Human Induced Pluripotent Stem-Cell-Derived Cardiomyocytes as Models for Genetic Cardiomyopathies

 ,
, 

Abstract
1. Introduction
2. Clinical Background
3. Genetic Basis of Inherited Cardiomyopathies
4. Generation of Patient-Specific-Induced Pluripotent Stem Cells Via Reprogramming
5. Genetic Modification of Induced Pluripotent Stem Cell Lines
6. Differentiation of Human Induced Pluripotent Stem Cells into Cardiomyocytes
7. Methods for the Functional Analysis of Cardiomyocytes Derived from Induced Pluripotent Stem Cells
8. Overview about Existing iPSC Lines Carrying Cardiomyopathy Associated Mutations
9. Limitations of Human Induced Pluripotent Stem-Cell-Derived Cardiomyocytes
9.1. Genomic Instability
9.2. Heterogeneity of iPSC-Derived Cardiomyocytes
9.3. Cellular, Molecular, and Functional Differences of Adult Ventricular Cardiomyocytes and iPSC-Derived Cardiomyocytes
10. Testing of Gene Therapies Using iPSC-Derived Cardiomyocytes as in Vitro Models
11. Summary
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACMG | American College of Medical Genetics and Genomics Institute |
| ACM | Arrhythmogenic Cardiomyopathy |
| CM | Cardiomyopathy |
| DCM | Dilated Cardiomyopathy |
| ESC | Embryonic Stem Cell |
| HCM | Hypertrophic Cardiomyopathy |
| iPSC | Induced Pluripotent Stem Cell |
| HTx | Heart Transplantation |
| NCCM | Non-Compaction Cardiomyopathy |
| MP | Myopathy |
| NMD | Nonsense Mediated RNA Decay |
| RCM | Restrictive Cardiomyopathy |
References
- Green, E.D.; Watson, J.D.; Collins, F.S. Human genome project: Twenty-five years of big biology. Nat. News 2015, 526, 29–31. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Ebbinghaus, H.; Gaertner-Rommel, A.; Stanasiuk, C.; Klauke, B.; Milting, H. Functional analysis of DES-p.L398P and RBM20-p.R636C. Genet. Med. 2019, 21, 1246–1247. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genet. and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Towbin, J.A.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; Moss, A.J.; Seidman, C.E.; Young, J.B.; American Heart, A.; et al. Contemporary definitions and classification of the cardiomyopathies: An american heart association scientific statement from the council on clinical cardiology, heart failure and transplantation committee; Quality of care and outcomes research and functional genomics and translational biology interdisciplinary working groups; and council on epidemiology and prevention. Circulation 2006, 113, 1807–1816. [Google Scholar] [PubMed]
- Alraies, M.C.; Eckman, P. Adult heart transplant: Indications and outcomes. J. Thorac. Dis. 2014, 6, 1120–1128. [Google Scholar] [PubMed]
- Minoche, A.E.; Horvat, C.; Johnson, R.; Gayevskiy, V.; Morton, S.U.; Drew, A.P.; Woo, K.; Statham, A.L.; Lundie, B.; Bagnall, R.D.; et al. Genome sequencing as a first-line genetic test in familial dilated cardiomyopathy. Genet. Med. 2019, 21, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Kolokotronis, K.; Kuhnisch, J.; Klopocki, E.; Dartsch, J.; Rost, S.; Huculak, C.; Mearini, G.; Stork, S.; Carrier, L.; Klaassen, S.; et al. Biallelic mutation in MYH7 and MYBPC3 leads to severe cardiomyopathy with left ventricular noncompaction phenotype. Hum. Mutat. 2019, 40, 1101–1114. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.P.; Kantor, P.F.; McBride, K.L.; Morales, A.; Taylor, M.R.G.; Vatta, M.; Ware, S.M.; et al. Genetic evaluation of cardiomyopathy: A clinical practice resource of the American College of Medical Genet. and Genomics (ACMG). Genet. Med. 2018, 20, 899–909. [Google Scholar] [CrossRef]
- Merner, N.D.; Hodgkinson, K.A.; Haywood, A.F.; Connors, S.; French, V.M.; Drenckhahn, J.D.; Kupprion, C.; Ramadanova, K.; Thierfelder, L.; McKenna, W.; et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am. J. Hum. Genet. 2008, 82, 809–821. [Google Scholar] [CrossRef] [PubMed]
- Milting, H.; Klauke, B.; Christensen, A.H.; Musebeck, J.; Walhorn, V.; Grannemann, S.; Munnich, T.; Saric, T.; Rasmussen, T.B.; Jensen, H.K.; et al. The TMEM43 Newfoundland mutation p.S358L causing ARVC-5 was imported from Europe and increases the stiffness of the cell nucleus. Eur. Heart J. 2015, 36, 872–881. [Google Scholar] [CrossRef]
- Christensen, A.H.; Andersen, C.B.; Tybjaerg-Hansen, A.; Haunso, S.; Svendsen, J.H. Mutation analysis and evaluation of the cardiac localization of TMEM43 in arrhythmogenic right ventricular cardiomyopathy. Clin. Genet. 2011, 80, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Stroud, M.J.; Fang, X.; Zhang, J.; Guimaraes-Camboa, N.; Veevers, J.; Dalton, N.D.; Gu, Y.; Bradford, W.H.; Peterson, K.L.; Evans, S.M.; et al. Luma is not essential for murine cardiac development and function. Cardiovasc. Res. 2018, 114, 378–388. [Google Scholar] [CrossRef]
- Figulla, H.R.; Wiegand, V.; Kreuzer, H. Dilated cardiomyopathy. Dtsch. Med. Wochenschr. 1989, 114, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Seidman, C.E.; Seidman, J.G. Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: A personal history. Circ. Res. 2011, 108, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Muchtar, E.; Blauwet, L.A.; Gertz, M.A. Restrictive cardiomyopathy: Genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ. Res. 2017, 121, 819–837. [Google Scholar] [CrossRef]
- Shariati, J.; Schlosser, T.; Erbel, R. Noncompaction cardiomyopathy. Herz 2015, 40, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Fazio, G.; Corrado, G.; Novo, G.; Zachara, E.; Rapezzi, C.; Sulafa, A.K.; Sutera, L.; D’Angelo, L.; Visconti, C.; Stollberger, C.; et al. Ventricular dysfunction and number of non compacted segments in non compaction: Non-independent predictors. Int. J. Cardiol. 2010, 141, 250–253. [Google Scholar] [CrossRef] [PubMed]
- Gandjbakhch, E.; Redheuil, A.; Pousset, F.; Charron, P.; Frank, R. Clinical diagnosis, imaging, and genetics of arrhythmogenic right ventricular cardiomyopathy/dysplasia: JACC state-of-the-art review. J. Am. Coll. Cardiol. 2018, 72, 784–804. [Google Scholar] [CrossRef]
- Basso, C.; Corrado, D.; Marcus, F.I.; Nava, A.; Thiene, G. Arrhythmogenic right ventricular cardiomyopathy. Lancet 2009, 373, 1289–1300. [Google Scholar] [CrossRef]
- Marcus, F.I.; Fontaine, G.H.; Guiraudon, G.; Frank, R.; Laurenceau, J.L.; Malergue, C.; Grosgogeat, Y. Right ventricular dysplasia: A report of 24 adult cases. Circulation 1982, 65, 384–398. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Thiene, G.; McKenna, W.J.; Davies, M.J.; Fontaliran, F.; Nava, A.; Silvestri, F.; Blomstrom-Lundqvist, C.; Wlodarska, E.K.; et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: A multicenter study. J. Am. Coll. Cardiol. 1997, 30, 1512–1520. [Google Scholar] [CrossRef]
- Geisterfer-Lowrance, A.A.; Kass, S.; Tanigawa, G.; Vosberg, H.P.; McKenna, W.; Seidman, C.E.; Seidman, J.G. A molecular basis for familial hypertrophic cardiomyopathy: A beta cardiac myosin heavy chain gene missense mutation. Cell 1990, 62, 999–1006. [Google Scholar] [CrossRef]
- Li, D.; Tapscoft, T.; Gonzalez, O.; Burch, P.E.; Quinones, M.A.; Zoghbi, W.A.; Hill, R.; Bachinski, L.L.; Mann, D.L.; Roberts, R. Desmin mutation responsible for idiopathic dilated cardiomyopathy. Circulation 1999, 100, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Dieding, M.; Biere, N.; Unger, A.; Klauke, B.; Walhorn, V.; Gummert, J.; Schulz, U.; Linke, W.A.; Gerull, B.; et al. Functional characterization of the novel DES mutation p.L136P associated with dilated cardiomyopathy reveals a dominant filament assembly defect. J. Mol. Cell. Cardiol. 2016, 91, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Harada, H.; Hayashi, T.; Nishi, H.; Kusaba, K.; Koga, Y.; Koga, Y.; Nonaka, I.; Kimura, A. Phenotypic expression of a novel desmin gene mutation: Hypertrophic cardiomyopathy followed by systemic myopathy. J. Hum. Genet. 2018, 63, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Klauke, B.; Kossmann, S.; Gaertner, A.; Brand, K.; Stork, I.; Brodehl, A.; Dieding, M.; Walhorn, V.; Anselmetti, D.; Gerdes, D.; et al. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum. Mol. Genet. 2010, 19, 4595–4607. [Google Scholar] [CrossRef] [PubMed]
- Bermudez-Jimenez, F.J.; Carriel, V.; Brodehl, A.; Alaminos, M.; Campos, A.; Schirmer, I.; Milting, H.; Abril, B.A.; Alvarez, M.; Lopez-Fernandez, S.; et al. Novel desmin mutation p.Glu401Asp impairs filament formation, disrupts cell membrane integrity, and causes severe arrhythmogenic left ventricular cardiomyopathy/dysplasia. Circulation 2018, 137, 1595–1610. [Google Scholar] [CrossRef] [PubMed]
- Pruszczyk, P.; Kostera-Pruszczyk, A.; Shatunov, A.; Goudeau, B.; Draminska, A.; Takeda, K.; Sambuughin, N.; Vicart, P.; Strelkov, S.V.; Goldfarb, L.G.; et al. Restrictive cardiomyopathy with atrioventricular conduction block resulting from a desmin mutation. Int. J. Cardiol. 2007, 117, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.; Lu, C.X.; Dong, X.Q.; Zhu, D.; Yang, K.Q.; Liu, K.Q.; Zhang, D.; Zhang, Y.; Meng, X.; Tan, H.Q.; et al. A novel phenotype with splicing mutation identified in a Chinese family with desminopathy. Chin. Med. J. 2019, 132, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Marakhonov, A.V.; Brodehl, A.; Myasnikov, R.P.; Sparber, P.A.; Kiseleva, A.V.; Kulikova, O.V.; Meshkov, A.N.; Zharikova, A.A.; Koretsky, S.N.; Kharlap, M.S.; et al. Non-compaction cardiomyopathy is caused by a novel in-frame desmin (DES) deletion mutation within the 1A coiled-coil rod segment leading to a severe filament assembly defect. Hum. Mutat. 2019. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Gaertner-Rommel, A.; Milting, H. Molecular insights into cardiomyopathies associated with desmin (DES) mutations. Biophys. Rev. 2018, 10, 983–1006. [Google Scholar] [CrossRef] [PubMed]
- Gerull, B.; Gramlich, M.; Atherton, J.; McNabb, M.; Trombitas, K.; Sasse-Klaassen, S.; Seidman, J.G.; Seidman, C.; Granzier, H.; Labeit, S.; et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat. Genet. 2002, 30, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Graw, S.; Sinagra, G.; Barnes, C.; Slavov, D.; Brun, F.; Pinamonti, B.; Salcedo, E.E.; Sauer, W.; Pyxaras, S.; et al. Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation 2011, 124, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Peled, Y.; Gramlich, M.; Yoskovitz, G.; Feinberg, M.S.; Afek, A.; Polak-Charcon, S.; Pras, E.; Sela, B.A.; Konen, E.; Weissbrod, O.; et al. Titin mutation in familial restrictive cardiomyopathy. Int. J. Cardiol. 2014, 171, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Haas, J.; Frese, K.S.; Peil, B.; Kloos, W.; Keller, A.; Nietsch, R.; Feng, Z.; Muller, S.; Kayvanpour, E.; Vogel, B.; et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur. Heart J. 2015, 36, 1123–1135. [Google Scholar] [CrossRef] [PubMed]
- Pugh, T.J.; Kelly, M.A.; Gowrisankar, S.; Hynes, E.; Seidman, M.A.; Baxter, S.M.; Bowser, M.; Harrison, B.; Aaron, D.; Mahanta, L.M.; et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet. Med. 2014, 16, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Klauke, B.; Gaertner-Rommel, A.; Schulz, U.; Kassner, A.; Zu Knyphausen, E.; Laser, T.; Kececioglu, D.; Paluszkiewicz, L.; Blanz, U.; Sandica, E.; et al. High proportion of genetic cases in patients with advanced cardiomyopathy including a novel homozygous Plakophilin 2-gene mutation. PLoS ONE 2017, 12, e0189489. [Google Scholar] [CrossRef] [PubMed]
- Thierfelder, L.; Watkins, H.; MacRae, C.; Lamas, R.; McKenna, W.; Vosberg, H.P.; Seidman, J.G.; Seidman, C.E. Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: A disease of the sarcomere. Cell 1994, 77, 701–712. [Google Scholar] [CrossRef]
- Landstrom, A.P.; Parvatiyar, M.S.; Pinto, J.R.; Marquardt, M.L.; Bos, J.M.; Tester, D.J.; Ommen, S.R.; Potter, J.D.; Ackerman, M.J. Molecular and functional characterization of novel hypertrophic cardiomyopathy susceptibility mutations in TNNC1-encoded troponin C. J. Mol. Cell. Cardiol. 2008, 45, 281–288. [Google Scholar] [CrossRef]
- Kimura, A.; Harada, H.; Park, J.E.; Nishi, H.; Satoh, M.; Takahashi, M.; Hiroi, S.; Sasaoka, T.; Ohbuchi, N.; Nakamura, T.; et al. Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat. Genet. 1997, 16, 379–382. [Google Scholar] [CrossRef]
- Knoblauch, H.; Geier, C.; Adams, S.; Budde, B.; Rudolph, A.; Zacharias, U.; Schulz-Menger, J.; Spuler, A.; Yaou, R.B.; Nurnberg, P.; et al. Contractures and hypertrophic cardiomyopathy in a novel FHL1 mutation. Ann. Neurol. 2010, 67, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Gaertner-Rommel, A.; Tiesmeier, J.; Jakob, T.; Strickmann, B.; Veit, G.; Bachmann-Mennenga, B.; Paluszkiewicz, L.; Klingel, K.; Schulz, U.; Laser, K.T.; et al. Molecular autopsy and family screening in a young case of sudden cardiac death reveals an unusually severe case of FHL1 related hypertrophic cardiomyopathy. Mol. Genet. Genom. Med. 2019, 7, e841. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, J.; Klausen, I.C.; Pedersen, A.K.; Egeblad, H.; Bross, P.; Kruse, T.A.; Gregersen, N.; Hansen, P.S.; Baandrup, U.; Borglum, A.D. Alpha-cardiac actin is a novel disease gene in familial hypertrophic cardiomyopathy. J. Clin. Investig. 1999, 103, R39–R43. [Google Scholar] [CrossRef] [PubMed]
- Haywood, N.J.; Wolny, M.; Rogers, B.; Trinh, C.H.; Shuping, Y.; Edwards, T.A.; Peckham, M. Hypertrophic cardiomyopathy mutations in the calponin-homology domain of ACTN2 affect actin binding and cardiomyocyte Z-disc incorporation. Biochem. J. 2016, 473, 2485–2493. [Google Scholar] [CrossRef] [PubMed]
- Valdes-Mas, R.; Gutierrez-Fernandez, A.; Gomez, J.; Coto, E.; Astudillo, A.; Puente, D.A.; Reguero, J.R.; Alvarez, V.; Moris, C.; Leon, D.; et al. Mutations in filamin C cause a new form of familial hypertrophic cardiomyopathy. Nat. Commun. 2014, 5, 5326. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.; Tebo, M.; Ingles, J.; Yeates, L.; Arthur, J.W.; Lind, J.M.; Semsarian, C. Genetic screening of calcium regulation genes in familial hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2007, 43, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Daehmlow, S.; Erdmann, J.; Knueppel, T.; Gille, C.; Froemmel, C.; Hummel, M.; Hetzer, R.; Regitz-Zagrosek, V. Novel mutations in sarcomeric protein genes in dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 2002, 298, 116–120. [Google Scholar] [CrossRef]
- Fatkin, D.; MacRae, C.; Sasaki, T.; Wolff, M.R.; Porcu, M.; Frenneaux, M.; Atherton, J.; Vidaillet, H.J., Jr.; Spudich, S.; De Girolami, U.; et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med. 1999, 341, 1715–1724. [Google Scholar] [CrossRef]
- McNair, W.P.; Ku, L.; Taylor, M.R.; Fain, P.R.; Dao, D.; Wolfel, E.; Mestroni, L.; Familial cardiomyopathy registry research, G. SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation 2004, 110, 2163–2167. [Google Scholar] [CrossRef]
- Schonberger, J.; Wang, L.; Shin, J.T.; Kim, S.D.; Depreux, F.F.; Zhu, H.; Zon, L.; Pizard, A.; Kim, J.B.; Macrae, C.A.; et al. Mutation in the transcriptional coactivator EYA4 causes dilated cardiomyopathy and sensorineural hearing loss. Nat. Genet. 2005, 37, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Brauch, K.M.; Karst, M.L.; Herron, K.J.; de Andrade, M.; Pellikka, P.A.; Rodeheffer, R.J.; Michels, V.V.; Olson, T.M. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 2009, 54, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Hey, T.M.; Rasmussen, T.B.; Madsen, T.; Aagaard, M.M.; Harbo, M.; Molgaard, H.; Moller, J.E.; Eiskjaer, H.; Mogensen, J. Pathogenic RBM20-variants are associated with a severe disease expression in male patients with dilated cardiomyopathy. Circ. Heart Fail. 2019, 12, e005700. [Google Scholar] [CrossRef] [PubMed]
- Nugent, A.W.; Daubeney, P.E.; Chondros, P.; Carlin, J.B.; Cheung, M.; Wilkinson, L.C.; Davis, A.M.; Kahler, S.G.; Chow, C.W.; Wilkinson, J.L.; et al. The epidemiology of childhood cardiomyopathy in Australia. N. Engl. J. Med. 2003, 348, 1639–1646. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, J.L.; Wilkinson, J.D.; Sleeper, L.A.; Colan, S.D.; Lu, M.; Pahl, E.; Kantor, P.F.; Everitt, M.D.; Webber, S.A.; Kaufman, B.D.; et al. Pediatric cardiomyopathy registry, I. Cardiomyopathy phenotypes and outcomes for children with left ventricular myocardial noncompaction: Results from the pediatric cardiomyopathy registry. J. Card. Fail. 2015, 21, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Marziliano, N.; Mannarino, S.; Nespoli, L.; Diegoli, M.; Pasotti, M.; Malattia, C.; Grasso, M.; Pilotto, A.; Porcu, E.; Raisaro, A.; et al. Barth syndrome associated with compound hemizygosity and heterozygosity of the TAZ and LDB3 genes. Am. J. Med. Genet. Part A 2007, 143A, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Probst, S.; Oechslin, E.; Schuler, P.; Greutmann, M.; Boye, P.; Knirsch, W.; Berger, F.; Thierfelder, L.; Jenni, R.; Klaassen, S. Sarcomere gene mutations in isolated left ventricular noncompaction cardiomyopathy do not predict clinical phenotype. Circ. Cardiovasc. Genet. 2011, 4, 367–374. [Google Scholar] [CrossRef] [PubMed]
- McKoy, G.; Protonotarios, N.; Crosby, A.; Tsatsopoulou, A.; Anastasakis, A.; Coonar, A.; Norman, M.; Baboonian, C.; Jeffery, S.; McKenna, W.J. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 2000, 355, 2119–2124. [Google Scholar] [CrossRef]
- Gerull, B.; Heuser, A.; Wichter, T.; Paul, M.; Basson, C.T.; McDermott, D.A.; Lerman, B.B.; Markowitz, S.M.; Ellinor, P.T.; MacRae, C.A.; et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat. Genet. 2004, 36, 1162–1164. [Google Scholar] [CrossRef]
- Patel, D.M.; Green, K.J. Desmosomes in the heart: A review of clinical and mechanistic analyses. Cell Commun. Adhes. 2014, 21, 109–128. [Google Scholar] [CrossRef]
- Xu, T.; Yang, Z.; Vatta, M.; Rampazzo, A.; Beffagna, G.; Pilichou, K.; Scherer, S.E.; Saffitz, J.; Kravitz, J.; Zareba, W.; et al. Multidisciplinary study of right ventricular dysplasia, I. Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J. Am. Coll. Cardiol. 2010, 55, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Hedde, P.N.; Dieding, M.; Fatima, A.; Walhorn, V.; Gayda, S.; Saric, T.; Klauke, B.; Gummert, J.; Anselmetti, D.; et al. Dual color photoactivation localization microscopy of cardiomyopathy-associated desmin mutants. J. Biol. Chem. 2012, 287, 16047–16057. [Google Scholar] [CrossRef] [PubMed]
- Quarta, G.; Syrris, P.; Ashworth, M.; Jenkins, S.; Zuborne Alapi, K.; Morgan, J.; Muir, A.; Pantazis, A.; McKenna, W.J.; Elliott, P.M. Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2012, 33, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Abdelfatah, N.; Chen, R.; Duff, H.J.; Seifer, C.M.; Buffo, I.; Huculak, C.; Clarke, S.; Clegg, R.; Jassal, D.S.; Gordon, P.M.K.; et al. Characterization of a unique form of arrhythmic cardiomyopathy caused by recessive mutation in LEMD2. JACC Basic Transl. Sci. 2019, 4, 204–221. [Google Scholar] [CrossRef] [PubMed]
- Tiso, N.; Stephan, D.A.; Nava, A.; Bagattin, A.; Devaney, J.M.; Stanchi, F.; Larderet, G.; Brahmbhatt, B.; Brown, K.; Bauce, B.; et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum. Mol. Genet. 2001, 10, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Milting, H.; Lukas, N.; Klauke, B.; Korfer, R.; Perrot, A.; Osterziel, K.J.; Vogt, J.; Peters, S.; Thieleczek, R.; Varsanyi, M. Composite polymorphisms in the ryanodine receptor 2 gene associated with arrhythmogenic right ventricular cardiomyopathy. Cardiovasc. Res. 2006, 71, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Van der Zwaag, P.A.; van Rijsingen, I.A.; Asimaki, A.; Jongbloed, J.D.; van Veldhuisen, D.J.; Wiesfeld, A.C.; Cox, M.G.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 1199–1207. [Google Scholar] [CrossRef]
- Brodehl, A.; Rezazadeh, S.; Williams, T.; Munsie, N.M.; Liedtke, D.; Oh, T.; Ferrier, R.; Shen, Y.; Jones, S.J.M.; Stiegler, A.L.; et al. Mutations in ILK, encoding integrin-linked kinase, are associated with arrhythmogenic cardiomyopathy. Transl. Res. 2019, 208, 15–29. [Google Scholar] [CrossRef]
- Kostareva, A.; Kiselev, A.; Gudkova, A.; Frishman, G.; Ruepp, A.; Frishman, D.; Smolina, N.; Tarnovskaya, S.; Nilsson, D.; Zlotina, A.; et al. Genetic spectrum of idiopathic restrictive cardiomyopathy uncovered by next-generation sequencing. PLoS ONE 2016, 11, e0163362. [Google Scholar] [CrossRef]
- Gallego-Delgado, M.; Delgado, J.F.; Brossa-Loidi, V.; Palomo, J.; Marzoa-Rivas, R.; Perez-Villa, F.; Salazar-Mendiguchia, J.; Ruiz-Cano, M.J.; Gonzalez-Lopez, E.; Padron-Barthe, L.; et al. Idiopathic restrictive cardiomyopathy Is primarily a genetic disease. J. Am. Coll. Cardiol. 2016, 67, 3021–3023. [Google Scholar] [CrossRef]
- Mogensen, J.; Kubo, T.; Duque, M.; Uribe, W.; Shaw, A.; Murphy, R.; Gimeno, J.R.; Elliott, P.; McKenna, W.J. Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. J. Clin. Investig. 2003, 111, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Ferrier, R.A.; Hamilton, S.J.; Greenway, S.C.; Brundler, M.A.; Yu, W.; Gibson, W.T.; McKinnon, M.L.; McGillivray, B.; Alvarez, N.; et al. Mutations in FLNC are associated with familial restrictive cardiomyopathy. Hum. Mutat. 2016, 37, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Tucker, N.R.; McLellan, M.A.; Hu, D.; Ye, J.; Parsons, V.A.; Mills, R.W.; Clauss, S.; Dolmatova, E.; Shea, M.A.; Milan, D.J.; et al. Novel mutation in FLNC (Filamin C) causes familial restrictive cardiomyopathy. Circ. Cardiovasc. Genet. 2017, 10, e001780. [Google Scholar] [CrossRef] [PubMed]
- Kiselev, A.; Vaz, R.; Knyazeva, A.; Khudiakov, A.; Tarnovskaya, S.; Liu, J.; Sergushichev, A.; Kazakov, S.; Frishman, D.; Smolina, N.; et al. De novo mutations in FLNC leading to early-onset restrictive cardiomyopathy and congenital myopathy. Hum. Mutat. 2018, 39, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Schubert, J.; Tariq, M.; Geddes, G.; Kindel, S.; Miller, E.M.; Ware, S.M. Novel pathogenic variants in filamin C identified in pediatric restrictive cardiomyopathy. Hum. Mutat. 2018, 39, 2083–2096. [Google Scholar] [CrossRef] [PubMed]
- Roldan-Sevilla, A.; Palomino-Doza, J.; de Juan, J.; Sanchez, V.; Dominguez-Gonzalez, C.; Salguero-Bodes, R.; Arribas-Ynsaurriaga, F. Missense mutations in the FLNC gene causing familial restrictive cardiomyopathy. Circ. Genom. Precis. Med. 2019, 12, e002388. [Google Scholar] [CrossRef]
- Bienengraeber, M.; Olson, T.M.; Selivanov, V.A.; Kathmann, E.C.; O’Cochlain, F.; Gao, F.; Karger, A.B.; Ballew, J.D.; Hodgson, D.M.; Zingman, L.V.; et al. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat. Genet. 2004, 36, 382–387. [Google Scholar] [CrossRef]
- Collet, M.; Assouline, Z.; Bonnet, D.; Rio, M.; Iserin, F.; Sidi, D.; Goldenberg, A.; Lardennois, C.; Metodiev, M.D.; Haberberger, B.; et al. High incidence and variable clinical outcome of cardiac hypertrophy due to ACAD9 mutations in childhood. Eur. J. Hum. Genet. 2016, 24, 1112–1116. [Google Scholar] [CrossRef]
- Carlus, S.J.; Almuzaini, I.S.; Karthikeyan, M.; Loganathan, L.; Al-Harbi, G.S.; Abdallah, A.M.; Al-Harbi, K.M. Next-generation sequencing identifies a homozygous mutation in ACADVL associated with pediatric familial dilated cardiomyopathy. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 1710–1721. [Google Scholar]
- Monserrat, L.; Hermida-Prieto, M.; Fernandez, X.; Rodriguez, I.; Dumont, C.; Cazon, L.; Cuesta, M.G.; Gonzalez-Juanatey, C.; Peteiro, J.; Alvarez, N.; et al. Mutation in the alpha-cardiac actin gene associated with apical hypertrophic cardiomyopathy, left ventricular non-compaction, and septal defects. Eur. Heart J. 2007, 28, 1953–1961. [Google Scholar] [CrossRef]
- Olson, T.M.; Michels, V.V.; Thibodeau, S.N.; Tai, Y.S.; Keating, M.T. Actin mutations in dilated cardiomyopathy, a heritable form of heart failure. Science 1998, 280, 750–752. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, S.; Probst, S.; Oechslin, E.; Gerull, B.; Krings, G.; Schuler, P.; Greutmann, M.; Hurlimann, D.; Yegitbasi, M.; Pons, L.; et al. Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation 2008, 117, 2893–2901. [Google Scholar] [CrossRef] [PubMed]
- Kaski, J.P.; Syrris, P.; Burch, M.; Tome-Esteban, M.T.; Fenton, M.; Christiansen, M.; Andersen, P.S.; Sebire, N.; Ashworth, M.; Deanfield, J.E.; et al. Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart 2008, 94, 1478–1484. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.; Bagnall, R.D.; Ingles, J.; Yeates, L.; Kennerson, M.; Donald, J.A.; Jormakka, M.; Lind, J.M.; Semsarian, C. Mutations in alpha-actinin-2 cause hypertrophic cardiomyopathy: A genome-wide analysis. J. Am. Coll. Cardiol. 2010, 55, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, B.; Jimenez, S.; Lin, J.H.; Bowles, K.R.; Coveler, K.J.; Marx, J.G.; Chrisco, M.A.; Murphy, R.T.; Lurie, P.R.; Schwartz, R.J.; et al. Mutations in the muscle LIM protein and alpha-actinin-2 genes in dilated cardiomyopathy and endocardial fibroelastosis. Mol. Genet. Metab. 2003, 80, 207–215. [Google Scholar] [CrossRef]
- Bagnall, R.D.; Molloy, L.K.; Kalman, J.M.; Semsarian, C. Exome sequencing identifies a mutation in the ACTN2 gene in a family with idiopathic ventricular fibrillation, left ventricular noncompaction, and sudden death. BMC Med. Genet. 2014, 15, 99. [Google Scholar] [CrossRef] [PubMed]
- Waldmuller, S.; Schroeder, C.; Sturm, M.; Scheffold, T.; Imbrich, K.; Junker, S.; Frische, C.; Hofbeck, M.; Bauer, P.; Bonin, M.; et al. Targeted 46-gene and clinical exome sequencing for mutations causing cardiomyopathies. Mol. Cell. Probes 2015, 29, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Forleo, C.; D’Erchia, A.M.; Sorrentino, S.; Manzari, C.; Chiara, M.; Iacoviello, M.; Guaricci, A.I.; De Santis, D.; Musci, R.L.; La Spada, A.; et al. Targeted next-generation sequencing detects novel gene-phenotype associations and expands the mutational spectrum in cardiomyopathies. PLoS ONE 2017, 12, e0181842. [Google Scholar] [CrossRef] [PubMed]
- Nerakh, G.; Ranganath, P. Alstrom syndrome presenting as isolated dilated cardiomyopathy. Indian J. Pediatr. 2019, 86, 296–298. [Google Scholar] [CrossRef]
- Almomani, R.; Verhagen, J.M.; Herkert, J.C.; Brosens, E.; van Spaendonck-Zwarts, K.Y.; Asimaki, A.; van der Zwaag, P.A.; Frohn-Mulder, I.M.; Bertoli-Avella, A.M.; Boven, L.G.; et al. Biallelic truncating mutations in ALPK3 cause severe pediatric cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 515–525. [Google Scholar] [CrossRef]
- Lopes, L.R.; Syrris, P.; Guttmann, O.P.; O’Mahony, C.; Tang, H.C.; Dalageorgou, C.; Jenkins, S.; Hubank, M.; Monserrat, L.; McKenna, W.J.; et al. Novel genotype-phenotype associations demonstrated by high-throughput sequencing in patients with hypertrophic cardiomyopathy. Heart 2015, 101, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.D.; Murphy, N.P.; Hamilton, R.M.; Lubbers, E.R.; James, C.A.; Kline, C.F.; Gollob, M.H.; Krahn, A.D.; Sturm, A.C.; Musa, H.; et al. Ankyrin-B dysfunction predisposes to arrhythmogenic cardiomyopathy and is amenable to therapy. J. Clin. Investig. 2019, 129, 3171–3184. [Google Scholar] [CrossRef] [PubMed]
- Arimura, T.; Bos, J.M.; Sato, A.; Kubo, T.; Okamoto, H.; Nishi, H.; Harada, H.; Koga, Y.; Moulik, M.; Doi, Y.L.; et al. Cardiac ankyrin repeat protein gene (ANKRD1) mutations in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2009, 54, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Duboscq-Bidot, L.; Charron, P.; Ruppert, V.; Fauchier, L.; Richter, A.; Tavazzi, L.; Arbustini, E.; Wichter, T.; Maisch, B.; Komajda, M.; et al. Mutations in the ANKRD1 gene encoding CARP are responsible for human dilated cardiomyopathy. Eur. Heart J. 2009, 30, 2128–2136. [Google Scholar] [CrossRef] [PubMed]
- Moulik, M.; Vatta, M.; Witt, S.H.; Arola, A.M.; Murphy, R.T.; McKenna, W.J.; Boriek, A.M.; Oka, K.; Labeit, S.; Bowles, N.E.; et al. ANKRD1, the gene encoding cardiac ankyrin repeat protein, is a novel dilated cardiomyopathy gene. J. Am. Coll. Cardiol. 2009, 54, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Norton, N.; Li, D.; Rieder, M.J.; Siegfried, J.D.; Rampersaud, E.; Zuchner, S.; Mangos, S.; Gonzalez-Quintana, J.; Wang, L.; McGee, S.; et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am. J. Hum. Genet. 2011, 88, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Schanzer, A.; Rupp, S.; Graf, S.; Zengeler, D.; Jux, C.; Akinturk, H.; Gulatz, L.; Mazhari, N.; Acker, T.; Van Coster, R.; et al. Dysregulated autophagy in restrictive cardiomyopathy due to Pro209Leu mutation in BAG3. Mol. Genet. Metab. 2018, 123, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Gripp, K.W.; Lin, A.E.; Nicholson, L.; Allen, W.; Cramer, A.; Jones, K.L.; Kutz, W.; Peck, D.; Rebolledo, M.A.; Wheeler, P.G.; et al. Further delineation of the phenotype resulting from BRAF or MEK1 germline mutations helps differentiate cardio-facio-cutaneous syndrome from Costello syndrome. Am. J. Med. Genet. Part A 2007, 143A, 1472–1480. [Google Scholar] [CrossRef] [PubMed]
- Shakeel, M.; Irfan, M.; Khan, I.A. Rare genetic mutations in Pakistani patients with dilated cardiomyopathy. Gene 2018, 673, 134–139. [Google Scholar] [CrossRef]
- Boczek, N.J.; Ye, D.; Jin, F.; Tester, D.J.; Huseby, A.; Bos, J.M.; Johnson, A.J.; Kanter, R.; Ackerman, M.J. Identification and functional characterization of a novel CACNA1C-mediated cardiac disorder characterized by prolonged QT intervals with hypertrophic cardiomyopathy, congenital heart defects, and sudden cardiac death. Circ. Arrhythm. Electrophysiol. 2015, 8, 1122–1132. [Google Scholar] [CrossRef]
- Friedrich, F.W.; Bausero, P.; Sun, Y.; Treszl, A.; Kramer, E.; Juhr, D.; Richard, P.; Wegscheider, K.; Schwartz, K.; Brito, D.; et al. A new polymorphism in human calmodulin III gene promoter is a potential modifier gene for familial hypertrophic cardiomyopathy. Eur. Heart J. 2009, 30, 1648–1655. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.B.; Qu, X.K.; Li, R.G.; Liu, H.; Xu, Y.J.; Zhang, M.; Shi, H.Y.; Hou, X.M.; Liu, X.; Yuan, F.; et al. CASZ1 loss-of-function mutation contributes to familial dilated cardiomyopathy. Clin. Chem. Lab. Med. 2017, 55, 1417–1425. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Li, Z.; Hao, C.; Guo, R.; Hu, X.; Qian, S.; Zeng, J.; Gao, H.; Li, W. A novel de novo CASZ1 heterozygous frameshift variant causes dilated cardiomyopathy and left ventricular noncompaction cardiomyopathy. Mol. Genet. Genom. Med. 2019, 7, e828. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Arimura, T.; Ueda, K.; Shibata, H.; Hohda, S.; Takahashi, M.; Hori, H.; Koga, Y.; Oka, N.; Imaizumi, T.; et al. Identification and functional analysis of a caveolin-3 mutation associated with familial hypertrophic cardiomyopathy. Biochem. Biophys. Res. Commun. 2004, 313, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, G.; Ueyama, T.; Ogata, T.; Czernuszewicz, G.; Tan, Y.; Dorn, G.W., II; Bogaev, R.; Amano, K.; Oh, H.; Matsubara, H.; et al. Molecular genetic and functional characterization implicate muscle-restricted coiled-coil gene (MURC) as a causal gene for familial dilated cardiomyopathy. Circ. Cardiovasc. Genet. 2011, 4, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Mayosi, B.M.; Fish, M.; Shaboodien, G.; Mastantuono, E.; Kraus, S.; Wieland, T.; Kotta, M.C.; Chin, A.; Laing, N.; Ntusi, N.B.; et al. Identification of Cadherin 2 (CDH2) mutations in arrhythmogenic right ventricular cardiomyopathy. Circ. Cardiovasc. Genet. 2017, 10, e001605. [Google Scholar] [CrossRef]
- Turkowski, K.L.; Tester, D.J.; Bos, J.M.; Haugaa, K.H.; Ackerman, M.J. Whole exome sequencing with genomic triangulation implicates CDH2-encoded N-cadherin as a novel pathogenic substrate for arrhythmogenic cardiomyopathy. Congenit. Heart Dis. 2017, 12, 226–235. [Google Scholar] [CrossRef]
- Zhang, L.; Hu, A.; Yuan, H.; Cui, L.; Miao, G.; Yang, X.; Wang, L.; Liu, J.; Liu, X.; Wang, S.; et al. A missense mutation in the CHRM2 gene is associated with familial dilated cardiomyopathy. Circ. Res. 2008, 102, 1426–1432. [Google Scholar] [CrossRef]
- Lan, N.S.R.; Fietz, M.; Pachter, N.; Paul, V.; Playford, D. A case of vascular Ehlers-Danlos Syndrome with a cardiomyopathy and multi-system involvement. Cardiovasc. Pathol. 2018, 35, 48–51. [Google Scholar] [CrossRef]
- Antonicka, H.; Mattman, A.; Carlson, C.G.; Glerum, D.M.; Hoffbuhr, K.C.; Leary, S.C.; Kennaway, N.G.; Shoubridge, E.A. Mutations in COX15 produce a defect in the mitochondrial heme biosynthetic pathway, causing early-onset fatal hypertrophic cardiomyopathy. Am. J. Hum. Genet. 2003, 72, 101–114. [Google Scholar] [CrossRef]
- Inagaki, N.; Hayashi, T.; Arimura, T.; Koga, Y.; Takahashi, M.; Shibata, H.; Teraoka, K.; Chikamori, T.; Yamashina, A.; Kimura, A. αB-crystallin mutation in dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 2006, 342, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Gaertner-Rommel, A.; Klauke, B.; Grewe, S.A.; Schirmer, I.; Peterschroder, A.; Faber, L.; Vorgerd, M.; Gummert, J.; Anselmetti, D.; et al. The novel αB-crystallin (CRYAB) mutation p.D109G causes restrictive cardiomyopathy. Hum. Mutat. 2017, 38, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Newman, B.; Cescon, D.; Woo, A.; Rakowski, H.; Erikkson, M.J.; Sole, M.; Wigle, E.D.; Siminovitch, K.A. W4R variant in CSRP3 encoding muscle LIM protein in a patient with hypertrophic cardiomyopathy. Mol. Genet. Metab. 2005, 84, 374–375. [Google Scholar] [CrossRef] [PubMed]
- Geier, C.; Gehmlich, K.; Ehler, E.; Hassfeld, S.; Perrot, A.; Hayess, K.; Cardim, N.; Wenzel, K.; Erdmann, B.; Krackhardt, F.; et al. Beyond the sarcomere: CSRP3 mutations cause hypertrophic cardiomyopathy. Hum. Mol. Genet. 2008, 17, 2753–2765. [Google Scholar] [CrossRef] [PubMed]
- Janin, A.; Bessiere, F.; Chauveau, S.; Chevalier, P.; Millat, G. First identification of homozygous truncating CSRP3 variants in two unrelated cases with hypertrophic cardiomyopathy. Gene 2018, 676, 110–116. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Parks, S.B.; Kushner, J.D.; Li, D.; Ludwigsen, S.; Jakobs, P.; Nauman, D.; Burgess, D.; Partain, J.; Litt, M. Coding sequence mutations identified in MYH7, TNNT2, SCN5A, CSRP3, LBD3, and TCAP from 313 patients with familial or idiopathic dilated cardiomyopathy. Clin. Transl. Sci. 2008, 1, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, J.; Hassfeld, S.; Kallisch, H.; Fleck, E.; Regitz-Zagrose, V. Genetic variants in the promoter (g983G>T) and coding region (A92T) of the human cardiotrophin-1 gene (CTF1) in patients with dilated cardiomyopathy. Hum. Mutat. 2000, 16, 448. [Google Scholar] [CrossRef]
- Van Hengel, J.; Calore, M.; Bauce, B.; Dazzo, E.; Mazzotti, E.; De Bortoli, M.; Lorenzon, A.; Li Mura, I.E.; Beffagna, G.; Rigato, I.; et al. Mutations in the area composita protein alphaT-catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2013, 34, 201–210. [Google Scholar] [CrossRef]
- Taylor, M.R.; Slavov, D.; Ku, L.; Di Lenarda, A.; Sinagra, G.; Carniel, E.; Haubold, K.; Boucek, M.M.; Ferguson, D.; Graw, S.L.; et al. Prevalence of desmin mutations in dilated cardiomyopathy. Circulation 2007, 115, 1244–1251. [Google Scholar] [CrossRef]
- Towbin, J.A.; Hejtmancik, J.F.; Brink, P.; Gelb, B.; Zhu, X.M.; Chamberlain, J.S.; McCabe, E.R.; Swift, M. X-linked dilated cardiomyopathy. Molecular genetic evidence of linkage to the Duchenne muscular dystrophy (dystrophin) gene at the Xp21 locus. Circulation 1993, 87, 1854–1865. [Google Scholar] [CrossRef]
- Ojala, T.; Polinati, P.; Manninen, T.; Hiippala, A.; Rajantie, J.; Karikoski, R.; Suomalainen, A.; Tyni, T. New mutation of mitochondrial DNAJC19 causing dilated and noncompaction cardiomyopathy, anemia, ataxia, and male genital anomalies. Pediatr. Res. 2012, 72, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Rush, E.T.; Baker, C.V.; Rizzo, W.B. Dolichol kinase deficiency (DOLK-CDG): Two new cases and expansion of phenotype. Am. J. Med. Genet. Part A 2017, 173, 2428–2434. [Google Scholar] [CrossRef] [PubMed]
- Svahn, J.; Laforet, P.; Vial, C.; Streichenberger, N.; Romero, N.; Bouchet-Seraphin, C.; Bruneel, A.; Dupre, T.; Seta, N.; Menassa, R.; et al. Dilated cardiomyopathy and limb-girdle muscular dystrophy-dystroglycanopathy due to novel pathogenic variants in the DPM3 gene. Neuromuscul. Disord. 2019, 29, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Heuser, A.; Plovie, E.R.; Ellinor, P.T.; Grossmann, K.S.; Shin, J.T.; Wichter, T.; Basson, C.T.; Lerman, B.B.; Sasse-Klaassen, S.; Thierfelder, L.; et al. Mutant desmocollin-2 causes arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2006, 79, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Posch, M.G.; Posch, M.J.; Geier, C.; Erdmann, B.; Mueller, W.; Richter, A.; Ruppert, V.; Pankuweit, S.; Maisch, B.; Perrot, A.; et al. A missense variant in desmoglein-2 predisposes to dilated cardiomyopathy. Mol. Genet. Metab. 2008, 95, 74–80. [Google Scholar] [CrossRef]
- Awad, M.M.; Dalal, D.; Cho, E.; Amat-Alarcon, N.; James, C.; Tichnell, C.; Tucker, A.; Russell, S.D.; Bluemke, D.A.; Dietz, H.C.; et al. DSG2 mutations contribute to arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am. J. Hum. Genet. 2006, 79, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Pilichou, K.; Nava, A.; Basso, C.; Beffagna, G.; Bauce, B.; Lorenzon, A.; Frigo, G.; Vettori, A.; Valente, M.; Towbin, J.; et al. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation 2006, 113, 1171–1179. [Google Scholar] [CrossRef]
- Norgett, E.E.; Hatsell, S.J.; Carvajal-Huerta, L.; Cabezas, J.C.; Common, J.; Purkis, P.E.; Whittock, N.; Leigh, I.M.; Stevens, H.P.; Kelsell, D.P. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum. Mol. Genet. 2000, 9, 2761–2766. [Google Scholar] [CrossRef]
- Williams, T.; Machann, W.; Kuhler, L.; Hamm, H.; Muller-Hocker, J.; Zimmer, M.; Ertl, G.; Ritter, O.; Beer, M.; Schonberger, J. Novel desmoplakin mutation: Juvenile biventricular cardiomyopathy with left ventricular non-compaction and acantholytic palmoplantar keratoderma. Clin. Res. Cardiol. 2011, 100, 1087–1093. [Google Scholar] [CrossRef]
- Rampazzo, A.; Nava, A.; Malacrida, S.; Beffagna, G.; Bauce, B.; Rossi, V.; Zimbello, R.; Simionati, B.; Basso, C.; Thiene, G.; et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2002, 71, 1200–1206. [Google Scholar] [CrossRef]
- Cao, Q.; Shen, Y.; Liu, X.; Yu, X.; Yuan, P.; Wan, R.; Liu, X.; Peng, X.; He, W.; Pu, J.; et al. Phenotype and functional analyses in a transgenic mouse model of left ventricular noncompaction caused by a DTNA mutation. Int. Heart J. 2017, 58, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Haack, T.B.; Kopajtich, R.; Freisinger, P.; Wieland, T.; Rorbach, J.; Nicholls, T.J.; Baruffini, E.; Walther, A.; Danhauser, K.; Zimmermann, F.A.; et al. ELAC2 mutations cause a mitochondrial RNA processing defect associated with hypertrophic cardiomyopathy. Am. J. Hum. Genet. 2013, 93, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Chen, J.; Si, D.; Zheng, Y.; Jiao, H.; Feng, Z.; Hu, Z.; Duan, R. Whole exome sequencing identifies a novel EMD mutation in a Chinese family with dilated cardiomyopathy. BMC Med. Genet. 2014, 15, 77. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Hoshida, H.; Kamisago, M.; Yagi, H.; Momma, K.; Matsuoka, R. Phenotype-genotype correlation in a patient with co-occurrence of Marfan and LEOPARD syndromes. Am. J. Med. Genet. Part A 2009, 149A, 2216–2219. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, A.M.; Toudjarska, I.; Racine, A.; Tsipouras, P.; Kilpatrick, M.W.; Shanske, A. A recurring FBN1 gene mutation in neonatal Marfan syndrome. Arch. Pediatr. Adolesc. Med. 2002, 156, 1081–1085. [Google Scholar] [CrossRef] [PubMed]
- Parent, J.J.; Towbin, J.A.; Jefferies, J.L. Fibrillin-1 gene mutations in left ventricular non-compaction cardiomyopathy. Pediatr. Cardiol. 2016, 37, 1123–1126. [Google Scholar] [CrossRef]
- Al-Yacoub, N.; Shaheen, R.; Awad, S.M.; Kunhi, M.; Dzimiri, N.; Nguyen, H.C.; Xiong, Y.; Al-Buraiki, J.; Al-Habeeb, W.; Alkuraya, F.S.; et al. FBXO32, encoding a member of the SCF complex, is mutated in dilated cardiomyopathy. Genome Biol. 2016, 17, 2. [Google Scholar] [CrossRef]
- Al-Hassnan, Z.N.; Shinwari, Z.M.; Wakil, S.M.; Tulbah, S.; Mohammed, S.; Rahbeeni, Z.; Alghamdi, M.; Rababh, M.; Colak, D.; Kaya, N.; et al. A substitution mutation in cardiac ubiquitin ligase, FBXO32, is associated with an autosomal recessive form of dilated cardiomyopathy. BMC Med. Genet. 2016, 17, 3. [Google Scholar] [CrossRef] [PubMed]
- Arimura, T.; Hayashi, T.; Matsumoto, Y.; Shibata, H.; Hiroi, S.; Nakamura, T.; Inagaki, N.; Hinohara, K.; Takahashi, M.; Manatsu, S.I.; et al. Structural analysis of four and half LIM protein-2 in dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 2007, 357, 162–167. [Google Scholar] [CrossRef]
- Ochoa, J.P.; Sabater-Molina, M.; Garcia-Pinilla, J.M.; Mogensen, J.; Restrepo-Cordoba, A.; Palomino-Doza, J.; Villacorta, E.; Martinez-Moreno, M.; Ramos-Maqueda, J.; Zorio, E.; et al. Formin Homology 2 Domain Containing 3 (FHOD3) Is a genetic basis for hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2018, 72, 2457–2467. [Google Scholar] [CrossRef]
- Arimura, T.; Takeya, R.; Ishikawa, T.; Yamano, T.; Matsuo, A.; Tatsumi, T.; Nomura, T.; Sumimoto, H.; Kimura, A. Dilated cardiomyopathy-associated FHOD3 variant impairs the ability to induce activation of transcription factor serum response factor. Circ. J. 2013, 77, 2990–2996. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.; Krasnianski, M.; Witthaut, R.; Deschauer, M.; Zierz, S. Dilated cardiomyopathy may be an early sign of the C826A Fukutin-related protein mutation. Neuromuscul. Disord. 2005, 15, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Amiya, E.; Morita, H.; Hatano, M.; Nitta, D.; Hosoya, Y.; Maki, H.; Motozawa, Y.; Sato, N.; Ishiura, H.; Numakura, S.; et al. Fukutin gene mutations that cause left ventricular noncompaction. Int. J. Cardiol. 2016, 222, 727–729. [Google Scholar] [CrossRef] [PubMed]
- Begay, R.L.; Tharp, C.A.; Martin, A.; Graw, S.L.; Sinagra, G.; Miani, D.; Sweet, M.E.; Slavov, D.B.; Stafford, N.; Zeller, M.J.; et al. FLNC gene splice mutations cause dilated cardiomyopathy. JACC Basic Transl. Sci. 2016, 1, 344–359. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Genga, M.F.; Cuenca, S.; Dal Ferro, M.; Zorio, E.; Salgado-Aranda, R.; Climent, V.; Padron-Barthe, L.; Duro-Aguado, I.; Jimenez-Jaimez, J.; Hidalgo-Olivares, V.M.; et al. Truncating FLNC mutations are associated with high-risk dilated and arrhythmogenic cardiomyopathies. J. Am. Coll. Cardiol. 2016, 68, 2440–2451. [Google Scholar] [CrossRef] [PubMed]
- Minoretti, P.; Arra, M.; Emanuele, E.; Olivieri, V.; Aldeghi, A.; Politi, P.; Martinelli, V.; Pesenti, S.; Falcone, C. A W148R mutation in the human FOXD4 gene segregating with dilated cardiomyopathy, obsessive-compulsive disorder, and suicidality. Int. J. Mol. Med. 2007, 19, 369–372. [Google Scholar] [CrossRef]
- Payne, R.M.; Wagner, G.R. Cardiomyopathy in Friedreich ataxia: Clinical findings and research. J. Child Neurol. 2012, 27, 1179–1186. [Google Scholar] [CrossRef]
- Campuzano, V.; Montermini, L.; Molto, M.D.; Pianese, L.; Cossee, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef]
- Li, R.G.; Li, L.; Qiu, X.B.; Yuan, F.; Xu, L.; Li, X.; Xu, Y.J.; Jiang, W.F.; Jiang, J.Q.; Liu, X.; et al. GATA4 loss-of-function mutation underlies familial dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 2013, 439, 591–596. [Google Scholar] [CrossRef][Green Version]
- Tang, V.T.; Arscott, P.; Helms, A.S.; Day, S.M. Whole-exome sequencing reveals GATA4 and PTEN mutations as a potential digenic cause of left ventricular noncompaction. Circ. Genom. Precis. Med. 2018, 11, e001966. [Google Scholar] [CrossRef]
- Zhang, X.L.; Dai, N.; Tang, K.; Chen, Y.Q.; Chen, W.; Wang, J.; Zhao, C.M.; Yuan, F.; Qiu, X.B.; Qu, X.K.; et al. GATA5 loss-of-function mutation in familial dilated cardiomyopathy. Int. J. Mol. Med. 2015, 35, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Theis, J.L.; Sharpe, K.M.; Matsumoto, M.E.; Chai, H.S.; Nair, A.A.; Theis, J.D.; de Andrade, M.; Wieben, E.D.; Michels, V.V.; Olson, T.M. Homozygosity mapping and exome sequencing reveal GATAD1 mutation in autosomal recessive dilated cardiomyopathy. Circ. Cardiovasc. Genet. 2011, 4, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Arad, M.; Penas-Lado, M.; Monserrat, L.; Maron, B.J.; Sherrid, M.; Ho, C.Y.; Barr, S.; Karim, A.; Olson, T.M.; Kamisago, M.; et al. Gene mutations in apical hypertrophic cardiomyopathy. Circulation 2005, 112, 2805–2811. [Google Scholar] [CrossRef] [PubMed]
- Kopajtich, R.; Nicholls, T.J.; Rorbach, J.; Metodiev, M.D.; Freisinger, P.; Mandel, H.; Vanlander, A.; Ghezzi, D.; Carrozzo, R.; Taylor, R.W.; et al. Mutations in GTPBP3 cause a mitochondrial translation defect associated with hypertrophic cardiomyopathy, lactic acidosis, and encephalopathy. Am. J. Hum. Genet. 2014, 95, 708–720. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.M.; Dai, X.Y.; Qiu, X.B.; Yuan, F.; Li, R.G.; Xu, Y.J.; Qu, X.K.; Huang, R.T.; Xue, S.; Yang, Y.Q. HAND1 loss-of-function mutation associated with familial dilated cardiomyopathy. Clin. Chem. Lab. Med. 2016, 54, 1161–1167. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xu, Y.J.; Li, R.G.; Wang, Z.S.; Zhang, M.; Qu, X.K.; Qiao, Q.; Li, X.M.; Di, R.M.; Qiu, X.B.; et al. HAND2 loss-of-function mutation causes familial dilated cardiomyopathy. Eur. J. Med. Genet. 2018. [Google Scholar] [CrossRef] [PubMed]
- Milano, A.; Vermeer, A.M.; Lodder, E.M.; Barc, J.; Verkerk, A.O.; Postma, A.V.; van der Bilt, I.A.; Baars, M.J.; van Haelst, P.L.; Caliskan, K.; et al. HCN4 mutations in multiple families with bradycardia and left ventricular noncompaction cardiomyopathy. J. Am. Coll. Cardiol. 2014, 64, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Hiippala, A.; Vasilescu, C.; Tallila, J.; Alastalo, T.P.; Paetau, A.; Tyni, T.; Suomalainen, A.; Euro, L.; Ojala, T. The rare Costello variant HRAS c.173C>T (p.T58I) with severe neonatal hypertrophic cardiomyopathy. Am. J. Med. Genet. Part A 2016, 170, 1433–1438. [Google Scholar] [CrossRef] [PubMed]
- Knoll, R.; Postel, R.; Wang, J.; Kratzner, R.; Hennecke, G.; Vacaru, A.M.; Vakeel, P.; Schubert, C.; Murthy, K.; Rana, B.K.; et al. Laminin-alpha4 and integrin-linked kinase mutations cause human cardiomyopathy via simultaneous defects in cardiomyocytes and endothelial cells. Circulation 2007, 116, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Meder, B.; Haas, J.; Keller, A.; Heid, C.; Just, S.; Borries, A.; Boisguerin, V.; Scharfenberger-Schmeer, M.; Stahler, P.; Beier, M.; et al. Targeted next-generation sequencing for the molecular genetic diagnostics of cardiomyopathies. Circ. Cardiovasc. Genet. 2011, 4, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.J.; Wang, Z.S.; Yang, C.X.; Di, R.M.; Qiao, Q.; Li, X.M.; Gu, J.N.; Guo, X.J.; Yang, Y.Q. Identification and functional characterization of an ISL1 mutation predisposing to dilated cardiomyopathy. J. Cardiovasc. Transl. Res. 2019, 12, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Esposito, T.; Sampaolo, S.; Limongelli, G.; Varone, A.; Formicola, D.; Diodato, D.; Farina, O.; Napolitano, F.; Pacileo, G.; Gianfrancesco, F.; et al. Digenic mutational inheritance of the integrin alpha 7 and the myosin heavy chain 7B genes causes congenital myopathy with left ventricular non-compact cardiomyopathy. Orphanet J. Rare Dis. 2013, 8, 91. [Google Scholar] [CrossRef] [PubMed]
- Handley, M.T.; Reddy, K.; Wills, J.; Rosser, E.; Kamath, A.; Halachev, M.; Falkous, G.; Williams, D.; Cox, P.; Meynert, A.; et al. ITPase deficiency causes a Martsolf-like syndrome with a lethal infantile dilated cardiomyopathy. PLoS Genet. 2019, 15, e1007605. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, Y.; Furukawa, T.; Kasanuki, H.; Nishibatake, M.; Kurihara, Y.; Ikeda, A.; Kamatani, N.; Takeshima, H.; Matsuoka, R. Mutation of junctophilin type 2 associated with hypertrophic cardiomyopathy. J. Hum. Genet. 2007, 52, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Sabater-Molina, M.; Navarro, M.; Garcia-Molina Saez, E.; Garrido, I.; Pascual-Figal, D.; Gonzalez Carrillo, J.; Gimeno Blanes, J.R. Mutation in JPH2 cause dilated cardiomyopathy. Clin. Genet. 2016, 90, 468–469. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Q.; Cao, Q.; Zhou, Q.; Xie, J.; Shen, Y.; Wan, R.; Yu, J.; Yan, S.; Marian, A.J.; Hong, K. Arrhythmogenic cardiomyopathy in a patient with a rare loss-of-function KCNQ1 mutation. J. Am. Heart Assoc. 2015, 4, e001526. [Google Scholar] [CrossRef] [PubMed]
- Hedberg-Oldfors, C.; Abramsson, A.; Osborn, D.P.S.; Danielsson, O.; Fazlinezhad, A.; Nilipour, Y.; Hubbert, L.; Nennesmo, I.; Visuttijai, K.; Bharj, J.; et al. Cardiomyopathy with lethal arrhythmias associated with inactivation of KLHL24. Hum. Mol. Genet. 2019, 28, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Arad, M.; Maron, B.J.; Gorham, J.M.; Johnson, W.H., Jr.; Saul, J.P.; Perez-Atayde, A.R.; Spirito, P.; Wright, G.B.; Kanter, R.J.; Seidman, C.E.; et al. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N. Engl. J. Med. 2005, 352, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Fratev, F.; Mihaylova, E.; Pajeva, I. Combination of genetic screening and molecular dynamics as a useful tool for identification of disease-related mutations: ZASP PDZ domain G54S mutation case. J. Chem. Inf. Model. 2014, 54, 1524–1536. [Google Scholar] [CrossRef]
- Vatta, M.; Mohapatra, B.; Jimenez, S.; Sanchez, X.; Faulkner, G.; Perles, Z.; Sinagra, G.; Lin, J.H.; Vu, T.M.; Zhou, Q.; et al. Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction. J. Am. Coll. Cardiol. 2003, 42, 2014–2027. [Google Scholar] [CrossRef]
- Arimura, T.; Hayashi, T.; Terada, H.; Lee, S.Y.; Zhou, Q.; Takahashi, M.; Ueda, K.; Nouchi, T.; Hohda, S.; Shibutani, M.; et al. A Cypher/ZASP mutation associated with dilated cardiomyopathy alters the binding affinity to protein kinase C. J. Biol. Chem. 2004, 279, 6746–6752. [Google Scholar] [CrossRef] [PubMed]
- Shan, S.; He, X.; He, L.; Wang, M.; Liu, C. Coexistence of congenital left ventricular aneurysm and prominent left ventricular trabeculation in a patient with LDB3 mutation: A case report. J. Med. Case Rep. 2017, 11, 229. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Ayala, J.M.; Ortiz-Genga, M.; Gomez-Milanes, I.; Lopez-Cuenca, D.; Ruiz-Espejo, F.; Sanchez-Munoz, J.J.; Oliva-Sandoval, M.J.; Monserrat, L.; Gimeno, J.R. A mutation in the Z-line Cypher/ZASP protein is associated with arrhythmogenic right ventricular cardiomyopathy. Clin. Genet. 2015, 88, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Boone, P.M.; Yuan, B.; Gu, S.; Ma, Z.; Gambin, T.; Gonzaga-Jauregui, C.; Jain, M.; Murdock, T.J.; White, J.J.; Jhangiani, S.N.; et al. Hutterite-type cataract maps to chromosome 6p21.32-p21.31, cosegregates with a homozygous mutation in LEMD2, and is associated with sudden cardiac death. Mol. Genet. Genom. Med. 2016, 4, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Shan, H.; Huang, J.; Li, N.; Hou, C.; Pu, J. A novel lamin A/C gene missense mutation (445 V > E) in immunoglobulin-like fold associated with left ventricular non-compaction. Europace 2016, 18, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.K.; Yuan, F.; Li, R.G.; Xu, L.; Jing, W.F.; Liu, H.; Xu, Y.J.; Zhang, M.; Liu, X.; Fang, W.Y.; et al. Prevalence and spectrum of LRRC10 mutations associated with idiopathic dilated cardiomyopathy. Mol. Med. Rep. 2015, 12, 3718–3724. [Google Scholar] [CrossRef]
- Luxan, G.; Casanova, J.C.; Martinez-Poveda, B.; Prados, B.; D’Amato, G.; MacGrogan, D.; Gonzalez-Rajal, A.; Dobarro, D.; Torroja, C.; Martinez, F.; et al. Mutations in the NOTCH pathway regulator MIB1 cause left ventricular noncompaction cardiomyopathy. Nat. Med. 2013, 19, 193–201. [Google Scholar] [CrossRef]
- Piccolo, P.; Attanasio, S.; Secco, I.; Sangermano, R.; Strisciuglio, C.; Limongelli, G.; Miele, E.; Mutarelli, M.; Banfi, S.; Nigro, V.; et al. MIB2 variants altering NOTCH signalling result in left ventricle hypertrabeculation/non-compaction and are associated with Menetrier-like gastropathy. Hum. Mol. Genet. 2017, 26, 33–43. [Google Scholar]
- Galmiche, L.; Serre, V.; Beinat, M.; Assouline, Z.; Lebre, A.S.; Chretien, D.; Nietschke, P.; Benes, V.; Boddaert, N.; Sidi, D.; et al. Exome sequencing identifies MRPL3 mutation in mitochondrial cardiomyopathy. Hum. Mutat. 2011, 32, 1225–1231. [Google Scholar] [CrossRef]
- Carroll, C.J.; Isohanni, P.; Poyhonen, R.; Euro, L.; Richter, U.; Brilhante, V.; Gotz, A.; Lahtinen, T.; Paetau, A.; Pihko, H.; et al. Whole-exome sequencing identifies a mutation in the mitochondrial ribosome protein MRPL44 to underlie mitochondrial infantile cardiomyopathy. J. Med. Genet. 2013, 50, 151–159. [Google Scholar] [CrossRef]
- Distelmaier, F.; Haack, T.B.; Catarino, C.B.; Gallenmuller, C.; Rodenburg, R.J.; Strom, T.M.; Baertling, F.; Meitinger, T.; Mayatepek, E.; Prokisch, H.; et al. MRPL44 mutations cause a slowly progressive multisystem disease with childhood-onset hypertrophic cardiomyopathy. NeuroGenet. 2015, 16, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Watkins, H.; Conner, D.; Thierfelder, L.; Jarcho, J.A.; MacRae, C.; McKenna, W.J.; Maron, B.J.; Seidman, J.G.; Seidman, C.E. Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nat. Genet. 1995, 11, 434–437. [Google Scholar] [CrossRef] [PubMed]
- Carrier, L.; Bonne, G.; Bahrend, E.; Yu, B.; Richard, P.; Niel, F.; Hainque, B.; Cruaud, C.; Gary, F.; Labeit, S.; et al. Organization and sequence of human cardiac myosin binding protein C gene (MYBPC3) and identification of mutations predicted to produce truncated proteins in familial hypertrophic cardiomyopathy. Circ. Res. 1997, 80, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Ehlermann, P.; Weichenhan, D.; Zehelein, J.; Steen, H.; Pribe, R.; Zeller, R.; Lehrke, S.; Zugck, C.; Ivandic, B.T.; Katus, H.A. Adverse events in families with hypertrophic or dilated cardiomyopathy and mutations in the MYBPC3 gene. BMC Med. Genet. 2008, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- Dellefave, L.M.; Pytel, P.; Mewborn, S.; Mora, B.; Guris, D.L.; Fedson, S.; Waggoner, D.; Moskowitz, I.; McNally, E.M. Sarcomere mutations in cardiomyopathy with left ventricular hypertrabeculation. Circ. Cardiovasc. Genet. 2009, 2, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Lu, C.X.; Wang, Y.N.; Liu, F.; Chen, W.; Liu, Y.T.; Han, Y.C.; Cao, J.; Zhang, S.Y.; Zhang, X. Novel phenotype-genotype correlations of restrictive cardiomyopathy with Myosin-Binding Protein C (MYBPC3) gene mutations tested by next-generation sequencing. J. Am. Heart Assoc. 2015, 4, e001879. [Google Scholar] [CrossRef]
- Barefield, D.Y.; Puckelwartz, M.J.; Kim, E.Y.; Wilsbacher, L.D.; Vo, A.H.; Waters, E.A.; Earley, J.U.; Hadhazy, M.; Dellefave-Castillo, L.; Pesce, L.L.; et al. Experimental modeling supports a role for MyBP-HL as a novel myofilament component in arrhythmia and dilated cardiomyopathy. Circulation 2017, 136, 1477–1491. [Google Scholar] [CrossRef] [PubMed]
- Carniel, E.; Taylor, M.R.; Sinagra, G.; Di Lenarda, A.; Ku, L.; Fain, P.R.; Boucek, M.M.; Cavanaugh, J.; Miocic, S.; Slavov, D.; et al. α-myosin heavy chain: A sarcomeric gene associated with dilated and hypertrophic phenotypes of cardiomyopathy. Circulation 2005, 112, 54–59. [Google Scholar] [CrossRef]
- Greenway, S.C.; Wilson, G.J.; Wilson, J.; George, K.; Kantor, P.F. Sudden death in an infant with angina, restrictive cardiomyopathy, and coronary artery bridging: An unusual phenotype for a beta-myosin heavy chain (MYH7) sarcomeric protein mutation. Circ. Heart Fail. 2012, 5, e92–e93. [Google Scholar] [CrossRef]
- Flavigny, J.; Richard, P.; Isnard, R.; Carrier, L.; Charron, P.; Bonne, G.; Forissier, J.F.; Desnos, M.; Dubourg, O.; Komajda, M.; et al. Identification of two novel mutations in the ventricular regulatory myosin light chain gene (MYL2) associated with familial and classical forms of hypertrophic cardiomyopathy. J. Mol. Med. 1998, 76, 208–214. [Google Scholar] [CrossRef]
- Caleshu, C.; Sakhuja, R.; Nussbaum, R.L.; Schiller, N.B.; Ursell, P.C.; Eng, C.; De Marco, T.; McGlothlin, D.; Burchard, E.G.; Rame, J.E. Furthering the link between the sarcomere and primary cardiomyopathies: Restrictive cardiomyopathy associated with multiple mutations in genes previously associated with hypertrophic or dilated cardiomyopathy. Am. J. Med. Genet. Part A 2011, 155A, 2229–2235. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.O.; Yu, C.W.; Chun Nah, J.; Rang Park, J.; Lee, B.S.; Jeong Choi, Y.; Cho, B.R.; Lee, S.C.; Woo Park, S.; Kimura, A.; et al. Long-term outcome of 4 Korean families with hypertrophic cardiomyopathy caused by 4 different mutations. Clin. Cardiol. 2010, 33, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Tobita, T.; Nomura, S.; Morita, H.; Ko, T.; Fujita, T.; Toko, H.; Uto, K.; Hagiwara, N.; Aburatani, H.; Komuro, I. Identification of MYLK3 mutations in familial dilated cardiomyopathy. Sci. Rep. 2017, 7, 17495. [Google Scholar] [CrossRef] [PubMed]
- Arola, A.M.; Sanchez, X.; Murphy, R.T.; Hasle, E.; Li, H.; Elliott, P.M.; McKenna, W.J.; Towbin, J.A.; Bowles, N.E. Mutations in PDLIM3 and MYOZ1 encoding myocyte Z line proteins are infrequently found in idiopathic dilated cardiomyopathy. Mol. Genet. Metab. 2007, 90, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Osio, A.; Tan, L.; Chen, S.N.; Lombardi, R.; Nagueh, S.F.; Shete, S.; Roberts, R.; Willerson, J.T.; Marian, A.J. Myozenin 2 is a novel gene for human hypertrophic cardiomyopathy. Circ. Res. 2007, 100, 766–768. [Google Scholar] [CrossRef] [PubMed]
- Purevjav, E.; Arimura, T.; Augustin, S.; Huby, A.C.; Takagi, K.; Nunoda, S.; Kearney, D.L.; Taylor, M.D.; Terasaki, F.; Bos, J.M.; et al. Molecular basis for clinical heterogeneity in inherited cardiomyopathies due to myopalladin mutations. Hum. Mol. Genet. 2012, 21, 2039–2053. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.; Ruppert, V.; Ackermann, S.; Richter, A.; Perrot, A.; Sperling, S.R.; Posch, M.G.; Maisch, B.; Pankuweit, S. Novel mutations in the sarcomeric protein myopalladin in patients with dilated cardiomyopathy. Eur. J. Hum. Genet. 2013, 21, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Huby, A.C.; Mendsaikhan, U.; Takagi, K.; Martherus, R.; Wansapura, J.; Gong, N.; Osinska, H.; James, J.F.; Kramer, K.; Saito, K.; et al. Disturbance in Z-disk mechanosensitive proteins induced by a persistent mutant myopalladin causes familial restrictive cardiomyopathy. J. Am. Coll. Cardiol. 2014, 64, 2765–2776. [Google Scholar] [CrossRef]
- Roh, J.I.; Cheong, C.; Sung, Y.H.; Lee, J.; Oh, J.; Lee, B.S.; Lee, J.E.; Gho, Y.S.; Kim, D.K.; Park, C.B.; et al. Perturbation of NCOA6 leads to dilated cardiomyopathy. Cell Rep. 2014, 8, 991–998. [Google Scholar] [CrossRef][Green Version]
- Fassone, E.; Taanman, J.W.; Hargreaves, I.P.; Sebire, N.J.; Cleary, M.A.; Burch, M.; Rahman, S. Mutations in the mitochondrial complex I assembly factor NDUFAF1 cause fatal infantile hypertrophic cardiomyopathy. J. Med. Genet. 2011, 48, 691–697. [Google Scholar] [CrossRef]
- Benit, P.; Beugnot, R.; Chretien, D.; Giurgea, I.; De Lonlay-Debeney, P.; Issartel, J.P.; Corral-Debrinski, M.; Kerscher, S.; Rustin, P.; Rotig, A.; et al. Mutant NDUFV2 subunit of mitochondrial complex I causes early onset hypertrophic cardiomyopathy and encephalopathy. Hum. Mutat. 2003, 21, 582–586. [Google Scholar] [CrossRef] [PubMed]
- Benit, P.; Slama, A.; Cartault, F.; Giurgea, I.; Chretien, D.; Lebon, S.; Marsac, C.; Munnich, A.; Rotig, A.; Rustin, P. Mutant NDUFS3 subunit of mitochondrial complex I causes Leigh syndrome. J. Med. Genet. 2004, 41, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Perrot, A.; Tomasov, P.; Villard, E.; Faludi, R.; Melacini, P.; Lossie, J.; Lohmann, N.; Richard, P.; De Bortoli, M.; Angelini, A.; et al. Mutations in NEBL encoding the cardiac Z-disk protein nebulette are associated with various cardiomyopathies. Arch. Med. Sci. 2016, 12, 263–278. [Google Scholar] [CrossRef] [PubMed]
- Arimura, T.; Nakamura, T.; Hiroi, S.; Satoh, M.; Takahashi, M.; Ohbuchi, N.; Ueda, K.; Nouchi, T.; Yamaguchi, N.; Akai, J.; et al. Characterization of the human nebulette gene: A polymorphism in an actin-binding motif is associated with nonfamilial idiopathic dilated cardiomyopathy. Hum. Genet. 2000, 107, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, Z.; Wang, J.; Sun, K.; Cui, Q.; Song, L.; Zou, Y.; Wang, X.; Liu, X.; Hui, R.; et al. Mutations in NEXN, a Z-disc gene, are associated with hypertrophic cardiomyopathy. Am. J. Hum. Genet. 2010, 87, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Hassel, D.; Dahme, T.; Erdmann, J.; Meder, B.; Huge, A.; Stoll, M.; Just, S.; Hess, A.; Ehlermann, P.; Weichenhan, D.; et al. Nexilin mutations destabilize cardiac Z-disks and lead to dilated cardiomyopathy. Nat. Med. 2009, 15, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Yuen, T.; Borle, K.; Wenzel, K.; Eileen, P.; French, V.; Brodehl, A.; Lauzon, J.; Sharma, P.; Klaassen, S.; Gerull, B. Novel Variants in Nexilin are Associated with Left Ventricular Noncompaction Cardiomyopathy. Circulation 2014, 130, A17287. [Google Scholar]
- Xu, J.H.; Gu, J.Y.; Guo, Y.H.; Zhang, H.; Qiu, X.B.; Li, R.G.; Shi, H.Y.; Liu, H.; Yang, X.X.; Xu, Y.J.; et al. Prevalence and spectrum of NKX2-5 mutations associated with sporadic adult-onset dilated cardiomyopathy. Int. Heart J. 2017, 58, 521–529. [Google Scholar] [CrossRef]
- Marston, S.; Montgiraud, C.; Munster, A.B.; Copeland, O.; Choi, O.; Dos Remedios, C.; Messer, A.E.; Ehler, E.; Knoll, R. OBSCN mutations associated with dilated cardiomyopathy and haploinsufficiency. PLoS ONE 2015, 10, e0138568. [Google Scholar] [CrossRef] [PubMed]
- Biswas, A.; Raza, A.; Das, S.; Kapoor, M.; Jayarajan, R.; Verma, A.; Shamsudheen, K.V.; Murry, B.; Seth, S.; Bhargava, B.; et al. Loss of function mutation in the P2X7, a ligand-gated ion channel gene associated with hypertrophic cardiomyopathy. Purinergic Signall. 2019, 15, 205–210. [Google Scholar] [CrossRef]
- Ramond, F.; Janin, A.; Di Filippo, S.; Chanavat, V.; Chalabreysse, L.; Roux-Buisson, N.; Sanlaville, D.; Touraine, R.; Millat, G. Homozygous PKP2 deletion associated with neonatal left ventricle noncompaction. Clin. Genet. 2017, 91, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Gerull, B.; Kirchner, F.; Chong, J.X.; Tagoe, J.; Chandrasekharan, K.; Strohm, O.; Waggoner, D.; Ober, C.; Duff, H.J. Homozygous founder mutation in desmocollin-2 (DSC2) causes arrhythmogenic cardiomyopathy in the Hutterite population. Circ. Cardiovasc. Genet. 2013, 6, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Haghighi, K.; Kolokathis, F.; Pater, L.; Lynch, R.A.; Asahi, M.; Gramolini, A.O.; Fan, G.C.; Tsiapras, D.; Hahn, H.S.; Adamopoulos, S.; et al. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J. Clin. Investig. 2003, 111, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, J.P.; Kamisago, M.; Asahi, M.; Li, G.H.; Ahmad, F.; Mende, U.; Kranias, E.G.; MacLennan, D.H.; Seidman, J.G.; Seidman, C.E. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science 2003, 299, 1410–1413. [Google Scholar] [CrossRef] [PubMed]
- Iuso, A.; Wiersma, M.; Schuller, H.J.; Pode-Shakked, B.; Marek-Yagel, D.; Grigat, M.; Schwarzmayr, T.; Berutti, R.; Alhaddad, B.; Kanon, B.; et al. Mutations in PPCS, encoding phosphopantothenoylcysteine synthetase, cause autosomal-recessive dilated cardiomyopathy. Am. J. Hum. Genet. 2018, 102, 1018–1030. [Google Scholar] [CrossRef]
- Long, P.A.; Evans, J.M.; Olson, T.M. Diagnostic yield of whole exome sequencing in pediatric dilated cardiomyopathy. J. Cardiovasc. Dev. Dis. 2017, 4, 11. [Google Scholar] [CrossRef]
- Arndt, A.K.; Schafer, S.; Drenckhahn, J.D.; Sabeh, M.K.; Plovie, E.R.; Caliebe, A.; Klopocki, E.; Musso, G.; Werdich, A.A.; Kalwa, H.; et al. Fine mapping of the 1p36 deletion syndrome identifies mutation of PRDM16 as a cause of cardiomyopathy. Am. J. Hum. Genet. 2013, 93, 67–77. [Google Scholar] [CrossRef]
- Blair, E.; Redwood, C.; Ashrafian, H.; Oliveira, M.; Broxholme, J.; Kerr, B.; Salmon, A.; Ostman-Smith, I.; Watkins, H. Mutations in the γ(2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: Evidence for the central role of energy compromise in disease pathogenesis. Hum. Mol. Genet. 2001, 10, 1215–1220. [Google Scholar] [CrossRef]
- Vaughan, C.J.; Hom, Y.; Okin, D.A.; McDermott, D.A.; Lerman, B.B.; Basson, C.T. Molecular genetic analysis of PRKAG2 in sporadic Wolff-Parkinson-White syndrome. J. Cardiovasc. Electrophysiol. 2003, 14, 263–268. [Google Scholar] [CrossRef]
- Li, D.; Parks, S.B.; Kushner, J.D.; Nauman, D.; Burgess, D.; Ludwigsen, S.; Partain, J.; Nixon, R.R.; Allen, C.N.; Irwin, R.P.; et al. Mutations of presenilin genes in dilated cardiomyopathy and heart failure. Am. J. Hum. Genet. 2006, 79, 1030–1039. [Google Scholar] [CrossRef]
- Gianni, D.; Li, A.; Tesco, G.; McKay, K.M.; Moore, J.; Raygor, K.; Rota, M.; Gwathmey, J.K.; Dec, G.W.; Aretz, T.; et al. Protein aggregates and novel presenilin gene variants in idiopathic dilated cardiomyopathy. Circulation 2010, 121, 1216–1226. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, M.; Mehler, E.L.; Goldberg, R.; Zampino, G.; Brunner, H.G.; Kremer, H.; van der Burgt, I.; Crosby, A.H.; Ion, A.; Jeffery, S.; et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet. 2001, 29, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Razzaque, M.A.; Nishizawa, T.; Komoike, Y.; Yagi, H.; Furutani, M.; Amo, R.; Kamisago, M.; Momma, K.; Katayama, H.; Nakagawa, M.; et al. Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat. Genet. 2007, 39, 1013–1017. [Google Scholar] [CrossRef] [PubMed]
- Pandit, B.; Sarkozy, A.; Pennacchio, L.A.; Carta, C.; Oishi, K.; Martinelli, S.; Pogna, E.A.; Schackwitz, W.; Ustaszewska, A.; Landstrom, A.; et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat. Genet. 2007, 39, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Dhandapany, P.S.; Razzaque, M.A.; Muthusami, U.; Kunnoth, S.; Edwards, J.J.; Mulero-Navarro, S.; Riess, I.; Pardo, S.; Sheng, J.; Rani, D.S.; et al. RAF1 mutations in childhood-onset dilated cardiomyopathy. Nat. Genet. 2014, 46, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Schafer, S.; Greaser, M.L.; Radke, M.H.; Liss, M.; Govindarajan, T.; Maatz, H.; Schulz, H.; Li, S.; Parrish, A.M.; et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 2012, 18, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Sedaghat-Hamedani, F.; Haas, J.; Zhu, F.; Geier, C.; Kayvanpour, E.; Liss, M.; Lai, A.; Frese, K.; Pribe-Wolferts, R.; Amr, A.; et al. Clinical genetics and outcome of left ventricular non-compaction cardiomyopathy. Eur. Heart J. 2017, 38, 3449–3460. [Google Scholar] [CrossRef]
- Van den Hoogenhof, M.M.G.; Beqqali, A.; Amin, A.S.; van der Made, I.; Aufiero, S.; Khan, M.A.F.; Schumacher, C.A.; Jansweijer, J.A.; van Spaendonck-Zwarts, K.Y.; Remme, C.A.; et al. RBM20 mutations induce an arrhythmogenic dilated cardiomyopathy related to disturbed calcium handling. Circulation 2018, 138, 1330–1342. [Google Scholar] [CrossRef]
- Parikh, V.N.; Caleshu, C.; Reuter, C.; Lazzeroni, L.C.; Ingles, J.; Garcia, J.; McCaleb, K.; Adesiyun, T.; Sedaghat-Hamedani, F.; Kumar, S.; et al. Regional variation in RBM20 causes a highly penetrant arrhythmogenic cardiomyopathy. Circ. Heart Fail. 2019, 12, e005371. [Google Scholar] [CrossRef] [PubMed]
- Long, P.A.; Zimmermann, M.T.; Kim, M.; Evans, J.M.; Xu, X.; Olson, T.M. De novo RRAGC mutation activates mTORC1 signaling in syndromic fetal dilated cardiomyopathy. Hum. Genet. 2016, 135, 909–917. [Google Scholar] [CrossRef]
- Mann, S.A.; Castro, M.L.; Ohanian, M.; Guo, G.; Zodgekar, P.; Sheu, A.; Stockhammer, K.; Thompson, T.; Playford, D.; Subbiah, R.; et al. R222Q SCN5A mutation is associated with reversible ventricular ectopy and dilated cardiomyopathy. J. Am. Coll. Cardiol. 2012, 60, 1566–1573. [Google Scholar] [CrossRef] [PubMed]
- Erkapic, D.; Neumann, T.; Schmitt, J.; Sperzel, J.; Berkowitsch, A.; Kuniss, M.; Hamm, C.W.; Pitschner, H.F. Electrical storm in a patient with arrhythmogenic right ventricular cardiomyopathy and SCN5A mutation. Europace 2008, 10, 884–887. [Google Scholar] [CrossRef] [PubMed]
- Jaksch, M.; Ogilvie, I.; Yao, J.; Kortenhaus, G.; Bresser, H.G.; Gerbitz, K.D.; Shoubridge, E.A. Mutations in SCO2 are associated with a distinct form of hypertrophic cardiomyopathy and cytochrome c oxidase deficiency. Hum. Mol. Genet. 2000, 9, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Levitas, A.; Muhammad, E.; Harel, G.; Saada, A.; Caspi, V.C.; Manor, E.; Beck, J.C.; Sheffield, V.; Parvari, R. Familial neonatal isolated cardiomyopathy caused by a mutation in the flavoprotein subunit of succinate dehydrogenase. Eur. J. Hum. Genet. 2010, 18, 1160–1165. [Google Scholar] [CrossRef] [PubMed]
- Van Spaendonck-Zwarts, K.Y.; van Rijsingen, I.A.; van den Berg, M.P.; Lekanne Deprez, R.H.; Post, J.G.; van Mil, A.M.; Asselbergs, F.W.; Christiaans, I.; van Langen, I.M.; Wilde, A.A.; et al. Genetic analysis in 418 index patients with idiopathic dilated cardiomyopathy: Overview of 10 years’ experience. Eur. J. Heart Fail. 2013, 15, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Tsubata, S.; Bowles, K.R.; Vatta, M.; Zintz, C.; Titus, J.; Muhonen, L.; Bowles, N.E.; Towbin, J.A. Mutations in the human δ-sarcoglycan gene in familial and sporadic dilated cardiomyopathy. J. Clin. Investig. 2000, 106, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Hoban, R.; Roberts, A.E.; Demmer, L.; Jethva, R.; Shephard, B. Noonan syndrome due to a SHOC2 mutation presenting with fetal distress and fatal hypertrophic cardiomyopathy in a premature infant. Am. J. Med. Genet. Part A 2012, 158A, 1411–1413. [Google Scholar] [CrossRef]
- Sandra, M.; Maria Pia, L.; Stefano, C.; Pietro, P.; Crociani, P.; Aldo, R.; Giuseppe, D.S.; Massimo, C. Emery-dreifuss muscular dystrophy type 4: A new SYNE1 mutation associated with hypertrophic cardiomyopathy masked by a perinatal distress-related spastic diplegia. Clin. Case Rep. 2019, 7, 1078–1082. [Google Scholar] [CrossRef]
- Puckelwartz, M.J.; Kessler, E.J.; Kim, G.; Dewitt, M.M.; Zhang, Y.; Earley, J.U.; Depreux, F.F.; Holaska, J.; Mewborn, S.K.; Pytel, P.; et al. Nesprin-1 mutations in human and murine cardiomyopathy. J. Mol. Cell. Cardiol. 2010, 48, 600–608. [Google Scholar] [CrossRef]
- Bione, S.; D’Adamo, P.; Maestrini, E.; Gedeon, A.K.; Bolhuis, P.A.; Toniolo, D. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat. Genet. 1996, 12, 385–389. [Google Scholar] [CrossRef]
- Xing, Y.; Ichida, F.; Matsuoka, T.; Isobe, T.; Ikemoto, Y.; Higaki, T.; Tsuji, T.; Haneda, N.; Kuwabara, A.; Chen, R.; et al. Genetic analysis in patients with left ventricular noncompaction and evidence for genetic heterogeneity. Mol. Genet. Metab. 2006, 88, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Hata, Y.; Hirono, K.; Takasaki, A.; Ozawa, S.W.; Nakaoka, H.; Saito, K.; Miyao, N.; Okabe, M.; Ibuki, K.; et al. A wide and specific spectrum of genetic variants and genotype-phenotype correlations revealed by next-generation sequencing in patients with left ventricular noncompaction. J. Am. Heart Assoc. 2017, 6, e006210. [Google Scholar] [CrossRef] [PubMed]
- Kirk, E.P.; Sunde, M.; Costa, M.W.; Rankin, S.A.; Wolstein, O.; Castro, M.L.; Butler, T.L.; Hyun, C.; Guo, G.; Otway, R.; et al. Mutations in cardiac T-box factor gene TBX20 are associated with diverse cardiac pathologies, including defects of septation and valvulogenesis and cardiomyopathy. Am. J. Hum. Genet. 2007, 81, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.M.; Bing, S.; Song, H.M.; Wang, J.; Xu, W.J.; Jiang, J.F.; Qiu, X.B.; Yuan, F.; Xu, J.H.; Yang, Y.Q. TBX20 loss-of-function mutation associated with familial dilated cardiomyopathy. Clin. Chem. Lab. Med. 2016, 54, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Arimura, T.; Itoh-Satoh, M.; Ueda, K.; Hohda, S.; Inagaki, N.; Takahashi, M.; Hori, H.; Yasunami, M.; Nishi, H.; et al. Tcap gene mutations in hypertrophic cardiomyopathy and dilated cardiomyopathy. J. Am. Coll. Cardiol. 2004, 44, 2192–2201. [Google Scholar] [CrossRef] [PubMed]
- Beffagna, G.; Occhi, G.; Nava, A.; Vitiello, L.; Ditadi, A.; Basso, C.; Bauce, B.; Carraro, G.; Thiene, G.; Towbin, J.A.; et al. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc. Res. 2005, 65, 366–373. [Google Scholar] [CrossRef] [PubMed]
- De Bortoli, M.; Postma, A.V.; Poloni, G.; Calore, M.; Minervini, G.; Mazzotti, E.; Rigato, I.; Ebert, M.; Lorenzon, A.; Vazza, G.; et al. Whole-exome sequencing identifies pathogenic variants in TJP1 gene associated with arrhythmogenic cardiomyopathy. Circ. Genom. Precis. Med. 2018, 11, e002123. [Google Scholar] [CrossRef]
- Yu, H.C.; Coughlin, C.R.; Geiger, E.A.; Salvador, B.J.; Elias, E.R.; Cavanaugh, J.L.; Chatfield, K.C.; Miyamoto, S.D.; Shaikh, T.H. Discovery of a potentially deleterious variant in TMEM87B in a patient with a hemizygous 2q13 microdeletion suggests a recessive condition characterized by congenital heart disease and restrictive cardiomyopathy. Cold Spring Harb. Mol. Case Stud. 2016, 2, a000844. [Google Scholar] [CrossRef]
- Mogensen, J.; Murphy, R.T.; Shaw, T.; Bahl, A.; Redwood, C.; Watkins, H.; Burke, M.; Elliott, P.M.; McKenna, W.J. Severe disease expression of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 2004, 44, 2033–2040. [Google Scholar] [CrossRef]
- Ploski, R.; Rydzanicz, M.; Ksiazczyk, T.M.; Franaszczyk, M.; Pollak, A.; Kosinska, J.; Michalak, E.; Stawinski, P.; Ziolkowska, L.; Bilinska, Z.T.; et al. Evidence for troponin C (TNNC1) as a gene for autosomal recessive restrictive cardiomyopathy with fatal outcome in infancy. Am. J. Med. Genet. Part A 2016, 170, 3241–3248. [Google Scholar] [CrossRef]
- Murphy, R.T.; Mogensen, J.; Shaw, A.; Kubo, T.; Hughes, S.; McKenna, W.J. Novel mutation in cardiac troponin I in recessive idiopathic dilated cardiomyopathy. Lancet 2004, 363, 371–372. [Google Scholar] [CrossRef]
- Fujino, M.; Tsuda, E.; Hirono, K.; Nakata, M.; Ichida, F.; Hata, Y.; Nishida, N.; Kurosaki, K. The TNNI3 Arg192His mutation in a 13-year-old girl with left ventricular noncompaction. J. Cardiol. Cases 2018, 18, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.L.; Huang, H.; Jin, J.Y.; Li, J.J.; Chen, Y.Q.; Zhao, S.P.; Xiang, R. Whole exome sequencing identifies a novel mutation (c.333 + 2T > C) of TNNI3K in a Chinese family with dilated cardiomyopathy and cardiac conduction disease. Gene 2018, 648, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Hanson, E.L.; Jakobs, P.M.; Keegan, H.; Coates, K.; Bousman, S.; Dienel, N.H.; Litt, M.; Hershberger, R.E. Cardiac troponin T lysine 210 deletion in a family with dilated cardiomyopathy. J. Card. Fail. 2002, 8, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Luedde, M.; Ehlermann, P.; Weichenhan, D.; Will, R.; Zeller, R.; Rupp, S.; Muller, A.; Steen, H.; Ivandic, B.T.; Ulmer, H.E.; et al. Severe familial left ventricular non-compaction cardiomyopathy due to a novel troponin T (TNNT2) mutation. Cardiovasc. Res. 2010, 86, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Poloni, G.; Calore, M.; Rigato, I.; Marras, E.; Minervini, G.; Mazzotti, E.; Lorenzon, A.; Li Mura, I.E.A.; Telatin, A.; Zara, I.; et al. A targeted next-generation gene panel reveals a novel heterozygous nonsense variant in the TP63 gene in patients with arrhythmogenic cardiomyopathy. Heart Rhythm 2019, 16, 773–780. [Google Scholar] [CrossRef]
- Karibe, A.; Tobacman, L.S.; Strand, J.; Butters, C.; Back, N.; Bachinski, L.L.; Arai, A.E.; Ortiz, A.; Roberts, R.; Homsher, E.; et al. Hypertrophic cardiomyopathy caused by a novel α-tropomyosin mutation (V95A) is associated with mild cardiac phenotype, abnormal calcium binding to troponin, abnormal myosin cycling, and poor prognosis. Circulation 2001, 103, 65–71. [Google Scholar] [CrossRef]
- Lakdawala, N.K.; Dellefave, L.; Redwood, C.S.; Sparks, E.; Cirino, A.L.; Depalma, S.; Colan, S.D.; Funke, B.; Zimmerman, R.S.; Robinson, P.; et al. Familial dilated cardiomyopathy caused by an α-tropomyosin mutation: The distinctive natural history of sarcomeric dilated cardiomyopathy. J. Am. Coll. Cardiol. 2010, 55, 320–329. [Google Scholar] [CrossRef]
- Chang, B.; Nishizawa, T.; Furutani, M.; Fujiki, A.; Tani, M.; Kawaguchi, M.; Ibuki, K.; Hirono, K.; Taneichi, H.; Uese, K.; et al. Identification of a novel TPM1 mutation in a family with left ventricular noncompaction and sudden death. Mol. Genet. Metab. 2011, 102, 200–206. [Google Scholar] [CrossRef]
- Chen, S.N.; Czernuszewicz, G.; Tan, Y.; Lombardi, R.; Jin, J.; Willerson, J.T.; Marian, A.J. Human molecular genetic and functional studies identify TRIM63, encoding muscle RING finger protein 1, as a novel gene for human hypertrophic cardiomyopathy. Circ. Res. 2012, 111, 907–919. [Google Scholar] [CrossRef]
- Saito, Y.; Nakamura, K.; Nishi, N.; Igawa, O.; Yoshida, M.; Miyoshi, T.; Watanabe, A.; Morita, H.; Ito, H. TRPM4 mutation in patients with ventricular noncompaction and cardiac conduction disease. Circ. Genom. Precis. Med. 2018, 11, e002103. [Google Scholar] [CrossRef] [PubMed]
- Perli, E.; Pisano, A.; Glasgow, R.I.C.; Carbo, M.; Hardy, S.A.; Falkous, G.; He, L.; Cerbelli, B.; Pignataro, M.G.; Zacara, E.; et al. Novel compound mutations in the mitochondrial translation elongation factor (TSFM) gene cause severe cardiomyopathy with myocardial fibro-adipose replacement. Sci. Rep. 2019, 9, 5108. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Takahashi, M.; Sakamoto, T.; Hiroe, M.; Marumo, F.; Kimura, A. Structural analysis of the titin gene in hypertrophic cardiomyopathy: Identification of a novel disease gene. Biochem. Biophys. Res. Commun. 1999, 262, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Itoh-Satoh, M.; Hayashi, T.; Nishi, H.; Koga, Y.; Arimura, T.; Koyanagi, T.; Takahashi, M.; Hohda, S.; Ueda, K.; Nouchi, T.; et al. Titin mutations as the molecular basis for dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 2002, 291, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, C.; Liu, N.; Bai, H.; Hou, C.; Song, L.; Pu, J. Titin-truncating variants are associated with heart failure events in patients with left ventricular noncompaction cardiomyopathy. Clin. Cardiol. 2019, 42, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, M.J.; Almeida Mdo, R.; Sherman, W.; Gawinowicz, M.; Costa, P.; Costa, P.P.; Goodman, D.S. A new transthyretin mutation associated with amyloid cardiomyopathy. Am. J. Hum. Genet. 1992, 50, 1027–1030. [Google Scholar]
- Holmgren, G.; Hellman, U.; Anan, I.; Lundgren, H.E.; Jonasson, J.; Stafberg, C.; Fahoum, S.; Suhr, O.B. Cardiomyopathy in Swedish patients with the Gly53Glu and His88Arg transthyretin variants. Amyloid 2005, 12, 184–188. [Google Scholar] [CrossRef]
- Sibbing, D.; Pfeufer, A.; Perisic, T.; Mannes, A.M.; Fritz-Wolf, K.; Unwin, S.; Sinner, M.F.; Gieger, C.; Gloeckner, C.J.; Wichmann, H.E.; et al. Mutations in the mitochondrial thioredoxin reductase gene TXNRD2 cause dilated cardiomyopathy. Eur. Heart J. 2011, 32, 1121–1133. [Google Scholar] [CrossRef]
- Vasile, V.C.; Will, M.L.; Ommen, S.R.; Edwards, W.D.; Olson, T.M.; Ackerman, M.J. Identification of a metavinculin missense mutation, R975W, associated with both hypertrophic and dilated cardiomyopathy. Mol. Genet. Metab. 2006, 87, 169–174. [Google Scholar] [CrossRef]
- Vasile, V.C.; Ommen, S.R.; Edwards, W.D.; Ackerman, M.J. A missense mutation in a ubiquitously expressed protein, vinculin, confers susceptibility to hypertrophic cardiomyopathy. Biochem. Biophys. Res. Commun. 2006, 345, 998–1003. [Google Scholar] [CrossRef]
- Olson, T.M.; Illenberger, S.; Kishimoto, N.Y.; Huttelmaier, S.; Keating, M.T.; Jockusch, B.M. Metavinculin mutations alter actin interaction in dilated cardiomyopathy. Circulation 2002, 105, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Buyandelger, B.; Mansfield, C.; Kostin, S.; Choi, O.; Roberts, A.M.; Ware, J.S.; Mazzarotto, F.; Pesce, F.; Buchan, R.; Isaacson, R.L.; et al. ZBTB17 (MIZ1) is important for the cardiac stress response and a novel candidate gene for cardiomyopathy and heart failure. Circ. Cardiovasc. Genet. 2015, 8, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.M.; Wang, J.; Xu, Y.J.; Wang, X.H.; Yuan, F.; Liu, H.; Li, R.G.; Zhang, M.; Li, Y.J.; Shi, H.Y.; et al. ZBTB17 loss-of-function mutation contributes to familial dilated cardiomyopathy. Heart Vessels 2018, 33, 722–732. [Google Scholar] [CrossRef] [PubMed]
- Gurdon, J.B.; Elsdale, T.R.; Fischberg, M. Sexually mature individuals of Xenopus laevis from the transplantation of single somatic nuclei. Nature 1958, 182, 64–65. [Google Scholar] [CrossRef] [PubMed]
- Elsdale, T.R.; Gurdon, J.B.; Fischberg, M. A description of the technique for nuclear transplantation in Xenopus laevis. J. Embryol. Exp. Morphol. 1960, 8, 437–444. [Google Scholar] [PubMed]
- Holmes, D. Stem cell scientists share 2012 Nobel Prize for medicine. Lancet 2012, 380, 1295. [Google Scholar] [CrossRef]
- Wilmut, I.; Schnieke, A.E.; McWhir, J.; Kind, A.J.; Campbell, K.H. Viable offspring derived from fetal and adult mammalian cells. Nature 1997, 385, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Okita, K.; Ichisaka, T.; Yamanaka, S. Generation of germline-competent induced pluripotent stem cells. Nature 2007, 448, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 2008, 26, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Blelloch, R.; Venere, M.; Yen, J.; Ramalho-Santos, M. Generation of induced pluripotent stem cells in the absence of drug selection. Cell Stem Cell 2007, 1, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Brambrink, T.; Foreman, R.; Welstead, G.G.; Lengner, C.J.; Wernig, M.; Suh, H.; Jaenisch, R. Sequential expression of pluripotency markers during direct reprogramming of mouse somatic cells. Cell Stem Cell 2008, 2, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Soldner, F.; Hockemeyer, D.; Beard, C.; Gao, Q.; Bell, G.W.; Cook, E.G.; Hargus, G.; Blak, A.; Cooper, O.; Mitalipova, M.; et al. Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 2009, 136, 964–977. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; von Kalle, C.; Schmidt, M.; Le Deist, F.; Wulffraat, N.; McIntyre, E.; Radford, I.; Villeval, J.L.; Fraser, C.C.; Cavazzana-Calvo, M.; et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2003, 348, 255–256. [Google Scholar] [CrossRef] [PubMed]
- Kaji, K.; Norrby, K.; Paca, A.; Mileikovsky, M.; Mohseni, P.; Woltjen, K. Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature 2009, 458, 771–775. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Freed, C.R. Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells 2009, 27, 2667–2674. [Google Scholar] [CrossRef] [PubMed]
- Meraviglia, V.; Zanon, A.; Lavdas, A.A.; Schwienbacher, C.; Silipigni, R.; Di Segni, M.; Chen, H.S.; Pramstaller, P.P.; Hicks, A.A.; Rossini, A. Generation of induced pluripotent stem cells from frozen buffy coats using non-integrating episomal plasmids. J. Vis. Exp. 2015, 100, e52885. [Google Scholar] [CrossRef]
- Okita, K.; Nakagawa, M.; Hyenjong, H.; Ichisaka, T.; Yamanaka, S. Generation of mouse induced pluripotent stem cells without viral vectors. Science 2008, 322, 949–953. [Google Scholar] [CrossRef]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Acad. Ser. B 2009, 85, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Judson, R.L.; Babiarz, J.E.; Venere, M.; Blelloch, R. Embryonic stem cell-specific microRNAs promote induced pluripotency. Nat. Biotechnol. 2009, 27, 459–461. [Google Scholar] [CrossRef] [PubMed]
- Card, D.A.; Hebbar, P.B.; Li, L.; Trotter, K.W.; Komatsu, Y.; Mishina, Y.; Archer, T.K. Oct4/Sox2-regulated miR-302 targets cyclin D1 in human embryonic stem cells. Mol. Cell. Biol. 2008, 28, 6426–6438. [Google Scholar] [CrossRef] [PubMed]
- Anokye-Danso, F.; Trivedi, C.M.; Juhr, D.; Gupta, M.; Cui, Z.; Tian, Y.; Zhang, Y.; Yang, W.; Gruber, P.J.; Epstein, J.A.; et al. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell 2011, 8, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Schlaeger, T.M.; Daheron, L.; Brickler, T.R.; Entwisle, S.; Chan, K.; Cianci, A.; DeVine, A.; Ettenger, A.; Fitzgerald, K.; Godfrey, M.; et al. A comparison of non-integrating reprogramming methods. Nat. Biotechnol. 2015, 33, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Freberg, C.T.; Dahl, J.A.; Timoskainen, S.; Collas, P. Epigenetic reprogramming of OCT4 and NANOG regulatory regions by embryonal carcinoma cell extract. Mol. Biol. Cell 2007, 18, 1543–1553. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, C.H.; Moon, J.I.; Chung, Y.G.; Chang, M.Y.; Han, B.S.; Ko, S.; Yang, E.; Cha, K.Y.; Lanza, R.; et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell 2009, 4, 472–476. [Google Scholar] [CrossRef]
- Huangfu, D.; Maehr, R.; Guo, W.; Eijkelenboom, A.; Snitow, M.; Chen, A.E.; Melton, D.A. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat. Biotechnol. 2008, 26, 795–797. [Google Scholar] [CrossRef]
- Li, W.; Ding, S. Small molecules that modulate embryonic stem cell fate and somatic cell reprogramming. Trends Pharmacol. Sci. 2010, 31, 36–45. [Google Scholar] [CrossRef]
- Bru, T.; Clarke, C.; McGrew, M.J.; Sang, H.M.; Wilmut, I.; Blow, J.J. Rapid induction of pluripotency genes after exposure of human somatic cells to mouse ES cell extracts. Exp. Cell Res. 2008, 314, 2634–2642. [Google Scholar] [CrossRef]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Weltner, J.; Balboa, D.; Katayama, S.; Bespalov, M.; Krjutskov, K.; Jouhilahti, E.M.; Trokovic, R.; Kere, J.; Otonkoski, T. Human pluripotent reprogramming with CRISPR activators. Nat. Commun. 2018, 9, 2643. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Joung, J.K.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2013, 14, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Nemudryi, A.A.; Valetdinova, K.R.; Medvedev, S.P.; Zakian, S.M. TALEN and CRISPR/Cas genome editing systems: Tools of discovery. Acta Nat. 2014, 6, 19–40. [Google Scholar] [CrossRef]
- Debus, J.D.; Milting, H.; Brodehl, A.; Kassner, A.; Anselmetti, D.; Gummert, J.; Gaertner-Rommel, A. In vitro analysis of arrhythmogenic cardiomyopathy associated desmoglein-2 (DSG2) mutations reveals diverse glycosylation patterns. J. Mol. Cell. Cardiol. 2019, 129, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Toepfer, C.N.; Schmid, M.; Garfinkel, A.C.; Seidman, C.E. Differentiation and contractile analysis of GFP-sarcomere reporter hiPSC-cardiomyocytes. Curr. Protoc. Hum. Genet. 2018, 96, 21.12.1–21.12.12. [Google Scholar] [CrossRef] [PubMed]
- Matsa, E.; Dixon, J.E.; Medway, C.; Georgiou, O.; Patel, M.J.; Morgan, K.; Kemp, P.J.; Staniforth, A.; Mellor, I.; Denning, C. Allele-specific RNA interference rescues the long–QT syndrome phenotype in human-induced pluripotency stem cell cardiomyocytes. Eur. Heart J. 2014, 35, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Dzilic, E.; Lahm, H.; Dressen, M.; Deutsch, M.A.; Lange, R.; Wu, S.M.; Krane, M.; Doppler, S.A. Genome editing redefines precision medicine in the cardiovascular field. Stem Cells Int. 2018, 2018, 4136473. [Google Scholar] [CrossRef]
- Gallagher, D.N.; Haber, J.E. Repair of a site-specific DNA cleavage: Old-school lessons for Cas9-mediated gene editing. ACS Chem. Biol. 2018, 13, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Zhang, X.H.; Clift, C.; Yamaguchi, N.; Morad, M. CRISPR/Cas9 Gene editing of RyR2 in human stem cell-derived cardiomyocytes provides a novel approach in investigating dysfunctional Ca(2+) signaling. Cell Calcium 2018, 73, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.M.; Anderson, K.M.; Chang, C.L.; Makarewich, C.A.; Nelson, B.R.; McAnally, J.R.; Kasaragod, P.; Shelton, J.M.; Liou, J.; Bassel-Duby, R.; et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell 2015, 160, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.; Hendershott, M.C.; Arakaki, J.; Gerbin, K.A.; Malik, H.; Nelson, A.; Gehring, J.; Hookway, C.; Ludmann, S.A.; Yang, R.; et al. Fluorescent gene tagging of transcriptionally silent genes in hiPSCs. Stem Cell Rep. 2019, 12, 1145–1158. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Li, C.; Chen, X.; Dou, H.; Li, Y.; Zhang, X.; Luo, Y. CRISPR/Cas9-mediated gene tagging: A step-by-step protocol. Methods Mol. Biol. 2019, 1961, 255–269. [Google Scholar] [PubMed]
- Haupt, A.; Grancharova, T.; Arakaki, J.; Fuqua, M.A.; Roberts, B.; Gunawardane, R.N. Endogenous protein tagging in human induced pluripotent stem cells using CRISPR/Cas9. J. Vis. Exp. 2018, 138, e58130. [Google Scholar] [CrossRef]
- Liu, M.; Rehman, S.; Tang, X.; Gu, K.; Fan, Q.; Chen, D.; Ma, W. Methodologies for Improving HDR efficiency. Front. Genet. 2018, 9, 691. [Google Scholar] [CrossRef] [PubMed]
- Smirnikhina, S.A.; Anuchina, A.A.; Lavrov, A.V. Ways of improving precise knock-in by genome-editing technologies. Hum. Genet. 2019, 138, 1–19. [Google Scholar] [CrossRef]
- Pawelczak, K.S.; Gavande, N.S.; VanderVere-Carozza, P.S.; Turchi, J.J. Modulating DNA repair pathways to improve precision genome engineering. ACS Chem. Biol. 2018, 13, 389–396. [Google Scholar] [CrossRef]
- Gu, B.; Posfai, E.; Rossant, J. Efficient generation of targeted large insertions by microinjection into two-cell-stage mouse embryos. Nat. Biotechnol. 2018, 36, 632–637. [Google Scholar] [CrossRef]
- Savic, N.; Ringnalda, F.C.; Lindsay, H.; Berk, C.; Bargsten, K.; Li, Y.; Neri, D.; Robinson, M.D.; Ciaudo, C.; Hall, J.; et al. Covalent linkage of the DNA repair template to the CRISPR-Cas9 nuclease enhances homology-directed repair. eLife 2018, 7, e33761. [Google Scholar] [CrossRef] [PubMed]
- Savic, N.; Ringnalda, F.C.; Berk, C.; Bargsten, K.; Hall, J.; Jinek, M.; Schwank, G. In vitro generation of CRISPR-Cas9 complexes with covalently bound repair templates for genome editing in mammalian cells. Bio Protoc. 2019, 9, e3136. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Zafra, M.P.; Schatoff, E.M.; Katti, A.; Foronda, M.; Breinig, M.; Schweitzer, A.Y.; Simon, A.; Han, T.; Goswami, S.; Montgomery, E.; et al. Optimized base editors enable efficient editing in cells, organoids and mice. Nat. Biotechnol. 2018, 36, 888–893. [Google Scholar] [CrossRef] [PubMed]
- Villiger, L.; Grisch-Chan, H.M.; Lindsay, H.; Ringnalda, F.; Pogliano, C.B.; Allegri, G.; Fingerhut, R.; Haberle, J.; Matos, J.; Robinson, M.D.; et al. Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat. Med. 2018, 24, 1519–1525. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.B.; de Carvalho, A.C. Heart regeneration: Past, present and future. World J. Cardiol. 2010, 2, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Claycomb, W.C.; Lanson, N.A., Jr.; Stallworth, B.S.; Egeland, D.B.; Delcarpio, J.B.; Bahinski, A.; Izzo, N.J., Jr. HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. USA 1998, 95, 2979–2984. [Google Scholar] [CrossRef] [PubMed]
- Mazzotta, S.; Lynch, A.T.; Hoppler, S. Cardiomyocyte differentiation from human embryonic stem cells. Methods Mol. Biol. 2018, 1816, 67–78. [Google Scholar] [PubMed]
- Hatani, T.; Miki, K.; Yoshida, Y. Induction of human induced pluripotent stem cells to cardiomyocytes using embryoid bodies. Methods Mol. Biol. 2018, 1816, 79–92. [Google Scholar]
- Zhang, J.; Wilson, G.F.; Soerens, A.G.; Koonce, C.H.; Yu, J.; Palecek, S.P.; Thomson, J.A.; Kamp, T.J. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ. Res. 2009, 104, e30–e41. [Google Scholar] [CrossRef]
- Pesl, M.; Acimovic, I.; Pribyl, J.; Hezova, R.; Vilotic, A.; Fauconnier, J.; Vrbsky, J.; Kruzliak, P.; Skladal, P.; Kara, T.; et al. Forced aggregation and defined factors allow highly uniform–sized embryoid bodies and functional cardiomyocytes from human embryonic and induced pluripotent stem cells. Heart Vessels 2014, 29, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Yokoo, N.; Baba, S.; Kaichi, S.; Niwa, A.; Mima, T.; Doi, H.; Yamanaka, S.; Nakahata, T.; Heike, T. The effects of cardioactive drugs on cardiomyocytes derived from human induced pluripotent stem cells. Biochem. Biophys. Res. Commun. 2009, 387, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Zwi, L.; Caspi, O.; Arbel, G.; Huber, I.; Gepstein, A.; Park, I.H.; Gepstein, L. Cardiomyocyte differentiation of human induced pluripotent stem cells. Circulation 2009, 120, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Burridge, P.W.; Thompson, S.; Millrod, M.A.; Weinberg, S.; Yuan, X.; Peters, A.; Mahairaki, V.; Koliatsos, V.E.; Tung, L.; Zambidis, E.T. A universal system for highly efficient cardiac differentiation of human induced pluripotent stem cells that eliminates interline variability. PLoS ONE 2011, 6, e18293. [Google Scholar] [CrossRef] [PubMed]
- Mummery, C.L.; Zhang, J.; Ng, E.S.; Elliott, D.A.; Elefanty, A.G.; Kamp, T.J. Differentiation of human embryonic stem cells and induced pluripotent stem cells to cardiomyocytes: A methods overview. Circ. Res. 2012, 111, 344–358. [Google Scholar] [CrossRef] [PubMed]
- Besser, R.R.; Ishahak, M.; Mayo, V.; Carbonero, D.; Claure, I.; Agarwal, A. Engineered microenvironments for maturation of stem cell derived cardiac myocytes. Theranostics 2018, 8, 124–140. [Google Scholar] [CrossRef] [PubMed]
- Tzahor, E. Wnt/beta-catenin signaling and cardiogenesis: Timing does matter. Dev. Cell 2007, 13, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, Y.; Yamagishi, T.; Hokari, S.; Nakamura, H. Mechanisms involved in valvuloseptal endocardial cushion formation in early cardiogenesis: Roles of transforming growth factor (TGF)-β and bone morphogenetic protein (BMP). Anat. Rec. 2000, 258, 119–127. [Google Scholar] [CrossRef]
- Mably, J.D.; Liew, C.C. Factors involved in cardiogenesis and the regulation of cardiac-specific gene expression. Circ. Res. 1996, 79, 4–13. [Google Scholar] [CrossRef]
- Govindsamy, A.; Naidoo, S.; Cerf, M.E. Cardiac development and transcription factors: Insulin signalling, insulin resistance, and intrauterine nutritional programming of cardiovascular disease. J. Nutr. Metab. 2018, 2018, 8547976. [Google Scholar] [CrossRef]
- Yan, S.; Jiao, K. Functions of miRNAs during mammalian heart development. Int. J. Mol. Sci. 2016, 17, 789. [Google Scholar] [CrossRef]
- Rajala, K.; Pekkanen-Mattila, M.; Aalto-Setala, K. Cardiac differentiation of pluripotent stem cells. Stem Cells Int. 2011, 2011, 383709. [Google Scholar] [CrossRef] [PubMed]
- Yassa, M.E.; Mansour, I.A.; Sewelam, N.I.; Hamza, H.; Gaafar, T. The impact of growth factors on human induced pluripotent stem cells differentiation into cardiomyocytes. Life Sci. 2018, 196, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Horst, A.; Blinka, S.; Stamm, K.; Mahnke, D.; Schuman, J.; Gundry, R.; Tomita-Mitchell, A.; Lough, J. Activin-A and Bmp4 levels modulate cell type specification during CHIR-induced cardiomyogenesis. PLoS ONE 2015, 10, e0118670. [Google Scholar] [CrossRef] [PubMed]
- Fonoudi, H.; Ansari, H.; Abbasalizadeh, S.; Larijani, M.R.; Kiani, S.; Hashemizadeh, S.; Zarchi, A.S.; Bosman, A.; Blue, G.M.; Pahlavan, S.; et al. A universal and robust integrated platform for the scalable production of human cardiomyocytes from pluripotent stem cells. Stem Cells Transl. Med. 2015, 4, 1482–1494. [Google Scholar] [CrossRef]
- Hemmi, N.; Tohyama, S.; Nakajima, K.; Kanazawa, H.; Suzuki, T.; Hattori, F.; Seki, T.; Kishino, Y.; Hirano, A.; Okada, M.; et al. A massive suspension culture system with metabolic purification for human pluripotent stem cell-derived cardiomyocytes. Life Sci. 2014, 3, 1473–1483. [Google Scholar] [CrossRef] [PubMed]
- Burridge, P.W.; Holmstrom, A.; Wu, J.C. Chemically defined culture and cardiomyocyte differentiation of human pluripotent stem cells. Curr. Protoc. Hum. Genet. 2015, 87, 21.3.1–21.3.15. [Google Scholar]
- Zhao, Y.; Rafatian, N.; Feric, N.T.; Cox, B.J.; Aschar-Sobbi, R.; Wang, E.Y.; Aggarwal, P.; Zhang, B.; Conant, G.; Ronaldson-Bouchard, K.; et al. A platform for generation of chamber-specific cardiac tissues and disease modeling. Cell 2019, 176, 913–927.e18. [Google Scholar] [CrossRef]
- Talkhabi, M.; Aghdami, N.; Baharvand, H. Human cardiomyocyte generation from pluripotent stem cells: A state-of-art. Life Sci. 2016, 145, 98–113. [Google Scholar] [CrossRef]
- Fatima, A.; Xu, G.; Shao, K.; Papadopoulos, S.; Lehmann, M.; Arnaiz-Cot, J.J.; Rosa, A.O.; Nguemo, F.; Matzkies, M.; Dittmann, S.; et al. In vitro modeling of ryanodine receptor 2 dysfunction using human induced pluripotent stem cells. Cell. Physiol. Biochem. 2011, 28, 579–592. [Google Scholar] [CrossRef]
- Pradhapan, P.; Kuusela, J.; Viik, J.; Aalto-Setala, K.; Hyttinen, J. Cardiomyocyte MEA data analysis (CardioMDA)—A novel field potential data analysis software for pluripotent stem cell derived cardiomyocytes. PLoS ONE 2013, 8, e73637. [Google Scholar] [CrossRef] [PubMed]
- Penttinen, K.; Siirtola, H.; Avalos-Salguero, J.; Vainio, T.; Juhola, M.; Aalto-Setala, K. Novel analysis software for detecting and classifying Ca2+ transient abnormalities in stem cell-derived cardiomyocytes. PLoS ONE 2015, 10, e0135806. [Google Scholar] [CrossRef] [PubMed]
- Tsurumi, F.; Baba, S.; Yoshinaga, D.; Umeda, K.; Hirata, T.; Takita, J.; Heike, T. The intracellular Ca2+ concentration is elevated in cardiomyocytes differentiated from hiPSCs derived from a Duchenne muscular dystrophy patient. PLoS ONE 2019, 14, e0213768. [Google Scholar] [CrossRef]
- Ahola, A.; Polonen, R.P.; Aalto-Setala, K.; Hyttinen, J. Simultaneous measurement of contraction and calcium transients in stem cell derived cardiomyocytes. Ann. Biomed. Eng. 2018, 46, 148–158. [Google Scholar] [CrossRef]
- Hortigon-Vinagre, M.P.; Zamora, V.; Burton, F.L.; Green, J.; Gintant, G.A.; Smith, G.L. The use of ratiometric fluorescence measurements of the voltage sensitive dye Di-4-ANEPPS to examine action potential characteristics and drug effects on human induced pluripotent stem cell-derived cardiomyocytes. Toxicol. Sci. 2016, 154, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Wheelwright, M.; Win, Z.; Mikkila, J.L.; Amen, K.Y.; Alford, P.W.; Metzger, J.M. Investigation of human Ipsc-derived cardiac myocyte functional maturation by single cell traction force microscopy. PLoS ONE 2018, 13, e0194909. [Google Scholar] [CrossRef]
- Pesl, M.; Pribyl, J.; Acimovic, I.; Vilotic, A.; Jelinkova, S.; Salykin, A.; Lacampagne, A.; Dvorak, P.; Meli, A.C.; Skladal, P.; et al. Atomic force microscopy combined with human pluripotent stem cell derived cardiomyocytes for biomechanical sensing. Biosens. Bioelectron. 2016, 85, 751–757. [Google Scholar] [CrossRef]
- Liu, J.; Sun, N.; Bruce, M.A.; Wu, J.C.; Butte, M.J. Atomic force mechanobiology of pluripotent stem cell-derived cardiomyocytes. PLoS ONE 2012, 7, e37559. [Google Scholar] [CrossRef]
- Feaster, T.K.; Cadar, A.G.; Wang, L.; Williams, C.H.; Chun, Y.W.; Hempel, J.E.; Bloodworth, N.; Merryman, W.D.; Lim, C.C.; Wu, J.C.; et al. Matrigel mattress: A method for the generation of single contracting human-induced pluripotent stem cell-derived cardiomyocytes. Circ. Res. 2015, 117, 995–1000. [Google Scholar] [CrossRef]
- Carvajal-Vergara, X.; Sevilla, A.; D’Souza, S.L.; Ang, Y.S.; Schaniel, C.; Lee, D.F.; Yang, L.; Kaplan, A.D.; Adler, E.D.; Rozov, R.; et al. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature 2010, 465, 808–812. [Google Scholar] [CrossRef]
- Digilio, M.C.; Conti, E.; Sarkozy, A.; Mingarelli, R.; Dottorini, T.; Marino, B.; Pizzuti, A.; Dallapiccola, B. Grouping of multiple-lentigines/LEOPARD and Noonan syndromes on the PTPN11 gene. Am. J. Hum. Genet. 2002, 71, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Ganigara, M.; Prabhu, A.; Kumar, R.S. LEOPARD syndrome in an infant with severe hypertrophic cardiomyopathy and PTPN11 mutation. Ann. Pediatr. Cardiol. 2011, 4, 74–76. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, G.; Pacileo, G.; Russo, M.G.; Sarkozy, A.; Felicetti, M.; Di Salvo, G.; Morelli, C.; Calabro, P.; Paladini, D.; Marino, B.; et al. Severe, early onset hypertrophic cardiomyopathy in a family with LEOPARD syndrome. J. Prenat. Med. 2008, 2, 24–26. [Google Scholar]
- Molkentin, J.D. Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc. Res. 2004, 63, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Adamcova, M.; Skarkova, V.; Seifertova, J.; Rudolf, E. Cardiac troponins are among targets of doxorubicin-induced cardiotoxicity in hiPCS-CMs. Int. J. Mol. Sci. 2019, 20, 2638. [Google Scholar] [CrossRef]
- Lu, Y.; Bu, M.; Yun, H. Sevoflurane prevents hypoxia/reoxygenation-induced cardiomyocyte apoptosis by inhibiting PI3KC3-mediated autophagy. Hum. Cell 2019, 32, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Naftali-Shani, N.; Molotski, N.; Nevo-Caspi, Y.; Arad, M.; Kuperstein, R.; Amit, U.; Huber, I.; Zeltzer, L.A.; Levich, A.; Abbas, H.; et al. Modeling peripartum cardiomyopathy with human induced pluripotent stem cells reveals distinctive abnormal function of cardiomyocytes. Circulation 2018, 138, 2721–2723. [Google Scholar] [CrossRef]
- Graneli, C.; Hicks, R.; Brolen, G.; Synnergren, J.; Sartipy, P. Diabetic cardiomyopathy modelling using induced pluripotent stem cell derived cardiomyocytes: Recent advances and emerging models. Stem Cell Rev. 2019, 15, 13–22. [Google Scholar] [CrossRef]
- Drawnel, F.M.; Boccardo, S.; Prummer, M.; Delobel, F.; Graff, A.; Weber, M.; Gerard, R.; Badi, L.; Kam-Thong, T.; Bu, L.; et al. Disease modeling and phenotypic drug screening for diabetic cardiomyopathy using human induced pluripotent stem cells. Cell Rep. 2014, 9, 810–821. [Google Scholar] [CrossRef]
- Ng, K.M.; Lau, Y.M.; Dhandhania, V.; Cai, Z.J.; Lee, Y.K.; Lai, W.H.; Tse, H.F.; Siu, C.W. Empagliflozin ammeliorates high glucose induced-cardiac dysfuntion in human iPSC-derived cardiomyocytes. Sci. Rep. 2018, 8, 14872. [Google Scholar] [CrossRef]
- Pant, T.; Mishra, M.K.; Bai, X.; Ge, Z.D.; Bosnjak, Z.J.; Dhanasekaran, A. Microarray analysis of long non-coding RNA and mRNA expression profiles in diabetic cardiomyopathy using human induced pluripotent stem cell-derived cardiomyocytes. Diabetes Vasc. Dis. Res. 2019, 16, 57–68. [Google Scholar] [CrossRef]
- Bozzi, A.; Sayed, N.; Matsa, E.; Sass, G.; Neofytou, E.; Clemons, K.V.; Correa-Oliveira, R.; Stevens, D.A.; Wu, J.C. Using human induced pluripotent stem cell-derived cardiomyocytes as a model to study Trypanosoma cruzi infection. Stem Cell Rep. 2019, 12, 1232–1241. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Marceau, C.; Hamaguchi, R.; Burridge, P.W.; Rajarajan, K.; Churko, J.M.; Wu, H.; Sallam, K.I.; Matsa, E.; Sturzu, A.C.; et al. Human induced pluripotent stem cell-derived cardiomyocytes as an in vitro model for coxsackievirus B3-induced myocarditis and antiviral drug screening platform. Circ. Res. 2014, 115, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Judge, L.M.; Perez-Bermejo, J.A.; Truong, A.; Ribeiro, A.J.; Yoo, J.C.; Jensen, C.L.; Mandegar, M.A.; Huebsch, N.; Kaake, R.M.; So, P.L.; et al. A BAG3 chaperone complex maintains cardiomyocyte function during proteotoxic stress. JCI Insight 2017, 2, e94623. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Zhang, J.; Itzhaki, I.; Zhang, S.L.; Chen, H.; Haddad, F.; Kitani, T.; Wilson, K.D.; Tian, L.; Shrestha, R.; et al. Determining the pathogenicity of a genomic variant of uncertain significance using CRISPR/Cas9 and human-induced pluripotent stem cells. Circulation 2018, 138, 2666–2681. [Google Scholar] [CrossRef]
- Bezzerides, V.J.; Caballero, A.; Wang, S.; Ai, Y.; Hylind, R.J.; Lu, F.; Heims-Waldron, D.A.; Chambers, K.D.; Zhang, D.; Abrams, D.J.; et al. Gene therapy for catecholaminergic polymorphic ventricular tachycardia by inhibition of Ca(2+)/calmodulin-dependent kinase II. Circulation 2019, 140, 405–419. [Google Scholar] [CrossRef] [PubMed]
- Gramlich, M.; Pane, L.S.; Zhou, Q.; Chen, Z.; Murgia, M.; Schotterl, S.; Goedel, A.; Metzger, K.; Brade, T.; Parrotta, E.; et al. Antisense-mediated exon skipping: A therapeutic strategy for titin-based dilated cardiomyopathy. EMBO Mol. Med. 2015, 7, 562–576. [Google Scholar] [CrossRef]
- Van Heesch, S.; Witte, F.; Schneider-Lunitz, V.; Schulz, J.F.; Adami, E.; Faber, A.B.; Kirchner, M.; Maatz, H.; Blachut, S.; Sandmann, C.L.; et al. The translational landscape of the human heart. Cell 2019, 178, 242–260.e29. [Google Scholar] [CrossRef]
- Kyrychenko, V.; Kyrychenko, S.; Tiburcy, M.; Shelton, J.M.; Long, C.; Schneider, J.W.; Zimmermann, W.H.; Bassel-Duby, R.; Olson, E.N. Functional correction of dystrophin actin binding domain mutations by genome editing. JCI Insight 2017, 2, e95918. [Google Scholar] [CrossRef]
- Smith, J.G.W.; Owen, T.; Bhagwan, J.R.; Mosqueira, D.; Scott, E.; Mannhardt, I.; Patel, A.; Barriales-Villa, R.; Monserrat, L.; Hansen, A.; et al. Isogenic pairs of hiPSC-CMs with hypertrophic cardiomyopathy/LVNC-associated ACTC1 E99K mutation unveil differential functional deficits. Stem Cell Rep. 2018, 11, 1226–1243. [Google Scholar] [CrossRef]
- Phelan, D.G.; Anderson, D.J.; Howden, S.E.; Wong, R.C.; Hickey, P.F.; Pope, K.; Wilson, G.R.; Pebay, A.; Davis, A.M.; Petrou, S.; et al. ALPK3-deficient cardiomyocytes generated from patient-derived induced pluripotent stem cells and mutant human embryonic stem cells display abnormal calcium handling and establish that ALPK3 deficiency underlies familial cardiomyopathy. Eur. Heart J. 2016, 37, 2586–2590. [Google Scholar] [CrossRef] [PubMed]
- Josowitz, R.; Mulero-Navarro, S.; Rodriguez, N.A.; Falce, C.; Cohen, N.; Ullian, E.M.; Weiss, L.A.; Rauen, K.A.; Sobie, E.A.; Gelb, B.D. Autonomous and Non-autonomous defects underlie hypertrophic cardiomyopathy in BRAF-mutant hiPSC-derived cardiomyocytes. Stem Cell Rep. 2016, 7, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Meraviglia, V.; Benzoni, P.; Landi, S.; Murano, C.; Langione, M.; Motta, B.M.; Baratto, S.; Silipigni, R.; Di Segni, M.; Pramstaller, P.P.; et al. Generation of human induced pluripotent stem cells (EURACi001-A, EURACi002-A, EURACi003-A) from peripheral blood mononuclear cells of three patients carrying mutations in the CAV3 gene. Stem Cell Res. 2018, 27, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Mitzelfelt, K.A.; Limphong, P.; Choi, M.J.; Kondrat, F.D.; Lai, S.; Kolander, K.D.; Kwok, W.M.; Dai, Q.; Grzybowski, M.N.; Zhang, H.; et al. The human 343delT HSPB5 chaperone associated with early-onset skeletal myopathy causes defects in protein solubility. J. Biol. Chem. 2016, 291, 14939–14953. [Google Scholar] [CrossRef] [PubMed]
- Fatima, A.; Dittmann, S.; Kaifeng, S.; Linke, M.; Zechner, U.; Hennies, H.C.; Stork, I.; Rosenkranz, S.; Farr, M.; Milting, H.; et al. Derivation of induced pluripotent stem (iPS) cells from a patient with an arrhythmogenic right ventricular cardiomyopathy (ARVC). J. Stem Cells Regen. Med. 2010, 6, 97. [Google Scholar] [PubMed]
- Khudiakov, A.; Kostina, D.; Zlotina, A.; Nikulina, T.; Sergushichev, A.; Gudkova, A.; Tomilin, A.; Malashicheva, A.; Kostareva, A. Generation of iPSC line from desmin-related cardiomyopathy patient carrying splice site mutation of DES gene. Stem Cell Res. 2017, 24, 77–80. [Google Scholar] [CrossRef]
- Tse, H.F.; Ho, J.C.; Choi, S.W.; Lee, Y.K.; Butler, A.W.; Ng, K.M.; Siu, C.W.; Simpson, M.A.; Lai, W.H.; Chan, Y.C.; et al. Patient-specific induced-pluripotent stem cells-derived cardiomyocytes recapitulate the pathogenic phenotypes of dilated cardiomyopathy due to a novel DES mutation identified by whole exome sequencing. Hum. Mol. Genet. 2013, 22, 1395–1403. [Google Scholar] [CrossRef]
- Eisen, B.; Ben Jehuda, R.; Cuttitta, A.J.; Mekies, L.N.; Shemer, Y.; Baskin, P.; Reiter, I.; Willi, L.; Freimark, D.; Gherghiceanu, M.; et al. Electrophysiological abnormalities in induced pluripotent stem cell-derived cardiomyocytes generated from Duchenne muscular dystrophy patients. J. Cell. Mol. Med. 2019, 23, 2125–2135. [Google Scholar] [CrossRef]
- Pioner, J.M.; Guan, X.; Klaiman, J.M.; Racca, A.W.; Pabon, L.; Muskheli, V.; Macadangdang, J.; Ferrantini, C.; Hoopmann, M.R.; Moritz, R.L.; et al. Absence of full-length dystrophin impairs normal maturation and contraction of cardiomyocytes derived from human induced pluripotent stem cells. Cardiovasc. Res. 2019. [Google Scholar] [CrossRef]
- Guan, X.; Mack, D.L.; Moreno, C.M.; Strande, J.L.; Mathieu, J.; Shi, Y.; Markert, C.D.; Wang, Z.; Liu, G.; Lawlor, M.W.; et al. Dystrophin-deficient cardiomyocytes derived from human urine: New biologic reagents for drug discovery. Stem Cell Res. 2014, 12, 467–480. [Google Scholar] [CrossRef] [PubMed]
- El-Battrawy, I.; Zhao, Z.; Lan, H.; Cyganek, L.; Tombers, C.; Li, X.; Buljubasic, F.; Lang, S.; Tiburcy, M.; Zimmermann, W.H.; et al. Electrical dysfunctions in human-induced pluripotent stem cell-derived cardiomyocytes from a patient with an arrhythmogenic right ventricular cardiomyopathy. Europace 2018, 20, f46–f56. [Google Scholar] [CrossRef] [PubMed]
- Ng, R.; Manring, H.R.; Papoutsidakis, N.; Albertelli, T.; Tsai, N.; See, C.; Li, X.; Park, J.; Stevens, T.L.; Bobbili, P.J.; et al. Patient mutations linked to arrhythmogenic cardiomyopathy enhance calpain-mediated desmoplakin degradation. JCI Insight 2019, 4, e128643. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Dvornik, J.L.; Yarrabothula, A.R.; Schaniel, C. A Marfan syndrome human induced pluripotent stem cell line with a heterozygous FBN1 c.4082G > A mutation, ISMMSi002-B, for disease modeling. Stem Cell Res. 2017, 23, 73–76. [Google Scholar] [CrossRef] [PubMed]
- El-Battrawy, I.; Zhao, Z.; Lan, H.; Li, X.; Yucel, G.; Lang, S.; Sattler, K.; Schunemann, J.D.; Zimmermann, W.H.; Cyganek, L.; et al. Ion channel dysfunctions in dilated cardiomyopathy in limb-girdle muscular dystrophy. Circ. Genom. Precis. Med. 2018, 11, e001893. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Ho, P.W.; Schick, R.; Lau, Y.M.; Lai, W.H.; Zhou, T.; Li, Y.; Ng, K.M.; Ho, S.L.; Esteban, M.A.; et al. Modeling of Friedreich ataxia-related iron overloading cardiomyopathy using patient-specific-induced pluripotent stem cells. Pflugers Arch. Eur. J. Physiol. 2014, 466, 1831–1844. [Google Scholar] [CrossRef] [PubMed]
- Hick, A.; Wattenhofer-Donze, M.; Chintawar, S.; Tropel, P.; Simard, J.P.; Vaucamps, N.; Gall, D.; Lambot, L.; Andre, C.; Reutenauer, L.; et al. Neurons and cardiomyocytes derived from induced pluripotent stem cells as a model for mitochondrial defects in Friedreich’s ataxia. Dis. Models Mech. 2013, 6, 608–621. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.; Chou, S.J.; Chang, Y.L.; Leu, H.B.; Yang, Y.P.; Tsai, P.H.; Lai, Y.H.; Chen, K.H.; Chang, W.C.; Sung, S.H.; et al. Inhibition of arachidonate 12/15-lipoxygenase improves α-galactosidase efficacy in iPSC-derived cardiomyocytes from fabry patients. Int. J. Mol. Sci. 2018, 19, 1480. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.J.; Yu, W.C.; Chang, Y.L.; Chen, W.Y.; Chang, W.C.; Chien, Y.; Yen, J.C.; Liu, Y.Y.; Chen, S.J.; Wang, C.Y.; et al. Energy utilization of induced pluripotent stem cell–derived cardiomyocyte in Fabry disease. Int. J. Cardiol. 2017, 232, 255–263. [Google Scholar] [CrossRef]
- Yoshida, S.; Nakanishi, C.; Okada, H.; Mori, M.; Yokawa, J.; Yoshimuta, T.; Ohta, K.; Konno, T.; Fujino, N.; Kawashiri, M.A.; et al. Characteristics of induced pluripotent stem cells from clinically divergent female monozygotic twins with Danon disease. J. Mol. Cell. Cardiol. 2018, 114, 234–242. [Google Scholar] [CrossRef]
- Hashem, S.I.; Murphy, A.N.; Divakaruni, A.S.; Klos, M.L.; Nelson, B.C.; Gault, E.C.; Rowland, T.J.; Perry, C.N.; Gu, Y.; Dalton, N.D.; et al. Impaired mitophagy facilitates mitochondrial damage in Danon disease. J. Mol. Cell. Cardiol. 2017, 108, 86–94. [Google Scholar] [CrossRef]
- Chi, C.; Leonard, A.; Knight, W.E.; Beussman, K.M.; Zhao, Y.; Cao, Y.; Londono, P.; Aune, E.; Trembley, M.A.; Small, E.M.; et al. LAMP-2B regulates human cardiomyocyte function by mediating autophagosome-lysosome fusion. Proc. Natl. Acad. Sci. USA 2019, 116, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.; Virtanen, L.; Prajapati, C.; Kiamehr, M.; Gullmets, J.; West, G.; Kreutzer, J.; Pekkanen-Mattila, M.; Helio, T.; Kallio, P.; et al. Modeling of LMNA-related dilated cardiomyopathy using human induced pluripotent stem cells. Cells 2019, 8, 594. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.C.; Zhou, T.; Lai, W.H.; Huang, Y.; Chan, Y.C.; Li, X.; Wong, N.L.; Li, Y.; Au, K.W.; Guo, D.; et al. Generation of induced pluripotent stem cell lines from 3 distinct laminopathies bearing heterogeneous mutations in lamin A/C. Aging 2011, 3, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Siu, C.W.; Lee, Y.K.; Ho, J.C.; Lai, W.H.; Chan, Y.C.; Ng, K.M.; Wong, L.Y.; Au, K.W.; Lau, Y.M.; Zhang, J.; et al. Modeling of lamin A/C mutation premature cardiac aging using patient-specific induced pluripotent stem cells. Aging 2012, 4, 803–822. [Google Scholar] [CrossRef]
- Lee, Y.K.; Lau, Y.M.; Cai, Z.J.; Lai, W.H.; Wong, L.Y.; Tse, H.F.; Ng, K.M.; Siu, C.W. Modeling treatment response for lamin A/C related dilated cardiomyopathy in human induced pluripotent stem cells. J. Am. Heart Assoc. 2017, 6, e005677. [Google Scholar] [CrossRef]
- Salvarani, N.; Crasto, S.; Miragoli, M.; Bertero, A.; Paulis, M.; Kunderfranco, P.; Serio, S.; Forni, A.; Lucarelli, C.; Dal Ferro, M.; et al. The K219T-Lamin mutation induces conduction defects through epigenetic inhibition of SCN5A in human cardiac laminopathy. Nat. Commun. 2019, 10, 2267. [Google Scholar] [CrossRef]
- Li, S.; Pan, H.; Tan, C.; Sun, Y.; Song, Y.; Zhang, X.; Yang, W.; Wang, X.; Li, D.; Dai, Y.; et al. Mitochondrial dysfunctions contribute to hypertrophic cardiomyopathy in patient iPSC-derived cardiomyocytes with MT-RNR2 mutation. Stem Cell Rep. 2018, 10, 808–821. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Yang, H.; Rhee, J.W.; Zhang, J.Z.; Lam, C.K.; Sallam, K.; Chang, A.C.Y.; Ma, N.; Lee, J.; Zhang, H.; et al. Modelling diastolic dysfunction in induced pluripotent stem cell-derived cardiomyocytes from hypertrophic cardiomyopathy patients. Eur. Heart J. 2019. [Google Scholar] [CrossRef] [PubMed]
- Dementyeva, E.V.; Medvedev, S.P.; Kovalenko, V.R.; Vyatkin, Y.V.; Kretov, E.I.; Slotvitsky, M.M.; Shtokalo, D.N.; Pokushalov, E.A.; Zakian, S.M. Applying patient-specific induced pluripotent stem cells to create a model of hypertrophic cardiomyopathy. Biochemistry (Mosc) 2019, 84, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Dambrot, C.; Braam, S.R.; Tertoolen, L.G.; Birket, M.; Atsma, D.E.; Mummery, C.L. Serum supplemented culture medium masks hypertrophic phenotypes in human pluripotent stem cell derived cardiomyocytes. J. Cell. Mol. Med. 2014, 18, 1509–1518. [Google Scholar] [CrossRef]
- Birket, M.J.; Ribeiro, M.C.; Kosmidis, G.; Ward, D.; Leitoguinho, A.R.; van de Pol, V.; Dambrot, C.; Devalla, H.D.; Davis, R.P.; Mastroberardino, P.G.; et al. Contractile defect caused by mutation in MYBPC3 revealed under conditions optimized for human PSC-cardiomyocyte function. Cell Rep. 2015, 13, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Holliday, M.; Ross, S.B.; Lim, S.; Semsarian, C. Generation of an induced pluripotent stem cell line from a hypertrophic cardiomyopathy patient with a pathogenic myosin binding protein C (MYBPC3) p.Arg502Trp mutation. Stem Cell Res. 2018, 33, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Cohn, R.; Thakar, K.; Lowe, A.; Ladha, F.A.; Pettinato, A.M.; Romano, R.; Meredith, E.; Chen, Y.S.; Atamanuk, K.; Huey, B.D.; et al. A contraction stress model of hypertrophic cardiomyopathy due to sarcomere mutations. Stem Cell Rep. 2019, 12, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Davis, L.C.; Correll, R.N.; Makarewich, C.A.; Schwanekamp, J.A.; Moussavi-Harami, F.; Wang, D.; York, A.J.; Wu, H.; Houser, S.R.; et al. A tension-based model distinguishes hypertrophic versus dilated cardiomyopathy. Cell 2016, 165, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Seeger, T.; Shrestha, R.; Lam, C.K.; Chen, C.; McKeithan, W.L.; Lau, E.; Wnorowski, A.; McMullen, G.; Greenhaw, M.; Lee, J.; et al. A premature termination codon mutation in MYBPC3 causes hypertrophic cardiomyopathy via chronic activation of nonsense-mediated decay. Circulation 2019, 139, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Yuasa, S.; Mearini, G.; Egashira, T.; Seki, T.; Kodaira, M.; Kusumoto, D.; Kuroda, Y.; Okata, S.; Suzuki, T.; et al. Endothelin-1 induces myofibrillar disarray and contractile vector variability in hypertrophic cardiomyopathy-induced pluripotent stem cell-derived cardiomyocytes. J. Am. Heart Assoc. 2014, 3, e001263. [Google Scholar] [CrossRef] [PubMed]
- Ojala, M.; Prajapati, C.; Polonen, R.P.; Rajala, K.; Pekkanen-Mattila, M.; Rasku, J.; Larsson, K.; Aalto-Setala, K. Mutation-specific phenotypes in hiPSC-derived cardiomyocytes carrying either myosin-binding protein C or α-tropomyosin mutation for hypertrophic cardiomyopathy. Stem Cells Int. 2016, 2016, 1684792. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, C.; Ojala, M.; Aalto-Setala, K. Divergent effects of adrenaline in human induced pluripotent stem cell-derived cardiomyocytes obtained from hypertrophic cardiomyopathy. Dis. Models Mech. 2018, 11, dmm032896. [Google Scholar] [CrossRef]
- Prondzynski, M.; Kramer, E.; Laufer, S.D.; Shibamiya, A.; Pless, O.; Flenner, F.; Muller, O.J.; Munch, J.; Redwood, C.; Hansen, A.; et al. Evaluation of MYBPC3 trans-splicing and gene replacement as therapeutic options in human iPSC-derived cardiomyocytes. Mol. Ther. Nucleic Acids 2017, 7, 475–486. [Google Scholar] [CrossRef]
- Viswanathan, S.K.; Puckelwartz, M.J.; Mehta, A.; Ramachandra, C.J.A.; Jagadeesan, A.; Fritsche-Danielson, R.; Bhat, R.V.; Wong, P.; Kandoi, S.; Schwanekamp, J.A.; et al. Association of cardiomyopathy with MYBPC3 D389V and MYBPC3Delta25bpIntronic deletion in South Asian descendants. JAMA Cardiol. 2018, 3, 481–488. [Google Scholar] [CrossRef]
- Mosqueira, D.; Mannhardt, I.; Bhagwan, J.R.; Lis-Slimak, K.; Katili, P.; Scott, E.; Hassan, M.; Prondzynski, M.; Harmer, S.C.; Tinker, A.; et al. CRISPR/Cas9 editing in human pluripotent stem cell-cardiomyocytes highlights arrhythmias, hypocontractility, and energy depletion as potential therapeutic targets for hypertrophic cardiomyopathy. Eur. Heart J. 2018, 39, 3879–3892. [Google Scholar] [CrossRef]
- Ross, S.B.; Fraser, S.T.; Nowak, N.; Semsarian, C. Generation of induced pluripotent stem cells (iPSCs) from a hypertrophic cardiomyopathy patient with the pathogenic variant p.Val698Ala in β-myosin heavy chain (MYH7) gene. Stem Cell Res. 2017, 20, 88–90. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.C.; Breitbart, A.; De Lange, W.J.; Hofsteen, P.; Futakuchi-Tsuchida, A.; Xu, J.; Schopf, C.; Razumova, M.V.; Jiao, A.; Boucek, R.; et al. Novel adult-onset systolic cardiomyopathy due to MYH7 E848G mutation in patient-derived induced pluripotent stem cells. JACC Basic Transl. Sci. 2018, 3, 728–740. [Google Scholar] [CrossRef] [PubMed]
- Pioner, J.M.; Racca, A.W.; Klaiman, J.M.; Yang, K.C.; Guan, X.; Pabon, L.; Muskheli, V.; Zaunbrecher, R.; Macadangdang, J.; Jeong, M.Y.; et al. Isolation and mechanical measurements of myofibrils from human induced pluripotent stem cell-derived cardiomyocytes. Stem Cell Rep. 2016, 6, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Holliday, M.; Ross, S.B.; Lim, S.; Mangala, M.; Hill, A.; Szappanos, H.C.; Hool, L.; Semsarian, C. Development of induced pluripotent stem cells from a patient with hypertrophic cardiomyopathy who carries the pathogenic myosin heavy chain 7 mutation p.Arg403Gln. Stem Cell Res. 2018, 33, 269–273. [Google Scholar] [CrossRef]
- Lan, F.; Lee, A.S.; Liang, P.; Sanchez-Freire, V.; Nguyen, P.K.; Wang, L.; Han, L.; Yen, M.; Wang, Y.; Sun, N.; et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell 2013, 12, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Li, Y.; Tchao, J.; Kaplan, A.D.; Lin, B.; Li, Y.; Mich-Basso, J.; Lis, A.; Hassan, N.; London, B.; et al. Study familial hypertrophic cardiomyopathy using patient-specific induced pluripotent stem cells. Cardiovasc. Res. 2014, 104, 258–269. [Google Scholar] [CrossRef]
- Zhou, W.; Bos, J.M.; Ye, D.; Tester, D.J.; Hrstka, S.; Maleszewski, J.J.; Ommen, S.R.; Nishimura, R.A.; Schaff, H.V.; Kim, C.S.; et al. Induced pluripotent stem cell-derived cardiomyocytes from a patient with MYL2-R58Q-mediated apical hypertrophic cardiomyopathy show hypertrophy, myofibrillar disarray, and calcium perturbations. J. Cardiovasc. Transl. Res. 2019, 1–10. [Google Scholar] [CrossRef]
- Ma, D.; Wei, H.; Lu, J.; Ho, S.; Zhang, G.; Sun, X.; Oh, Y.; Tan, S.H.; Ng, M.L.; Shim, W.; et al. Generation of patient-specific induced pluripotent stem cell-derived cardiomyocytes as a cellular model of arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2013, 34(15), 1122–1133. [Google Scholar] [CrossRef]
- Kim, C.; Wong, J.; Wen, J.; Wang, S.; Wang, C.; Spiering, S.; Kan, N.G.; Forcales, S.; Puri, P.L.; Leone, T.C.; et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature 2013, 494, 105–110. [Google Scholar] [CrossRef]
- Martewicz, S.; Luni, C.; Serena, E.; Pavan, P.; Chen, H.V.; Rampazzo, A.; Elvassore, N. Transcriptomic characterization of a human in vitro model of arrhythmogenic cardiomyopathy under topological and mechanical stimuli. Ann. Biomed. Eng. 2019, 47, 852–865. [Google Scholar] [CrossRef] [PubMed]
- Akdis, D.; Saguner, A.M.; Shah, K.; Wei, C.; Medeiros-Domingo, A.; von Eckardstein, A.; Luscher, T.F.; Brunckhorst, C.; Chen, H.S.V.; Duru, F. Sex hormones affect outcome in arrhythmogenic right ventricular cardiomyopathy/dysplasia: From a stem cell derived cardiomyocyte-based model to clinical biomarkers of disease outcome. Eur. Heart J. 2017, 38, 1498–1508. [Google Scholar] [CrossRef]
- Caspi, O.; Huber, I.; Gepstein, A.; Arbel, G.; Maizels, L.; Boulos, M.; Gepstein, L. Modeling of arrhythmogenic right ventricular cardiomyopathy with human induced pluripotent stem cells. Circ. Cardiovasc. Genet. 2013, 6, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Khudiakov, A.; Kostina, D.; Zlotina, A.; Yany, N.; Sergushichev, A.; Pervunina, T.; Tomilin, A.; Kostareva, A.; Malashicheva, A. Generation of iPSC line from patient with arrhythmogenic right ventricular cardiomyopathy carrying mutations in PKP2 gene. Stem Cell Res. 2017, 24, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Ermon, B.; Volpato, C.B.; Cattelan, G.; Silipigni, R.; Di Segni, M.; Cantaloni, C.; Casella, M.; Pramstaller, P.P.; Pompilio, G.; Sommariva, E.; et al. Derivation of human induced pluripotent stem cell line EURACi004-A from skin fibroblasts of a patient with arrhythmogenic cardiomyopathy carrying the heterozygous PKP2 mutation c.2569_3018del50. Stem Cell Res. 2018, 32, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Ceholski, D.K.; Turnbull, I.C.; Kong, C.W.; Koplev, S.; Mayourian, J.; Gorski, P.A.; Stillitano, F.; Skodras, A.A.; Nonnenmacher, M.; Cohen, N.; et al. Functional and transcriptomic insights into pathogenesis of R9C phospholamban mutation using human induced pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell. Cardiol. 2018, 119, 147–154. [Google Scholar] [CrossRef]
- Karakikes, I.; Stillitano, F.; Nonnenmacher, M.; Tzimas, C.; Sanoudou, D.; Termglinchan, V.; Kong, C.W.; Rushing, S.; Hansen, J.; Ceholski, D.; et al. Correction of human phospholamban R14del mutation associated with cardiomyopathy using targeted nucleases and combination therapy. Nat. Commun. 2015, 6, 6955. [Google Scholar] [CrossRef]
- Stillitano, F.; Turnbull, I.C.; Karakikes, I.; Nonnenmacher, M.; Backeris, P.; Hulot, J.S.; Kranias, E.G.; Hajjar, R.J.; Costa, K.D. Genomic correction of familial cardiomyopathy in human engineered cardiac tissues. Eur. Heart J. 2016, 37, 3282–3284. [Google Scholar] [CrossRef]
- Ben Jehuda, R.; Eisen, B.; Shemer, Y.; Mekies, L.N.; Szantai, A.; Reiter, I.; Cui, H.; Guan, K.; Haron-Khun, S.; Freimark, D.; et al. CRISPR correction of the PRKAG2 gene mutation in the patient’s induced pluripotent stem cell-derived cardiomyocytes eliminates electrophysiological and structural abnormalities. Heart Rhythm 2018, 15, 267–276. [Google Scholar] [CrossRef]
- Hinson, J.T.; Chopra, A.; Lowe, A.; Sheng, C.C.; Gupta, R.M.; Kuppusamy, R.; O’Sullivan, J.; Rowe, G.; Wakimoto, H.; Gorham, J.; et al. Integrative analysis of PRKAG2 cardiomyopathy iPS and microtissue models identifies AMPK as a regulator of metabolism, survival, and fibrosis. Cell Rep. 2017, 19, 2410. [Google Scholar] [CrossRef]
- Li, R.; Baskfield, A.; Lin, Y.; Beers, J.; Zou, J.; Liu, C.; Jaffre, F.; Roberts, A.E.; Ottinger, E.A.; Kontaridis, M.I.; et al. Generation of an induced pluripotent stem cell line (TRNDi003-A) from a Noonan syndrome with multiple lentigines (NSML) patient carrying a p.Q510P mutation in the PTPN11 gene. Stem Cell Res. 2019, 34, 101374. [Google Scholar] [CrossRef] [PubMed]
- Jaffre, F.; Miller, C.L.; Schanzer, A.; Evans, T.; Roberts, A.E.; Hahn, A.; Kontaridis, M.I. iPSC-derived cardiomyocytes reveal aberrant ERK5 and MEK1/2 signaling concomitantly promote hypertrophic cardiomyopathy in RAF1-associated noonan syndrome. Circulation 2019, 140, 207–224. [Google Scholar] [CrossRef] [PubMed]
- Streckfuss-Bomeke, K.; Tiburcy, M.; Fomin, A.; Luo, X.; Li, W.; Fischer, C.; Ozcelik, C.; Perrot, A.; Sossalla, S.; Haas, J.; et al. Severe DCM phenotype of patient harboring RBM20 mutation S635A can be modeled by patient-specific induced pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell. Cardiol. 2017, 113, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Wyles, S.P.; Li, X.; Hrstka, S.C.; Reyes, S.; Oommen, S.; Beraldi, R.; Edwards, J.; Terzic, A.; Olson, T.M.; Nelson, T.J. Modeling structural and functional deficiencies of RBM20 familial dilated cardiomyopathy using human induced pluripotent stem cells. Hum. Mol. Genet. 2016, 25, 254–265. [Google Scholar] [CrossRef]
- Wyles, S.P.; Hrstka, S.C.; Reyes, S.; Terzic, A.; Olson, T.M.; Nelson, T.J. Pharmacological modulation of calcium homeostasis in familial dilated cardiomyopathy: An in vitro analysis from an RBM20 patient-derived iPSC model. Clin. Transl. Sci. 2016, 9, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Te Riele, A.S.; Agullo-Pascual, E.; James, C.A.; Leo–Macias, A.; Cerrone, M.; Zhang, M.; Lin, X.; Lin, B.; Sobreira, N.L.; Amat-Alarcon, N.; et al. Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non-canonical mechanisms for disease pathogenesis. Cardiovasc. Res. 2017, 113, 102–111. [Google Scholar] [CrossRef]
- Moreau, A.; Gosselin-Badaroudine, P.; Mercier, A.; Burger, B.; Keller, D.I.; Chahine, M. A leaky voltage sensor domain of cardiac sodium channels causes arrhythmias associated with dilated cardiomyopathy. Sci. Rep. 2018, 8, 13804. [Google Scholar] [CrossRef]
- Hallas, T.; Eisen, B.; Shemer, Y.; Ben Jehuda, R.; Mekies, L.N.; Naor, S.; Schick, R.; Eliyahu, S.; Reiter, I.; Vlodavsky, E.; et al. Investigating the cardiac pathology of SCO2-mediated hypertrophic cardiomyopathy using patients induced pluripotent stem cell-derived cardiomyocytes. J. Cell. Mol. Med. 2018, 22, 913–925. [Google Scholar] [CrossRef]
- Wang, G.; McCain, M.L.; Yang, L.; He, A.; Pasqualini, F.S.; Agarwal, A.; Yuan, H.; Jiang, D.; Zhang, D.; Zangi, L.; et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med. 2014, 20, 616–623. [Google Scholar] [CrossRef]
- Kodo, K.; Ong, S.G.; Jahanbani, F.; Termglinchan, V.; Hirono, K.; InanlooRahatloo, K.; Ebert, A.D.; Shukla, P.; Abilez, O.J.; Churko, J.M.; et al. iPSC-derived cardiomyocytes reveal abnormal TGF-β signalling in left ventricular non-compaction cardiomyopathy. Nat. Cell Biol. 2016, 18, 1031–1042. [Google Scholar] [CrossRef]
- Sun, N.; Yazawa, M.; Liu, J.; Han, L.; Sanchez-Freire, V.; Abilez, O.J.; Navarrete, E.G.; Hu, S.; Wang, L.; Lee, A.; et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci. Transl. Med. 2012, 4, 130ra47. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Lee, J.; Vincent, L.G.; Wang, Q.; Gu, M.; Lan, F.; Churko, J.M.; Sallam, K.I.; Matsa, E.; Sharma, A.; et al. Epigenetic regulation of phosphodiesterases 2A and 3A underlies compromised β-adrenergic signaling in an iPSC model of dilated cardiomyopathy. Cell Stem Cell 2015, 17, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Karakikes, I.; Termglinchan, V.; Cepeda, D.A.; Lee, J.; Diecke, S.; Hendel, A.; Itzhaki, I.; Ameen, M.; Shrestha, R.; Wu, H.; et al. A comprehensive TALEN-based knockout library for generating human-induced pluripotent stem cell-based models for cardiovascular diseases. Circ. Res. 2017, 120, 1561–1571. [Google Scholar] [CrossRef] [PubMed]
- Broughton, K.M.; Li, J.; Sarmah, E.; Warren, C.M.; Lin, Y.H.; Henze, M.P.; Sanchez-Freire, V.; Solaro, R.J.; Russell, B. A myosin activator improves actin assembly and sarcomere function of human-induced pluripotent stem cell-derived cardiomyocytes with a troponin T point mutation. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H107–H117. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Kryshtal, D.O.; Kim, K.; Parikh, S.; Cadar, A.G.; Bersell, K.R.; He, H.; Pinto, J.R.; Knollmann, B.C. Myofilament calcium-buffering dependent action potential triangulation in human-induced pluripotent stem cell model of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2017, 70, 2600–2602. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Kim, K.; Parikh, S.; Cadar, A.G.; Bersell, K.R.; He, H.; Pinto, J.R.; Kryshtal, D.O.; Knollmann, B.C. Hypertrophic cardiomyopathy-linked mutation in troponin T causes myofibrillar disarray and pro-arrhythmic action potential changes in human iPSC cardiomyocytes. J. Mol. Cell. Cardiol. 2018, 114, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Hinson, J.T.; Chopra, A.; Nafissi, N.; Polacheck, W.J.; Benson, C.C.; Swist, S.; Gorham, J.; Yang, L.; Schafer, S.; Sheng, C.C.; et al. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 2015, 349, 982–986. [Google Scholar] [CrossRef] [PubMed]
- Schick, R.; Mekies, L.N.; Shemer, Y.; Eisen, B.; Hallas, T.; Ben Jehuda, R.; Ben-Ari, M.; Szantai, A.; Willi, L.; Shulman, R.; et al. Functional abnormalities in induced pluripotent stem cell-derived cardiomyocytes generated from titin-mutated patients with dilated cardiomyopathy. PLoS ONE 2018, 13, e0205719. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.; Nah, S.K.; Reid, W.; Ebata, A.; Koch, C.M.; Monti, S.; Genereux, J.C.; Wiseman, R.L.; Wolozin, B.; Connors, L.H.; et al. Induced pluripotent stem cell modeling of multisystemic, hereditary transthyretin amyloidosis. Stem Cell Rep. 2013, 1, 451–463. [Google Scholar] [CrossRef]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting cardiac cellular composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef]
- Castro, L.; Geertz, B.; Reinsch, M.; Aksehirlioglu, B.; Hansen, A.; Eschenhagen, T.; Reichenspurner, H.; Weinberger, F.; Pecha, S. Implantation of hiPSC-derived cardiac-muscle patches after myocardial injury in a Guinea pig model. J. Vis. Exp. 2019, 145, e58810. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, M.D.; Mannhardt, I.; Breckwoldt, K.; Prondzynski, M.; Flenner, F.; Ulmer, B.; Hirt, M.N.; Neuber, C.; Horvath, A.; Kloth, B.; et al. Human iPSC-derived cardiomyocytes cultured in 3D engineered heart tissue show physiological upstroke velocity and sodium current density. Sci. Rep. 2017, 7, 5464. [Google Scholar] [CrossRef] [PubMed]
- Breckwoldt, K.; Letuffe-Breniere, D.; Mannhardt, I.; Schulze, T.; Ulmer, B.; Werner, T.; Benzin, A.; Klampe, B.; Reinsch, M.C.; Laufer, S.; et al. Differentiation of cardiomyocytes and generation of human engineered heart tissue. Nat. Protoc. 2017, 12, 1177–1197. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, F.; Mannhardt, I.; Eschenhagen, T. Engineering cardiac muscle tissue: A maturating field of research. Circ. Res. 2017, 120, 1487–1500. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.; Tiburcy, M.; Zimmermann, W.H. Cardiac macrotissues-on-a-plate models for phenotypic drug screens. Adv. Drug Deliv. Rev. 2019, 140, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Garber, K. RIKEN suspends first clinical trial involving induced pluripotent stem cells. Nat. Biotechnol. 2015, 33, 890–891. [Google Scholar] [CrossRef] [PubMed]
- Mayshar, Y.; Ben-David, U.; Lavon, N.; Biancotti, J.C.; Yakir, B.; Clark, A.T.; Plath, K.; Lowry, W.E.; Benvenisty, N. Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell 2010, 7, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, M.; Hayashizaki, Y.; Murakawa, Y. Genomic instability of iPSCs: Challenges towards their clinical applications. Stem Cell Rev. 2017, 13, 7–16. [Google Scholar] [CrossRef]
- Liu, P.; Kaplan, A.; Yuan, B.; Hanna, J.H.; Lupski, J.R.; Reiner, O. Passage number is a major contributor to genomic structural variations in mouse iPSCs. Stem Cells 2014, 32, 2657–2667. [Google Scholar] [CrossRef]
- Kempf, H.; Zweigerdt, R. Scalable cardiac differentiation of pluripotent stem cells using specific growth factors and small molecules. In Engineering and Application of Pluripotent Stem Cells; Springer: Cham, Switzerland, 2017; Volume 163, pp. 39–69. [Google Scholar]
- Goktepe, S.; Abilez, O.J.; Parker, K.K.; Kuhl, E. A multiscale model for eccentric and concentric cardiac growth through sarcomerogenesis. J. Theor. Biol. 2010, 265, 433–442. [Google Scholar] [CrossRef]
- Gherghiceanu, M.; Barad, L.; Novak, A.; Reiter, I.; Itskovitz-Eldor, J.; Binah, O.; Popescu, L.M. Cardiomyocytes derived from human embryonic and induced pluripotent stem cells: Comparative ultrastructure. J. Cell. Mol. Med. 2011, 15, 2539–2551. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Park, P.; Hong, S.M.; Ban, K. Maturation of cardiomyocytes derived from human pluripotent stem cells: Current strategies and limitations. Mol. Cells 2018, 41, 613–621. [Google Scholar] [PubMed]
- Koivumaki, J.T.; Naumenko, N.; Tuomainen, T.; Takalo, J.; Oksanen, M.; Puttonen, K.A.; Lehtonen, S.; Kuusisto, J.; Laakso, M.; Koistinaho, J.; et al. Structural immaturity of human iPSC-derived cardiomyocytes: In silico investigation of effects on function and disease modeling. Front. Physiol. 2018, 9, 80. [Google Scholar] [CrossRef]
- Bedada, F.B.; Wheelwright, M.; Metzger, J.M. Maturation status of sarcomere structure and function in human iPSC-derived cardiac myocytes. Biochim. Biophys. Acta 2016, 1863, 1829–1838. [Google Scholar] [CrossRef] [PubMed]
- Vermij, S.H.; Abriel, H.; van Veen, T.A. Refining the molecular organization of the cardiac intercalated disc. Cardiovasc. Res. 2017, 113, 259–275. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, M.; Yan, P.; Otsuji, T.G.; Narazaki, G.; Uosaki, H.; Fukushima, H.; Kuwahara, K.; Harada, M.; Matsuda, H.; Matsuoka, S.; et al. Induction and enhancement of cardiac cell differentiation from mouse and human induced pluripotent stem cells with cyclosporin-A. PLoS ONE 2011, 6, e16734. [Google Scholar] [CrossRef]
- Zuppinger, C.; Gibbons, G.; Dutta-Passecker, P.; Segiser, A.; Most, H.; Suter, T.M. Characterization of cytoskeleton features and maturation status of cultured human iPSC-derived cardiomyocytes. Eur. J. Histochem. 2017, 61, 2763. [Google Scholar] [CrossRef]
- Scuderi, G.J.; Butcher, J. Naturally engineered maturation of cardiomyocytes. Front. Cell Dev. Biol. 2017, 5, 50. [Google Scholar] [CrossRef]
- Brodsky, V.; Chernyaev, A.L.; Vasilyeva, I.A. Variability of the cardiomyocyte ploidy in normal human hearts. Virchows Archiv. B 1991, 61, 289–294. [Google Scholar] [CrossRef]
- Feric, N.T.; Radisic, M. Maturing human pluripotent stem cell-derived cardiomyocytes in human engineered cardiac tissues. Adv. Drug Deliv. Rev. 2016, 96, 110–134. [Google Scholar] [CrossRef]
- Veerman, C.C.; Kosmidis, G.; Mummery, C.L.; Casini, S.; Verkerk, A.O.; Bellin, M. Immaturity of human stem-cell-derived cardiomyocytes in culture: Fatal flaw or soluble problem? Stem Cells Dev. 2015, 24, 1035–1052. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.D.; Tinney, J.P.; Ye, F.; Elnakib, A.A.; Yuan, F.; El-Baz, A.; Sethu, P.; Keller, B.B.; Giridharan, G.A. Effects of physiologic mechanical stimulation on embryonic chick cardiomyocytes using a microfluidic cardiac cell culture model. Anal. Chem. 2015, 87, 2107–2113. [Google Scholar] [CrossRef] [PubMed]
- Sarig, U.; Sarig, H.; de-Berardinis, E.; Chaw, S.Y.; Nguyen, E.B.; Ramanujam, V.S.; Thang, V.D.; Al-Haddawi, M.; Liao, S.; Seliktar, D.; et al. Natural myocardial ECM patch drives cardiac progenitor based restoration even after scarring. Acta Biomater. 2016, 44, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.C.; Wang, Z.; Missinato, M.A.; Park, D.W.; Long, D.W.; Liu, H.J.; Zeng, X.; Yates, N.A.; Kim, K.; Wang, Y. Decellularized zebrafish cardiac extracellular matrix induces mammalian heart regeneration. Sci. Adv. 2016, 2, e1600844. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Majdi, M.; Xia, P.; Wei, K.A.; Talantova, M.; Spiering, S.; Nelson, B.; Mercola, M.; Chen, H.S. Non-cardiomyocytes influence the electrophysiological maturation of human embryonic stem cell–derived cardiomyocytes during differentiation. Stem Cells Dev. 2010, 19, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Kolanowski, T.J.; Antos, C.L.; Guan, K. Making human cardiomyocytes up to date: Derivation, maturation state and perspectives. Int. J. Cardiol. 2017, 241, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Glazier, A.A.; Thompson, A.; Day, S.M. Allelic imbalance and haploinsufficiency in MYBPC3-linked hypertrophic cardiomyopathy. Pflugers Archiv Eur. J. Physiol. 2019, 471, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Clemen, C.S.; Stockigt, F.; Strucksberg, K.H.; Chevessier, F.; Winter, L.; Schutz, J.; Bauer, R.; Thorweihe, J.M.; Wenzel, D.; Schlotzer-Schrehardt, U.; et al. The toxic effect of R350P mutant desmin in striated muscle of man and mouse. Acta Neuropathol. 2015, 129, 297–315. [Google Scholar] [CrossRef]
- Sugawara, M.; Kato, K.; Komatsu, M.; Wada, C.; Kawamura, K.; Shindo, P.S.; Yoshioka, P.N.; Tanaka, K.; Watanabe, S.; Toyoshima, I. A novel de novo mutation in the desmin gene causes desmin myopathy with toxic aggregates. Neurology 2000, 55, 986–990. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Marti-Gutierrez, N.; Park, S.W.; Wu, J.; Lee, Y.; Suzuki, K.; Koski, A.; Ji, D.; Hayama, T.; Ahmed, R.; et al. Correction of a pathogenic gene mutation in human embryos. Nature 2017, 548, 413–419. [Google Scholar] [CrossRef]
- Echigoya, Y.; Lim, K.R.Q.; Nakamura, A.; Yokota, T. Multiple Exon skipping in the duchenne muscular dystrophy hot spots: Prospects and challenges. J. Personal. Med. 2018, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; van Ommen, G.J. Antisense-mediated exon skipping: A versatile tool with therapeutic and research applications. Rna 2007, 13, 1609–1624. [Google Scholar] [CrossRef] [PubMed]
- Dick, E.; Kalra, S.; Anderson, D.; George, V.; Ritso, M.; Laval, S.H.; Barresi, R.; Aartsma-Rus, A.; Lochmuller, H.; Denning, C. Exon skipping and gene transfer restore dystrophin expression in human induced pluripotent stem cells-cardiomyocytes harboring DMD mutations. Stem Cells Dev. 2013, 22, 2714–2724. [Google Scholar] [CrossRef] [PubMed]
- Van Deutekom, J.C.; Janson, A.A.; Ginjaar, I.B.; Frankhuizen, W.S.; Aartsma-Rus, A.; Bremmer-Bout, M.; den Dunnen, J.T.; Koop, K.; van der Kooi, A.J.; Goemans, N.M.; et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N. Engl. J. Med. 2007, 357, 2677–2686. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Long, C.; Li, H.; McAnally, J.R.; Baskin, K.K.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. CRISPR-Cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice. Sci. Adv. 2017, 3, e1602814. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.L.; Li, H.; Rodriguez-Caycedo, C.; Mireault, A.A.; Huang, J.; Shelton, J.M.; McAnally, J.R.; Amoasii, L.; Mammen, P.P.A.; Bassel-Duby, R.; et al. CRISPR-Cas9 corrects Duchenne muscular dystrophy exon 44 deletion mutations in mice and human cells. Sci. Adv. 2019, 5, eaav4324. [Google Scholar] [CrossRef] [PubMed]
- White, J.A.; McAlpine, P.J.; Antonarakis, S.; Cann, H.; Eppig, J.T.; Frazer, K.; Frezal, J.; Lancet, D.; Nahmias, J.; Pearson, P.; et al. Guidelines for human gene nomenclature (1997). HUGO nomenclature committee. Genomics 1997, 45, 468–471. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
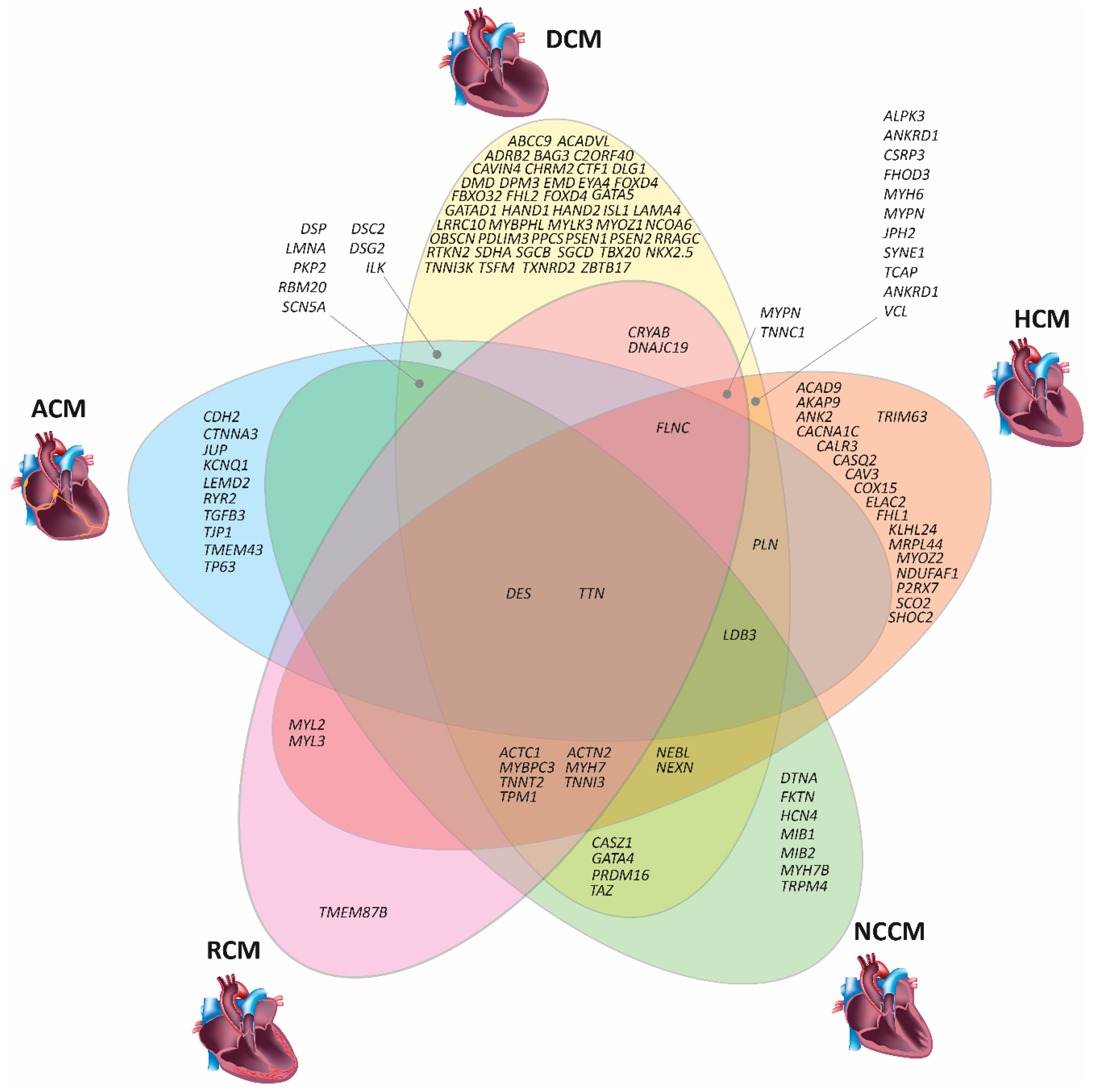{kind=link}
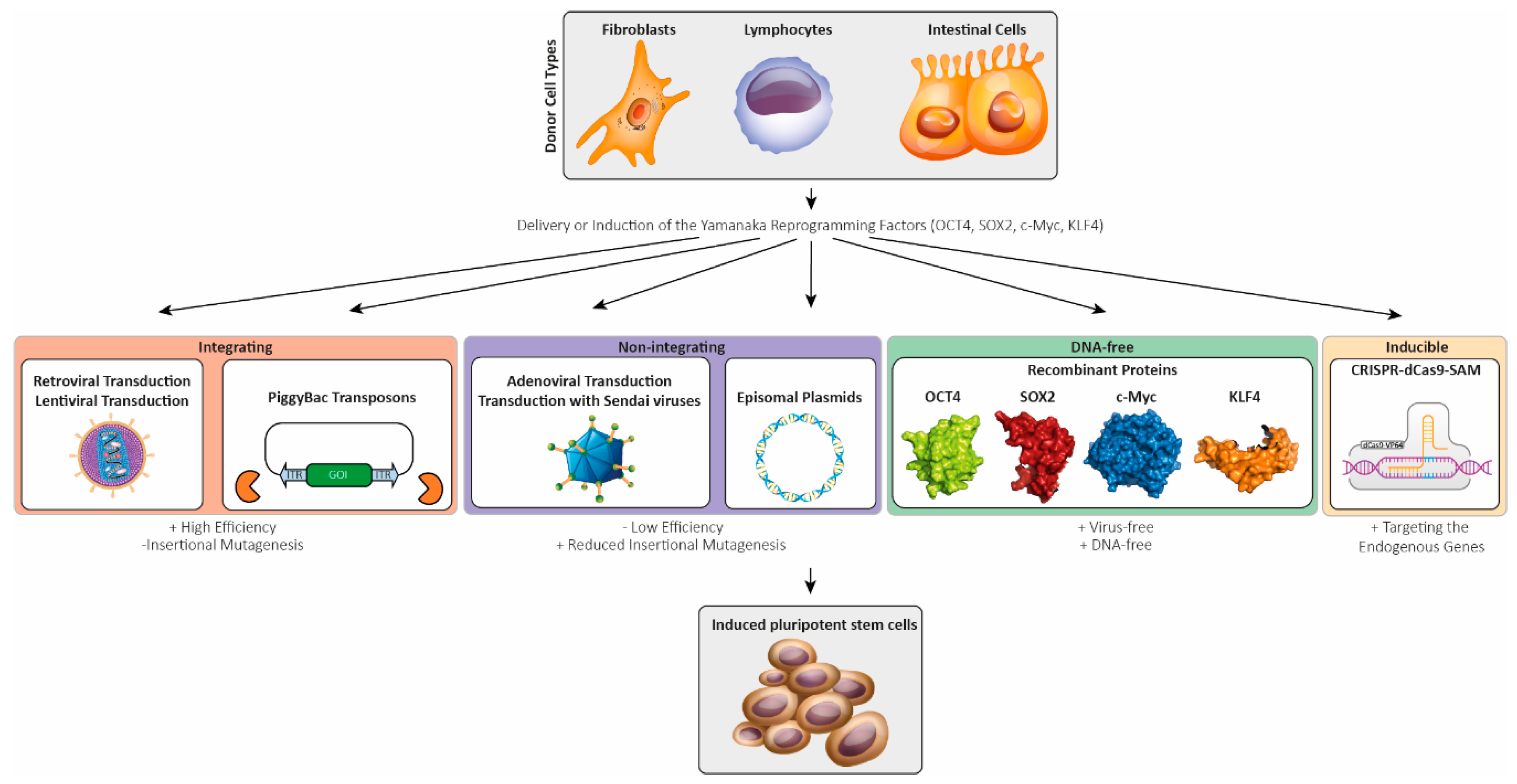{kind=link}
| Gene | Protein | Function | HCM | DCM | NCCM | ACM | RCM |
|---|---|---|---|---|---|---|---|
| ABCC9 | ATP Binding Cassette Subfamily C Member 9 | ABC transporter | [77] | ||||
| ACAD9 | Acyl-CoA Dehydrogenase Member 9 | Dehydrogenase | [78] | ||||
| ACADVL | Acyl-CoA Dehydrogenase Very Long Chain | Dehydrogenase | [79] | ||||
| ACTC1 | Cardiac Actin | Sarcomere protein | [43,80] | [81] | [82] | [83] | |
| ACTN2 | α-Actinin 2 | Z-band protein | [84] | [85] | [86] | [69] | |
| ADRB2 | Adrenoreceptor β2 | G-protein coupled receptor | [87] | ||||
| AKAP9 | A Kinase Anchoring Protein 9 | Scaffolding protein | [88] | ||||
| ALMS1 | Alstrom Syndrome Protein 1 | Microtubule organization | [89] 1 | ||||
| ALPK3 | α-Kinase 3 | Kinase | [90] | [90] | |||
| ANK2 | Ankyrin 2 | Cytoskeleton linker protein | [91] | [92] | |||
| ANKRD1 | Ankyrin Repeat Domain Containing Protein 1 | Transcription factor | [93] | [94,95] | |||
| BAG3 | Bcl-2 Associated Athanogene 3 | Co-chaperone | [96] | [69,97] | |||
| BRAF | B-Raf Proto-Oncogene, Serine/Threonine Kinase | Kinase | [98] 2 | ||||
| C2ORF40 | Chromosome 2 Open Reading Frame 40 | Hormone | [99] | ||||
| CACNA1C | Calcium Voltage-Gated Channel Subunit α1C | Calcium channel | [100] | ||||
| CALM3 | Calmodulin 3 | Calcium binding | [101] 3 | ||||
| CALR3 | Calreticulin 3 | Calcium binding chaperone | [46] | ||||
| CASQ2 | Calsequestrin 2 | Calcium binding | [46] | ||||
| CASZ1 | Castor Zinc Finger 1 | Transcription factor | [102] | [103] | |||
| CAV3 | Caveolin 3 | Scaffolding protein | [104] | ||||
| CAVIN4 | Muscle Restricted Coiled Coil Protein | Myofibrillar organization | [105] | ||||
| CDH2 | N-Cadherin | Cell–cell adhesion | [106,107] | ||||
| CHRM2 | Cholinergic Receptor Muscarinic 2 | G-protein coupled receptor | [108] | ||||
| COL3A1 | Collagen Type III Alpha 1 Chain | Extra cellular matrix protein | [109] 4 | ||||
| COX15 | Cytochrome C Oxidase Assembly Homolog COX15 | Mitochondrial respiratory chain | [110] | ||||
| CRYAB | αB-Crystallin | Chaperone-like activity | [111] | [112] | |||
| CSRP3 | Muscle LIM Protein | Scaffolding protein | [113,114,115] | [116] | |||
| CTF1 | Cardiotrophin 1 | Cytokine | [117] | ||||
| CTNNA3 | αT-Catenin | Cell–cell adhesion | [118] | ||||
| DES | Desmin | Intermediate filament protein | [25] | [24,119] | [30] | [26] | [28] |
| DLG1 | Discs Large MAGUK Scaffold Protein 1 | Scaffolding protein | [88] | ||||
| DMD | Dystrophin | Dystrophin–glycoprotein complex | [120] | ||||
| DNAJC19 | DNAJ Heat Shock Protein Family C19 | Co-chaperone | [121] | [121] | |||
| DOLK | Dolichol Kinase | Phosphorylation of dolichol | [122] 5 | ||||
| DPM3 | Dolichyl-Phosphate Mannosyltransferase Subunit 3 | Mannosyltransferase | [123] | ||||
| DSC2 | Desmocollin 2 | Cell–cell adhesion | [35] | [124] | |||
| DSG2 | Desmoglein 2 | Cell–cell adhesion | [125] | [126,127] | |||
| DSP | Desmoplakin | Cell–cell adhesion | [128] | [129] | [130] | ||
| DTNA | α-Dystrobrevin | Dystrophin-glycoprotein complex | [131] | ||||
| ELAC2 | ElaC Ribonuclease Z2 | 3′-tRNA endoribonuclease | [132] | ||||
| EMD | Emerin | Nuclear lamina associated protein | [133] | ||||
| EYA4 | Eyes Absent Homolog 4 | Transcription factor | [51] | ||||
| FBN1 | Fibrillin 1 | Extra cellular matrix protein | [134] 6 | [135] 7 | [136] 7 | ||
| FBXO32 | F-Box Only Protein 32 | Ubiquitin–protein ligase complex | [137,138] | ||||
| FHL1 | Four and a Half LIM Domain Protein 1 | Scaffolding protein | [41] | ||||
| FHL2 | Four and a Half LIM Domain Protein 2 | Scaffolding protein | [139] | ||||
| FHOD3 | Formin Homology 2 Domain Containing Protein 3 | Organization of actin-polymerization | [140] | [141] | |||
| FKRP | Fukutin Related Protein | Posttranslational modification of dystroglycan | [142] 8 | ||||
| FKTN | Fukutin | Glycosyltransferase of dystroglycan | [143] | ||||
| FLNC | Filamin C | Cell junction organization | [45] | [144,145] | [145] | [72] | |
| FOXD4 | Forkhead Box Protein D4 | Transcription factor | [146] | ||||
| FXN | Frataxin | Regulation of mitochondrial iron transport | [147] 9 | ||||
| GAA | α-Glucosidase | Glycogen metabolism | [148] 9 | ||||
| GATA4 | GATA Binding Protein 4 | Transcription factor | [149] | [150] 10 | |||
| GATA5 | GATA Binding Protein 5 | Transcription factor | [151] | ||||
| GATAD1 | GATA Zink Finger Domain Containing Protein 1 | Gene expression regulation | [152] | ||||
| GLA | Galactosidase α | Galactose metabolism | [153] 11 | ||||
| GTPBP3 | GTP Binding Protein 3, Mitochondrial | Mitochondrial tRNA modification | [154] 12 | ||||
| HAND1 | Heart and Neural Crest Derivatives Expressed 1 | Transcription factor | [155] | ||||
| HAND2 | Heart and Neural Crest Derivatives Expressed 2 | Transcription factor | [156] | ||||
| HCN4 | Hyperpolarization Activated Cyclic Nucleotide Gated Potassium Channel 4 | Potassium channel | [157] | ||||
| HRAS | HRas Proto-Oncogene GTPase | Signaling protein | [158]13 | ||||
| ILK | Integrin Linked Kinase | Scaffolding protein | [159,160] | [68] | |||
| ISL1 | ISL LIM Homeobox 1 | Transcription factor | [161] | ||||
| ITGA7 | Integrin Subunit A7 | Cell–cell and cell–matrix junction protein | [162] 14 | ||||
| ITPA | Inosine Triphosphate Pyrophosphatase | Nucleotide metabolism | [163] 15 | ||||
| JPH2 | Junctophilin 2 | Junctional complex | [164] | [165] | |||
| JUP | Plakoglobin | Cell–cell adhesion | [58] | ||||
| KCNQ1 | Potassium Channel Voltage Gated KQT-Like Subfamily Member 1 | Potassium channel | [166] | ||||
| KLHL24 | Kelch Like 24 | Ubiquitin ligase substrate receptor | [167] | ||||
| LAMA4 | Laminin α4 | Extra cellular matrix protein | [159] | ||||
| LAMP2 | Lysosomal Associated Membrane Protein 2 | Chaperone-mediated autophagy | [168] 16 | ||||
| LDB3 | LIM Domain Binding Protein 3 | Z-band protein | [169] | [170,171] | [170,172] | [173] | |
| LEMD2 | LEM Domain Containing Protein 2 | Nuclear lamina associated protein | [64,174] 17 | ||||
| LMNA | Lamin A/C | Nuclear lamina associated protein | [49] | [175] | [63] | ||
| LRRC10 | Leucine Rich Repeat Containing Protein 10 | Actin and α-actinin binding protein | [176] | ||||
| MIB1 | Mindbomb Drosophila Homolog 1 | Ubiquitin ligase | [177] | ||||
| MIB2 | Mindbomb Drosophila Homolog 2 | Ubiquitin ligase | [178] 18 | ||||
| MRPL3 | Mitochondrial Ribosomal Protein L3 | Mitochondrial ribosomal protein | [179] 19 | ||||
| MRPL44 | Mitochondrial Ribosomal Protein L44 | Mitochondrial ribosomal protein | [180,181] | ||||
| MYBPC3 | Myosin Binding Protein C3 | Sarcomere protein | [182,183] | [184] | [185] | [186] | |
| MYBPHL | Myosin Binding Protein H-Like | Sarcomere protein | [187] | ||||
| MYH6 | Myosin Heavy Chain 6 | Sarcomere protein | [188] | [188] | |||
| MYH7 | Myosin Heavy Chain 7 | Sarcomere protein | [22] | [48] | [7] | [189] | |
| MYH7B | Myosin Heavy Chain 7B | Sarcomere protein | [162] 20 | ||||
| MYL2 | Myosin Light Chain 2 | Sarcomere protein | [190] | [191] | |||
| MYL3 | Myosin Light Chain 3 | Sarcomere protein | [192] | [192] | |||
| MYLK3 | Myosin Light Chain Kinase 3 | Kinase | [193] | ||||
| MYOZ1 | Myozenin 1 | Calcineurin interacting protein | [194] | ||||
| MYOZ2 | Myozenin 2 | Calcineurin interacting protein | [195] | ||||
| MYPN | Myopalladin | Z-band protein | [196] | [94,197] | [196,198] | ||
| NCOA6 | Nuclear Receptor Coactivator 6 | Gene expression regulation | [199] | ||||
| NDUFAF1 | NADH: Ubiquinone Oxidoreductase Complex Assembly Factor 1 | Mitochondrial respiratory chain | [200] | ||||
| NDUFV2 | NADH: Ubiquinone Oxidoreductase Core Subunit V2 | Mitochondrial respiratory chain | [201,202] 21 | ||||
| NEBL | Nebulette | Z-band protein | [203] | [204] | [203] | ||
| NEXN | Nexilin | Sarcomere protein | [205] | [206] | [207] | ||
| NKX2.5 | NK2 Homeobox 5 | Transcription factor | [208] | ||||
| OBSCN | Obscurin | Scaffolding protein | [209] | ||||
| P2RX7 | Purinergic receptor P2X7 | ATP gated ion channel | [210] | ||||
| PDLIM3 | PDZ And LIM Domain 3 | Z-band protein | [194] | ||||
| PKP2 | Plakophilin 2 | Cell-cell adhesion | [35] | [211] | [212] | ||
| PLN | Phospholamban | Regulator of SERCA | [46] | [213,214] | [67] | ||
| PPCS | Phosphopantothenoylcystein Synthetase | Co-enzyme A synthesis | [215] | ||||
| PRDM16 | PR Domain Containing Protein 16 | Transcription factor | [216] | [217] | |||
| PRKAG2 | Protein Kinase AMP Activated Non-catalytic G2 | Energy sensor kinase | [218,219] 22 | ||||
| PSEN1 | Presenilin 1 | γ-Secretase | [220,221] | ||||
| PSEN2 | Presenilin 2 | γ-Secretase | [220] | ||||
| PTEN | Phosphatase and Tensin Homolog | Phosphatase | [150] 23 | ||||
| PTPN11 | Protein Tyrosine Phosphatase Non-Receptor Type 1 | Phosphatase | [222] 24 | ||||
| RAF1 | Raf-1 Proto-Oncogene, Serine/Threonine Kinase | Kinase | [223,224] 25 | [225] | |||
| RBM20 | RNA Binding Protein 20 | Splicing factor | [52,226] | [227] | [228,229] | ||
| RRAGC | Ras Related GTP Binding C | GTR/RAG GTP-binding protein | [230] | ||||
| RTKN2 | Rhotekin 2 | Scaffolding protein | [99] | ||||
| RYR2 | Ryanodine Receptor 2 | Calcium channel | [66] | ||||
| SCN5A | Sodium Channel Voltage Gated Type V Subunit A | Sodium channel | [50,231] | [232] | |||
| SCO2 | SCO2 Cytochrome C Oxidase Assembly Protein | Metallo-chaperone | [233] | ||||
| SDHA | Succinate Dehydrogenase Complex Subunit A | Mitochondrial respiratory chain | [234] | ||||
| SGCB | Sarcoglycan β | Dystrophin-glycoprotein complex | [235] | ||||
| SGCD | Sarcoglycan δ | Dystrophin-glycoprotein complex | [236] | ||||
| SHOC2 | Suppressor Of Clear, C. Elegans, Homolog | Scaffolding protein | [237] | ||||
| SYNE1 | Nesprin 1 | Component of the LINC complex | [238] | [239] | |||
| TAZ | Tafazzin | Cardiolipin metabolism | [240] 26 | [241,242] | |||
| TBX20 | T-Box Factor 20 | Transcription factor | [243,244] | ||||
| TCAP | Thelethonin | Titin binding | [245] | [244,245] | |||
| TGFB3 | Transforming Growth Factor β3 | Growth factor | [246] | ||||
| TJP1 | Zonula Occludens 1 | Tight junction adapter protein | [247] | ||||
| TMEM43 | Transmembrane Protein 43 | Nuclear lamina associated protein | [9,10] | ||||
| TMEM87B | Transmembrane Protein 87B | Endosome-to-trans-Golgi retrograde transport | [248] | ||||
| TNNC1 | Cardiac Troponin C | Sarcomere protein | [39] | [249] | [250] | ||
| TNNI3 | Cardiac Troponin I | Sarcomere protein | [40] | [251] | [252] | [71] | |
| TNNI3K | TNNI3 Interacting Kinase | Kinase | [253] | ||||
| TNNT2 | Cardiac Troponin T | Sarcomere protein | [38] | [254] | [255] | [83] | |
| TP63 | Tumor Protein 63 | Transcription factor | [256] | ||||
| TPM1 | Tropomyosin 1 | Sarcomere protein | [38,257] | [258] | [259] | [191] | |
| TRIM63 | Tripartite Motif Containing Protein 63 | Ubiquitin ligase | [260] | ||||
| TRPM4 | Transient Receptor Potential Cation Channel Subfamily M | Cation channel | [261] | ||||
| TSFM | Mitochondrial Translation Elongation Factor Ts | Translation elongation factor | [262] | ||||
| TTN | Titin | Sarcomere protein | [263] | [32,264] | [87,265] | [33] | [34] |
| TTR | Transthyretin | Carrier protein | [266,267] 27 | ||||
| TXNRD2 | Thioredoxin Reductase 2 | Reduces thioredoxins | [268] | ||||
| VCL | Vinculin | Cell–cell and cell–matrix junction protein | [269,270] | [271] | |||
| ZBTB17 | Zinc Finger and BTB Domain Containing Protein 17 | Transcription factor | [272,273] |
| Gene | Protein | Mutation(s) | Method of Generation | Main Phenotypic Findings | Associated Disease | References |
|---|---|---|---|---|---|---|
| ACTC1 | Cardiac Actin | p.E99K |
| Arrhythmias | HCM/LVNC | [380] |
| ALPK3 | α-Kinase 3 | p.W1264Xhom | Electroporation with episomal plasmids |
| HCM | [381] |
| BAG3 | Bcl-2 Associated Athanogene 3 |
|
|
| DCM | [374] |
| BRAF | B-Raf Proto-Oncogene, Serine/Threonine Kinase |
|
|
| CFCS/HCM | [382] |
| CAV | Caveolin |
| Electroporation with episomal plasmids | NA | MP | [383] |
| CRYAB | αB-Crystallin |
| Retroviral transduction and genome editing (zinc finger nucleases) |
| MFM | [384] |
| DES | Desmin | p.N116S | Lentiviral transduction | NA | ACM | [385] |
| DES | Desmin | c.735+1G > A | Sendai virus transduction | NA | DRC | [386] |
| DES | Desmin | p.A285V | Retroviral transduction |
| DCM | [387] |
| DMD | Dystrophin |
| Sendai virus transduction |
| DMD | [388] |
| DMD | Dystrophin |
| Sendai virus transduction in combination with CRISPR-Cas9 |
| DMD | [379] |
| DMD | Dystrophin |
| Lentiviral transduction CRISPR-Cas9 |
| DMD | [389,390] |
| DSG2 | Desmoglein-2 | p.G638R | Sendai virus transduction |
| ACM | [391] |
| DSP | Desmoplakin | p.R451G | Sendai virus transduction & genome editing for correction (CRISPR-Cas9) | Reduced desmoplakin expression | ACM | [392] |
| FBN1 | Fibrillin 1 | c.4028G > A | Sendai virus transduction | NA | Marfan Syndrome (HCM) | [393] |
| FKRP | Fukutin Related Protein | c.826C > Ahom | Lentiviral transduction |
| Limb-Girdle Muscular Dystrophy (DCM) | [394] |
| FXN | Frataxin | Expanded GAA repeats | Retroviral transduction |
| Friedreich Ataxia (HCM) | [395] |
| FXN | Frataxin | Expanded GAA repeats
| Lentiviral transduction |
| Friedreich Ataxia (HCM) | [396] |
| GLA | Galactosidase α | IVS4+919G > A | Retroviral transduction |
| Fabry Disease (HCM) | [397,398] |
| LAMP2 | Lysosomal Associated Membrane Protein 2 | IVS6+1_4delGTGA | Sendai virus transduction | Autophagy dysfunction | Danon Disease (CM) | [399] |
| LAMP2 | Lysosomal Associated Membrane Protein 2 |
| Unknown |
| Danon Disease (CM) | [400] |
| LAMP2 | Lysosomal Associated Membrane Protein 2 |
|
|
| Danon Disease (CM) | [401] |
| LMNA | Lamin A/C | p.S143P | Sendai virus transduction |
| DCM | [402] |
| LMNA | Lamin A/C | p.S18fsX | Combined lentiviral and retroviral transduction | Normal nuclear membrane morphology | DCM | [403] |
| LMNA | Lamin A/C | p.R225X | Lentiviral transduction |
| DCM | [404] |
| LMNA | Lamin A/C |
| Lentiviral transduction |
| DCM & conduction disorders | [405] |
| LMNA | Lamin A/C | p.K219T | Lentiviral transduction |
| DCM & conduction disorders | [406] |
| MT-RNR2 | Mitochondrially Encoded 16S rRNA | m.2336T > C | Retroviral transduction |
| HCM | [407] |
| MYBPC3 | Myosin Binding Protein C3 |
| Sendai virus transduction | Abnormal Ca2+ handling | HCM | [408] |
| MYBPC3 | Myosin Binding Protein C3 | p.R326Q | Electroporation with episomal plasmids | Ca2+ handling deficits | HCM | [409] |
| MYBPC3 | Myosin Binding Protein C3 | c.2373 | Lentiviral transduction |
| HCM | [410,411] |
| MYBPC3 | Myosin Binding Protein C3 | p.R502W | Electroporation with episomal plasmids | NA | HCM | [412] |
| MYBPC3 | Myosin Binding Protein C3 |
| CRISPR-Cas9 |
| HCM | [413] |
| MYBPC3 | Myosin Binding Protein C3 |
| Sendai virus transduction & genome editing for correction (CRISPR-Cas9) |
| HCM | [414,415] |
| MYBPC3 | Myosin Binding Protein C3 | p.G999-Q1004del | Sendai virus transduction |
| HCM | [416] |
| MYBPC3 | Myosin Binding Protein C3 | p.Q1061X |
| Arrhythmias | HCM | [417,418] |
| MYBPC3 | Myosin Binding Protein C3 | p.V454CfsX21 | Retroviral transduction |
| HCM | [419] |
| MYBPC3 | Myosin Binding Protein C3 | ∆25 bp in intron 32 including the splicing branch point & p.D389V (same allele) | Sendai virus transduction |
| HCM | [420] |
| MYBPHL | Myosin Binding Protein H-Like | p.R255X | Electroporation with episomal plasmids | Haploinsufficiency by nonsense mediated mRNA decay | DCM & conduction disorders | [187] |
| MYH7 | Myosin Heavy Chain 7 | p.R663H | Sendai virus transduction | Abnormal Ca2+ handling | HCM | [408] |
| MYH7 | Myosin Heavy Chain 7 |
| CRISPR-Cas9 |
| HCM | [421] |
| MYH7 | Myosin Heavy Chain 7 |
| CRISPR-Cas9 |
| HCM | [413] |
| MYH7 | Myosin Heavy Chain 7 | p.V698A | Electroporation with episomal plasmids | NA | HCM | [422] |
| MYH7 | Myosin Heavy Chain 7 | p.E848G | Electroporation with episomal plasmids | Reduced contractile function | HCM | [423,424] |
| MYH7 | Myosin Heavy Chain 7 | p.R403Q | Electroporation with episomal plasmids | NA | HCM | [425] |
| MYH7 | Myosin Heavy Chain 7 | p.R633H | Lentiviral transduction |
| HCM | [414,426] |
| MYH7 | Myosin Heavy Chain 7 | p.R442G | Retroviral transduction |
| HCM | [427] |
| MYL2 | Myosin Light Chain 2 | p.R58Q | Non-integrating mRNA/miRNA technology |
| HCM | [428] |
| MYL3 | Myosin Light Chain 3 |
| CRISPR-Cas9 |
| HCM | [375] |
| PKP2 | Plakophilin-2 | p.L614P | Retroviral transduction |
| ACM | [429] |
| PKP2 | Plakophilin-2 |
| Retroviral transduction |
| ACM | [430,431,432] |
| PKP2 | Plakophilin-2 | c.972insT | Retroviral transduction |
| ACM | [433] |
| PKP2 | Plakophilin-2 |
| Sendai virus transduction | NA | ACM | [434] |
| PKP2 | Plakophilin-2 | c.2569_3018del50 | Electroporation with episomal plasmids | NA | ACM | [435] |
| PLN | Phospholamban | p.R9C | CRISPR-Cas9 |
| DCM | [414,436] |
| PLN | Phospholamban | p.R14del | Transfection with mRNAs& genome editing (TALENs) for mutation correction |
| DCM | [437,438] |
| PRGAG2 | Protein Kinase AMP-Activated Non-Catalytic Subunit Gamma 2 | p.R302Q | Sendai virus transduction & genome editing for correction (CRISPR-Cas9) |
| Wolff–Parkinson–White Syndrome (HCM) | [439] |
| PRKAG2 | Protein Kinase AMP-Activated Non-Catalytic Subunit Gamma 2 | p.N488I | Lentiviral transduction & genome editing for correction (TALEN) |
| HCM | [440] |
| PTPN11 | Protein Tyrosine Phosphatase Non-Receptor Type 11 | p.T468M | Retroviral transduction |
| LEOPARD Syndrome (HCM) | [360] |
| PTPN11 | Protein Tyrosine Phosphatase Non-Receptor Type 11 | p.Q510P | Sendai virus transduction | NA | LEOPARD Syndrome (HCM) | [441] |
| RAF1 | Raf-1 Proto-Oncogene, Serine/Threonine Kinase | p.S257L | Electroporation of episomal plasmids & genome editing for correction (CRISPR-Cas9) |
| Noonan Syndrome (HCM) | [442] |
| RBM20 | RNA Binding Motif Protein 20 | p.S635A | Lentiviral transduction |
| DCM | [443] |
| RBM20 | RNA Binding Motif Protein 20 | p.R636S | Sendai virus transduction |
| DCM | [444,445] |
| RYR2 | Ryanodine Receptor 2 | p.F2483I | Retroviral transduction |
| CPVT | [350] |
| RYR2 | Ryanodine Receptor 2 |
| Sendai virus transduction |
| CPVT | [376] |
| SCN5A | Sodium Voltage-Gated Channel Alpha Subunit 5 |
| Sendai virus transduction & CRISPR-Cas9 for correction |
| ACM | [446] |
| SCN5A | Sodium Voltage-Gated Channel Alpha Subunit 5 | p.R219H | Sendai virus transduction |
| ACM/DCM | [447] |
| SCO2 | SCO2 Cytochrome C Oxidase Assembly Protein |
| Sendai virus transduction |
| HCM | [448] |
| TAZ | Tafazzin |
| Transfection with synthetic mRNAs & CRISPR-Cas9 for correction |
| Barth Syndrome | [449] |
| TBX20 | T-Box Factor 20 |
| Sendai virus transduction |
| LVNC | [450] |
| TNNT2 | Cardiac Troponin T | p.R92W | Sendai virus transduction & CRISPR-Cas9 for correction | Abnormal Ca2+ handling | HCM | [408] |
| TNNT2 | Cardiac Troponin T | p.R173W | Lentiviral transduction |
| DCM | [414,451,452,453,454] |
| TNNT2 | Cardiac Troponin T |
| TALEN |
| DCM/HCM | [453] |
| TNNT2 | Cardiac Troponin T | p.I79N | CRISPR-Cas9 |
| HCM | [455,456] |
| TPM1 | Tropomyosin-1 | p.D175N |
| Arrhythmias | HCM | [417,418] |
| TTN | Titin |
|
|
| DCM | [457] |
| TTN | Titin | p.S14450fsX4 | Sendai virus transduction | Antisense-mediated exon skipping restores titin expression | DCM | [377] |
| TTN | Titin |
| Lentiviral transduction |
| DCM | [458] |
| TTR | Transthyretin | p.L55P | Lentiviral transduction | Increased oxidative stress | Hereditary Transthyretin Amyloidosis | [459] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brodehl, A.; Ebbinghaus, H.; Deutsch, M.-A.; Gummert, J.; Gärtner, A.; Ratnavadivel, S.; Milting, H. Human Induced Pluripotent Stem-Cell-Derived Cardiomyocytes as Models for Genetic Cardiomyopathies. Int. J. Mol. Sci. 2019, 20, 4381. https://doi.org/10.3390/ijms20184381
Brodehl A, Ebbinghaus H, Deutsch M-A, Gummert J, Gärtner A, Ratnavadivel S, Milting H. Human Induced Pluripotent Stem-Cell-Derived Cardiomyocytes as Models for Genetic Cardiomyopathies. International Journal of Molecular Sciences. 2019; 20(18):4381. https://doi.org/10.3390/ijms20184381
Chicago/Turabian StyleBrodehl, Andreas, Hans Ebbinghaus, Marcus-André Deutsch, Jan Gummert, Anna Gärtner, Sandra Ratnavadivel, and Hendrik Milting. 2019. "Human Induced Pluripotent Stem-Cell-Derived Cardiomyocytes as Models for Genetic Cardiomyopathies" International Journal of Molecular Sciences 20, no. 18: 4381. https://doi.org/10.3390/ijms20184381
APA StyleBrodehl, A., Ebbinghaus, H., Deutsch, M.-A., Gummert, J., Gärtner, A., Ratnavadivel, S., & Milting, H. (2019). Human Induced Pluripotent Stem-Cell-Derived Cardiomyocytes as Models for Genetic Cardiomyopathies. International Journal of Molecular Sciences, 20(18), 4381. https://doi.org/10.3390/ijms20184381