRef]
- Gunning, P.W.; Hardeman, E.C.; Lappalainen, P.; Mulvihill, D.P. Tropomyosin—Master Regulator of Actin Filament Function in the Cytoskeleton. J. Cell Sci. 2015, 128, 2965–2974. [Google Scholar] [CrossRef]
- Escolar, D.M.; O’Carroll, P.; Leshner, R. Treatment and Management of Muscular Dystrophies. In Neuromuscular Disorders; Elsevier Inc.: Philadelphia, PA, USA, 2011; pp. 343–372. [Google Scholar] [CrossRef]
- Ojima, K.; Kawabata, Y.; Nakao, H.; Nakao, K.; Doi, N.; Kitamura, F.; Ono, Y.; Hata, S.; Suzuki, H.; Kawahara, H.; et al. Dynamic Distribution of Muscle-Specific Calpain in Mice Has a Key Role in Physical-Stress Adaptation and Is Impaired in Muscular Dystrophy. J. Clin. Investig. 2010, 120, 2672–2683. [Google Scholar] [CrossRef]
- Taveau, M.; Bourg, N.; Sillon, G.; Roudaut, C.; Bartoli, M.; Richard, I. Calpain 3 Is Activated through Autolysis within the Active Site and Lyses Sarcomeric and Sarcolemmal Components. Mol. Cell. Biol. 2003, 23, 9127–9135. [Google Scholar] [CrossRef] [PubMed]
- Zak, R.; Martin, A.F.; Prior, G.; Rabinowitz, M. Comparison of Turnover of Several Myofibrillar Proteins and Critical Evaluation of Double Isotope Method. J. Biol. Chem. 1977, 252, 3430–3435. [Google Scholar] [PubMed]
- Isaacs, W.B.; Kim, I.S.; Struve, A.; Fulton, A.B. Biosynthesis of Titin in Cultured Skeletal Muscle Cells. J. Cell Biol. 1989, 109, 2189–2195. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, J.S.; Spencer, M. Calpain 3, the “Gatekeeper” of Proper Sarcomere Assembly, Turnover and Maintenance. Neuromuscul. Disord. 2008, 18, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Richard, I.; Roudaut, C.; Marchand, S.; Baghdiguian, S.; Herasse, M.; Stockholm, D.; Ono, Y.; Suel, L.; Bourg, N.; Sorimachi, H.; et al. Loss of Calpain 3 Proteolytic Activity Leads to Muscular Dystrophy and to Apoptosis-Associated IκBα/Nuclear Factor ΚB Pathway Perturbation in Mice. J. Cell Biol. 2000, 151, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Spencer, M.J.; Guyon, J.R.; Sorimachi, H.; Potts, A.; Richard, I.; Herasse, M.; Chamberlain, J.; Dalkilic, I.; Kunkel, L.M.; Beckmann, J.S. Stable Expression of Calpain 3 from a Muscle Transgene in Vivo: Immature Muscle in Transgenic Mice Suggests a Role for Calpain 3 in Muscle Maturation. Proc. Natl. Acad. Sci. USA 2002, 99, 8874–8879. [Google Scholar] [CrossRef] [PubMed]
- Sacco, A.; Mourkioti, F.; Tran, R.; Choi, J.; Llewellyn, M.; Kraft, P.; Shkreli, M.; Delp, S.; Pomerantz, J.H.; Artandi, S.E.; et al. Short Telomeres and Stem Cell Exhaustion Model Duchenne Muscular Dystrophy in Mdx/MTR Mice. Cell 2010, 143, 1059–1071. [Google Scholar] [CrossRef] [PubMed]
- Jaka, O.; Casas-Fraile, L.; Azpitarte, M.; Aiastui, A.; López de Munain, A.; Sáenz, A. FRZB and Melusin, Overexpressed in LGMD2A, Regulate Integrin Β1D Isoform Replacement Altering Myoblast Fusion and the Integrin-Signalling Pathway. Expert Rev. Mol. Med. 2017, 19, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Amici, D.R.; Pinal-Fernandez, I.; Mázala, D.A.G.; Lloyd, T.E.; Corse, A.M.; Christopher-Stine, L.; Mammen, A.L.; Chin, E.R. Calcium Dysregulation, Functional Calpainopathy, and Endoplasmic Reticulum Stress in Sporadic Inclusion Body Myositis. Acta Neuropathol. Commun. 2017, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, P.; Szalai, P.; Olesen, C.; Praetorius, H.A.; Nissen, P.; Christensen, S.B.; Engedal, N.; Møller, J.V. Inhibition of the Sarco/Endoplasmic Reticulum (ER) Ca2-ATPase by Thapsigargin Analogs Induces Cell Death via ER Ca2 Depletion and the Unfolded Protein Response. J. Biol. Chem. 2017, 292, 19656–19673. [Google Scholar] [CrossRef] [PubMed]
- Mekahli, D.; Bultynck, G.; Parys, J.B.; De Smedt, H.; Missiaen, L. Endoplasmic-Reticulum Calcium Depletion and Disease. Cold Spring Harb. Perspect. Biol. 2011, 3, 131–154. [Google Scholar] [CrossRef] [PubMed]
- Michel, L.Y.M.; Hoenderop, J.G.J.; Bindels, R.J.M. Calpain-3-Mediated Regulation of the Na+-Ca2+ exchanger Isoform 3. Pflugers Arch. Eur. J. Physiol. 2016, 468, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Matsakas, A.; Patel, K. Intracellular Signalling Pathways Regulating the Adaptation of Skeletal Muscle to Exercise and Nutritional Changes. Histol. Histopathol. 2009, 24, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Cohen, N.; Kudryashova, E.; Kramerova, I.; Anderson, L.V.B.; Beckmann, J.S.; Bushby, K.; Spencer, M.J. Identification of Putative in Vivo Substrates of Calpain 3 by Comparative Proteomics of Overexpressing Transgenic and Nontransgenic Mice. Proteomics 2006, 6, 6075–6084. [Google Scholar] [CrossRef]
- Kramerova, I.; Beckmann, J.S.; Spencer, M.J. Molecular and Cellular Basis of Calpainopathy (Limb Girdle Muscular Dystrophy Type 2A). Biochim. Biophys. Acta 2007, 1772, 128–144. [Google Scholar] [CrossRef] [PubMed]
- Sáenz, A.; Azpitarte, M.; Armañanzas, R.; Leturcq, F.; Alzualde, A.; Inza, I.; García-Bragado, F.; De la Herran, G.; Corcuera, J.; Cabello, A.; et al. Gene Expression Profiling in Limb-Girdle Muscular Dystrophy 2A. PLoS ONE 2008, 3, e3750. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial Membrane Permeabilization in Cell Death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef]
- Handschin, C.; Rhee, J.; Lin, J.; Tarr, P.T.; Spiegelman, B.M. An Autoregulatory Loop Controls Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1alpha Expression in Muscle. Proc. Natl. Acad. Sci. USA 2003, 100, 7111–7116. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative Stress, Mitochondrial Damage and Neurodegenerative Diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef]
- Bellinger, A.M.; Reiken, S.; Carlson, C.; Mongillo, M.; Liu, X.; Rothman, L.; Matecki, S.; Lacampagne, A.; Marks, A.R. Hypernitrosylated Ryanodine Receptor/Calcium Release Channels Are Leaky in Dystrophic Muscle Andrew. Nat. Med. 2009, 15, 325–330. [Google Scholar] [CrossRef]
- Raney, M.A.; Turcotte, L.P. Evidence for the Involvement of CaMKII and AMPK in Ca2+-Dependent Signaling Pathways Regulating FA Uptake and Oxidation in Contracting Rodent Muscle. J. Appl. Physiol. 2008, 104, 1366–1373. [Google Scholar] [CrossRef] [PubMed]
- Shan, T.; Zhang, P.; Liang, X.; Bi, P.; Yue, F.; Kuang, S. Lkb1 Is Indispensable for Skeletal Muscle Development, Regeneration, and Satellite Cell Homeostasis. Stem Cells 2014, 32, 2893–2907. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Gravel, S.P.; Hulea, L.; Larsson, O.; Pollak, M.; St-Pierre, J.; Topisirovic, I. MTOR Coordinates Protein Synthesis, Mitochondrial Activity. Cell Cycle 2015, 14, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Gravel, S.P.; Chénard, V.; Sikström, K.; Zheng, L.; Alain, T.; Gandin, V.; Avizonis, D.; Arguello, M.; Zakaria, C.; et al. MTORC1 Controls Mitochondrial Activity and Biogenesis through 4E-BP-Dependent Translational Regulation. Cell Metab. 2013, 18, 698–711. [Google Scholar] [CrossRef] [PubMed]
- Rosales, X.Q.; Malik, V.; Sneh, A.; Chen, L.; Lewis, S.; Kota, J.; Gastier-Foster, J.M.; Astbury, C.; Pyatt, R.; Reshmi, S.; et al. Impaired Regeneration in LGMD2A Supported by Increased PAX7-Positive Satellite Cell Content and Muscle-Specific Microrna Dysregulation. Muscle Nerve 2013, 47, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Blomberg, K.E.M.; McClorey, G.; El Andaloussi, S.; Godfrey, C.; Betts, C.; Coursindel, T.; Gait, M.J.; Smith, C.E.; Wood, M.J. Expression Analysis in Multiple Muscle Groups and Serum Reveals Complexity in the MicroRNA Transcriptome of the Mdx Mouse with Implications for Therapy. Mol. Ther. Nucleic Acids 2012, 1, e39. [Google Scholar] [CrossRef]
- Chen, J.F.; Tao, Y.; Li, J.; Deng, Z.; Yan, Z.; Xiao, X.; Wang, D.Z. MicroRNA-1 and MicroRNA-206 Regulate Skeletal Muscle Satellite Cell Proliferation and Differentiation by Repressing Pax7. J. Cell Biol. 2010, 190, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Dey, B.K.; Gagan, J.; Dutta, A. MiR-206 and -486 Induce Myoblast Differentiation by Downregulating Pax7. Mol. Cell. Biol. 2011, 31, 1329. [Google Scholar] [CrossRef]
- Liu, N.; Bezprozvannaya, S.; Shelton, J.M.; Frisard, M.I.; Hulver, M.W.; McMillan, R.P.; Wu, Y.; Voelker, K.A.; Grange, R.W.; Richardson, J.A.; et al. Mice Lacking MicroRNA 133a Develop Dynamin 2-Dependent Centronuclear Myopathy. J. Clin. Investig. 2011, 121, 3258–3268. [Google Scholar] [CrossRef]
- Stuelsatz, P.; Pouzoulet, F.; Lamarre, Y.; Dargelos, E.; Poussard, S.; Leibovitch, S.; Cottin, P.; Veschambre, P. Down-Regulation of MyoD by Calpain 3 Promotes Generation of Reserve Cells in C2C12 Myoblasts. J. Biol. Chem. 2010, 285, 12670–12683. [Google Scholar] [CrossRef]
- Kramerova, I.; Kudryashova, E.; Wu, B.; Spencer, M.J. Regulation of the M-Cadherin-b-Catenin Complex by Calpain 3 during Terminal Stages of Myogenic Differentiation. Mol. Cell. Biol. 2006, 26, 8437–8447. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Malhotra, S.; Kumar, A. Nuclear Factor-Kappa B Signaling in Skeletal Muscle Atrophy. J. Mol. Med. 2008, 86, 1113–1126. [Google Scholar] [CrossRef] [PubMed]
- Benayoun, B.; Baghdiguian, S.; Lajmanovich, A.; Bartoli, M.; Daniele, N.; Gicquel, E.; Bourg, N.; Raynaud, F.; Pasquier, M.; Suel, L.; et al. NFkB-Dependent Expression of the Antiapoptotic Factor c-FLIP Is Regulated by Calpain 3, the Protein Involved in Limb-Girdle Muscular Dystrophy Type 2A Be. FASEB J. 2008, 22, 1521–1529. [Google Scholar] [CrossRef] [PubMed]
- Elbaz, M.; Yanay, N.; Laban, S.; Rabie, M.; Mitrani-Rosenbaum, S.; Nevo, Y. Life or Death by NFκB, Losartan Promotes Survival in Dy2J/Dy2J Mouse of MDC1A. Cell Death Dis. 2015, 6, e1690. [Google Scholar] [CrossRef] [PubMed]
- Groenendyk, J.; Lynch, J.; Michalak, M. Calreticulin, Ca2+, and Calcineurin-Signaling from the Endoplasmic Reticulum. Mol. Cells 2004, 17, 383–389. [Google Scholar] [PubMed]
- Vissing, J. Limb Girdle Muscular Dystrophies: Classification, Clinical Spectrum and Emerging Therapies. Curr. Opin. Neurol. 2016, 29, 635–641. [Google Scholar] [CrossRef]
- Sveen, M.L.; Andersen, S.P.; Ingelsrud, L.H.; Blichter, S.; Olsen, N.E.; Jønck, S.; Krag, T.O.; Vissing, J. Resistance Training in Patients with Limb-Girdle and Becker Muscular Dystrophies. Muscle Nerve 2013, 47, 163–169. [Google Scholar] [CrossRef]
- Sczesny-Kaiser, M.; Kowalewski, R.; Schildhauer, T.A.; Aach, M.; Jansen, O.; Grasmücke, D.; Güttsches, A.K.; Vorgerd, M.; Tegenthoff, M. Treadmill Training with HAL Exoskeleton-A Novel Approach for Symptomatic Therapy in Patients with Limb-Girdle Muscular Dystrophy-Preliminary Study. Front. Neurosci. 2017, 11, 1–9. [Google Scholar] [CrossRef]
- Bartoli, M.; Poupiot, J.; Vulin, A.; Fougerousse, F.; Arandel, L.; Daniele, N.; Roudaut, C.; Noulet, F.; Garcia, L.; Danos, O.; et al. AAV-Mediated Delivery of a Mutated Myostatin Propeptide Ameliorates Calpain 3 but Not α-Sarcoglycan Deficiency. Gene Ther. 2007, 14, 733–740. [Google Scholar] [CrossRef][Green Version]
- Wagner, K.R.; Fleckenstein, J.L.; Amato, A.A.; Barohn, R.J.; Bushby, K.; Escolar, D.M.; Flanigan, K.M.; Pestronk, A.; Tawil, R.; Wolfe, G.I.; et al. A Phase I/II Trial of MYO-029 in Adult Subjects with Muscular Dystrophy. Ann. Neurol. 2008, 63, 561–571. [Google Scholar] [CrossRef]
- Roudaut, C.; Le Roy, F.; Suel, L.; Poupiot, J.; Charton, K.; Bartoli, M.; Richard, I. Restriction of Calpain3 Expression to the Skeletal Muscle Prevents Cardiac Toxicity and Corrects Pathology in a Murine Model of Limb-Girdle Muscular Dystrophy. Circulation 2013, 128, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Coalition to Cure Calpain 3. Available online: http://www.curecalpain3.org (accessed on 15 May 2019).
- Sarepta Therapeutics. Available online: https://www.sarepta.com/ (accessed on 16 May 2019).
- Selvaraj, S.; Filareto, A.; Kiley, J.; Voytas, D.; Kyba, M.; Perlingeiro, R. Gene Correction of LGMD2A Patient-Specific IPS Cells for Targeted Autologous Cell Therapy. Mol. Ther. 2016, 24, S125–S126. [Google Scholar] [CrossRef]
- Straub, V.; Bertoli, M. Where Do We Stand in Trial Readiness for Autosomal Recessive Limb Girdle Muscular Dystrophies? Neuromuscul. Disord. 2015, 26, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Rodino-Klapac, L.R.; Janssen, P.M.L.; Shontz, K.M.; Canan, B.; Montgomery, C.L.; Griffin, D.; Heller, K.; Schmelzer, L.; Handy, C.; Clark, K.R.; et al. Micro-Dystrophin and Follistatin Co-Delivery Restores Muscle Function in Aged DMD Model. Hum. Mol. Genet. 2013, 22, 4929–4937. [Google Scholar] [CrossRef] [PubMed]
- Sondergaard, P.C.; Griffin, D.A.; Pozsgai, E.R.; Johnson, R.W.; Grose, W.E.; Heller, K.N.; Shontz, K.M.; Montgomery, C.L.; Liu, J.; Clark, K.R.; et al. AAV.Dysferlin Overlap Vectors Restore Function in Dysferlinopathy Animal Models. Ann. Clin. Transl. Neurol. 2015, 2, 256–270. [Google Scholar] [CrossRef] [PubMed]
- Gruntman, A.M.; Flotte, T.R. Delivery of Adeno-Associated Virus Gene Therapy by Intravascular Limb Infusion Methods. Hum. Gene Ther. Clin. Dev. 2015, 26, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.C.; Meli, A.C.; Reiken, S.; Betzenhauser, M.J.; Umanskaya, A.; Shiomi, T.; D’Armiento, J.; Marks, A.R. Leaky Ryanodine Receptors in β-Sarcoglycan Deficient Mice: A Potential Common Defect in Muscular Dystrophy. Skelet. Muscle 2012, 2, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mázala, D.A.G.; Pratt, S.J.P.; Chen, D.; Molkentin, J.D.; Lovering, R.M.; Chin, E.R. SERCA1 Overexpression Minimizes Skeletal Muscle Damage in Dystrophic Mouse Models. Am. J. Physiol. Physiol. 2015, 308, C699–C709. [Google Scholar] [CrossRef]
- Zsebo, K.; Yaroshinsky, A.; Rudy, J.J.; Wagner, K.; Greenberg, B.; Jessup, M.; Hajjar, R.J. Long-Term Effects of AAV1/SERCA2a Gene Transfer in Patients with Severe Heart Failure: Analysis of Recurrent Cardiovascular Events and Mortality. Circ. Res. 2014, 114, 101–108. [Google Scholar] [CrossRef]
- Yi, J.; Ma, C.; Li, Y.; Weisleder, N.; Ríos, E.; Ma, J.; Zhou, J. Mitochondrial Calcium Uptake Regulates Rapid Calcium Transients in Skeletal Muscle during Excitation-Contraction (E-C) Coupling. J. Biol. Chem. 2011, 286, 32436–32443. [Google Scholar] [CrossRef]
- Qaisar, R.; Bhaskaran, S.; Ranjit, R.; Sataranatarajan, K.; Premkumar, P.; Huseman, K.; Van Remmen, H. Restoration of SERCA ATPase Prevents Oxidative Stress-Related Muscle Atrophy and Weakness. Redox Biol. 2019, 20, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Voit, A.; Patel, V.; Pachon, R.; Shah, V.; Bakhutma, M.; Kohlbrenner, E.; McArdle, J.J.; Dell’Italia, L.J.; Mendell, J.R.; Xie, L.H.; et al. Reducing Sarcolipin Expression Mitigates Duchenne Muscular Dystrophy and Associated Cardiomyopathy in Mice. Nat. Commun. 2017, 8, 1068. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, T.; del Monte, F.; Yoshikawa, Y.; Abe, T.; Shimizu, J.; Nakajima-Takenaka, C.; Taniguchi, S.; Hajjar, R.J.; Takaki, M. Rescue of Ca2+ Overload-Induced Left Ventriclur Dysfunction by Targeted Ablation of Phospholamban. Am. J. Physiol. Circ. Physiol. 2008, 296, H310–H317. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, E.; Testoni, S.; Gentile, A.; Calì, T.; Ottolini, D.; Villa, A.; Brini, M.; Betto, R.; Mascarello, F.; Nissen, P.; et al. Inhibition of Ubiquitin Proteasome System Rescues the Defective Sarco(Endo)Plasmic Reticulum Ca2+-ATPase (SERCA1) Protein Causing Chianina Cattle Pseudomyotonia. J. Biol. Chem. 2014, 289, 33073–33082. [Google Scholar] [CrossRef] [PubMed]
- Katsetos, C.D.; Koutzaki, S.; Melvin, J.J. Mitochondrial Dysfunction in Neuromuscular Disorders. Semin. Pediatr. Neurol. 2013, 20, 202–215. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapy | Clinical-Pharmacological Use | State | Comments | Ref. |
|---|---|---|---|---|
| Pharmacological Therapy | ||||
| MYO-029 | Myostatin human recombinant neutralizing antibody | Competed I/II trial | Minimal improvement in muscle strength | [136] |
| Gene therapy | ||||
| AAV-delivered mutant myostatin propeptide | Prevention of the cleavage of myostatin propeptide | Preclinical | Increased muscle mass and force generation in mice | [135] |
| AAV-mediated transfer of calpain 3 | Increase of calpain 3 expression and function | Preclinical | Rescue of the contractile force deficits in mice | [137] |
| Plasmid DNA | Increase of calpain 3 expression and function | Active project | [138] | |
| AAVrh74 vector | Increase of calpain 3 expression and function | Active project | Systemic delivery to muscle | [139] |
| Cell therapy | ||||
| iPSC | Increase of calpain 3 expression and function | Active project | [140] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lasa-Elgarresta, J.; Mosqueira-Martín, L.; Naldaiz-Gastesi, N.; Sáenz, A.; López de Munain, A.; Vallejo-Illarramendi, A. Calcium Mechanisms in Limb-Girdle Muscular Dystrophy with CAPN3 Mutations. Int. J. Mol. Sci. 2019, 20, 4548. https://doi.org/10.3390/ijms20184548
Lasa-Elgarresta J, Mosqueira-Martín L, Naldaiz-Gastesi N, Sáenz A, López de Munain A, Vallejo-Illarramendi A. Calcium Mechanisms in Limb-Girdle Muscular Dystrophy with CAPN3 Mutations. International Journal of Molecular Sciences. 2019; 20(18):4548. https://doi.org/10.3390/ijms20184548
Chicago/Turabian StyleLasa-Elgarresta, Jaione, Laura Mosqueira-Martín, Neia Naldaiz-Gastesi, Amets Sáenz, Adolfo López de Munain, and Ainara Vallejo-Illarramendi. 2019. "Calcium Mechanisms in Limb-Girdle Muscular Dystrophy with CAPN3 Mutations" International Journal of Molecular Sciences 20, no. 18: 4548. https://doi.org/10.3390/ijms20184548
APA StyleLasa-Elgarresta, J., Mosqueira-Martín, L., Naldaiz-Gastesi, N., Sáenz, A., López de Munain, A., & Vallejo-Illarramendi, A. (2019). Calcium Mechanisms in Limb-Girdle Muscular Dystrophy with CAPN3 Mutations. International Journal of Molecular Sciences, 20(18), 4548. https://doi.org/10.3390/ijms20184548