Mismatch Recognition by Saccharomyces cerevisiae Msh2-Msh6: Role of Structure and Dynamics


Abstract
1. Introduction
2. Results
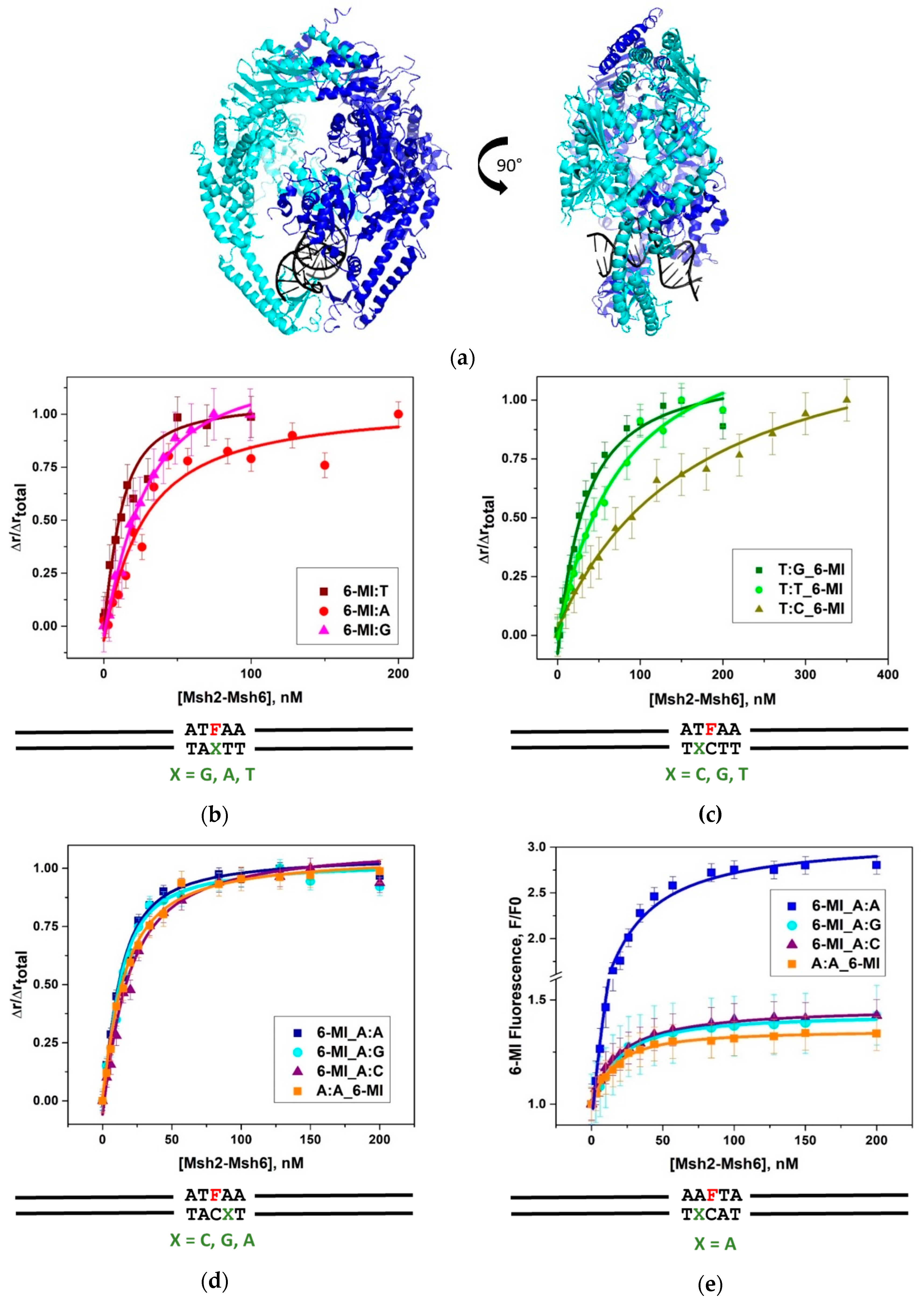
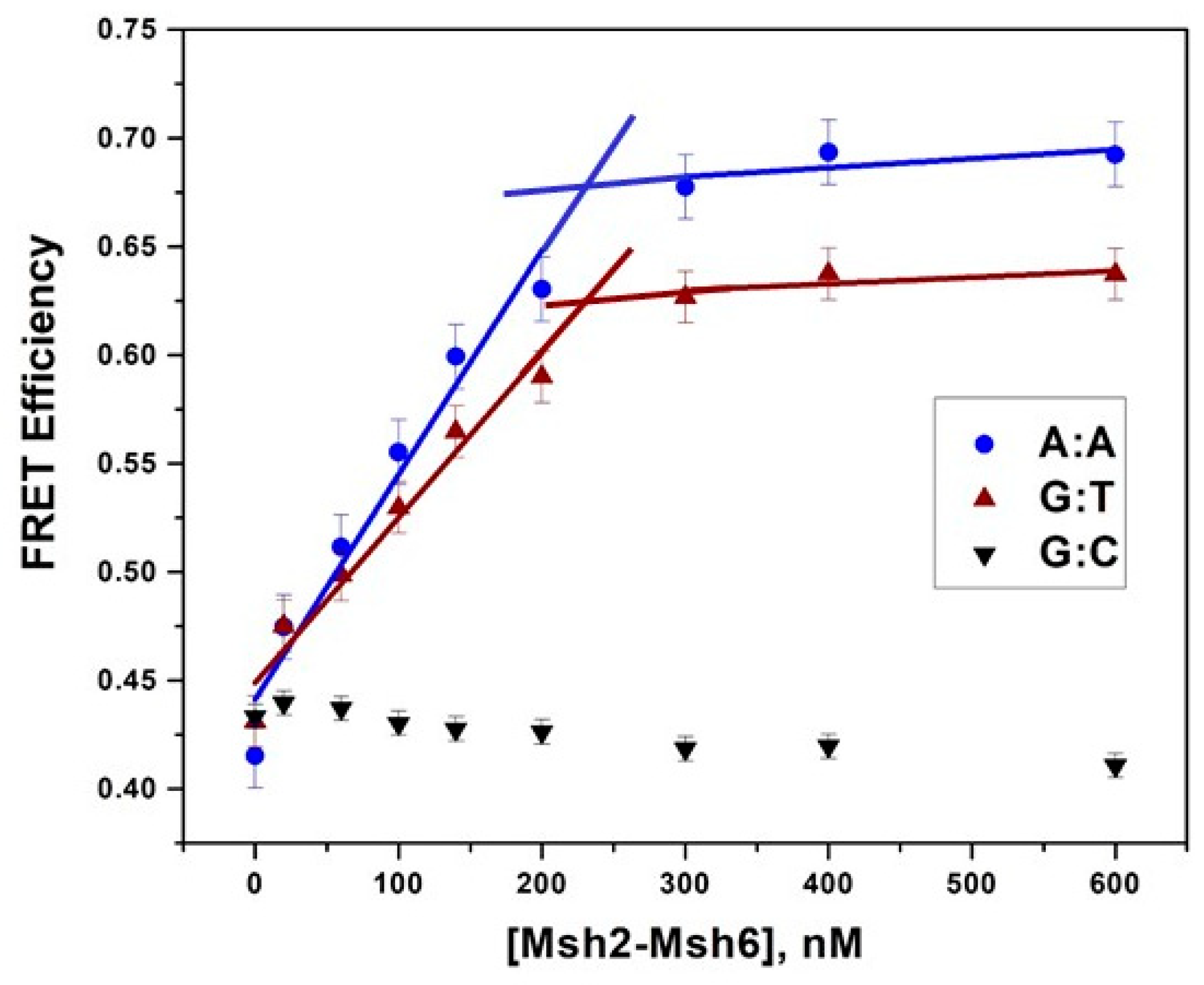
2.1. Msh2-Msh6 Recognizes Mismatched DNA Base Pairs with Different Affinities
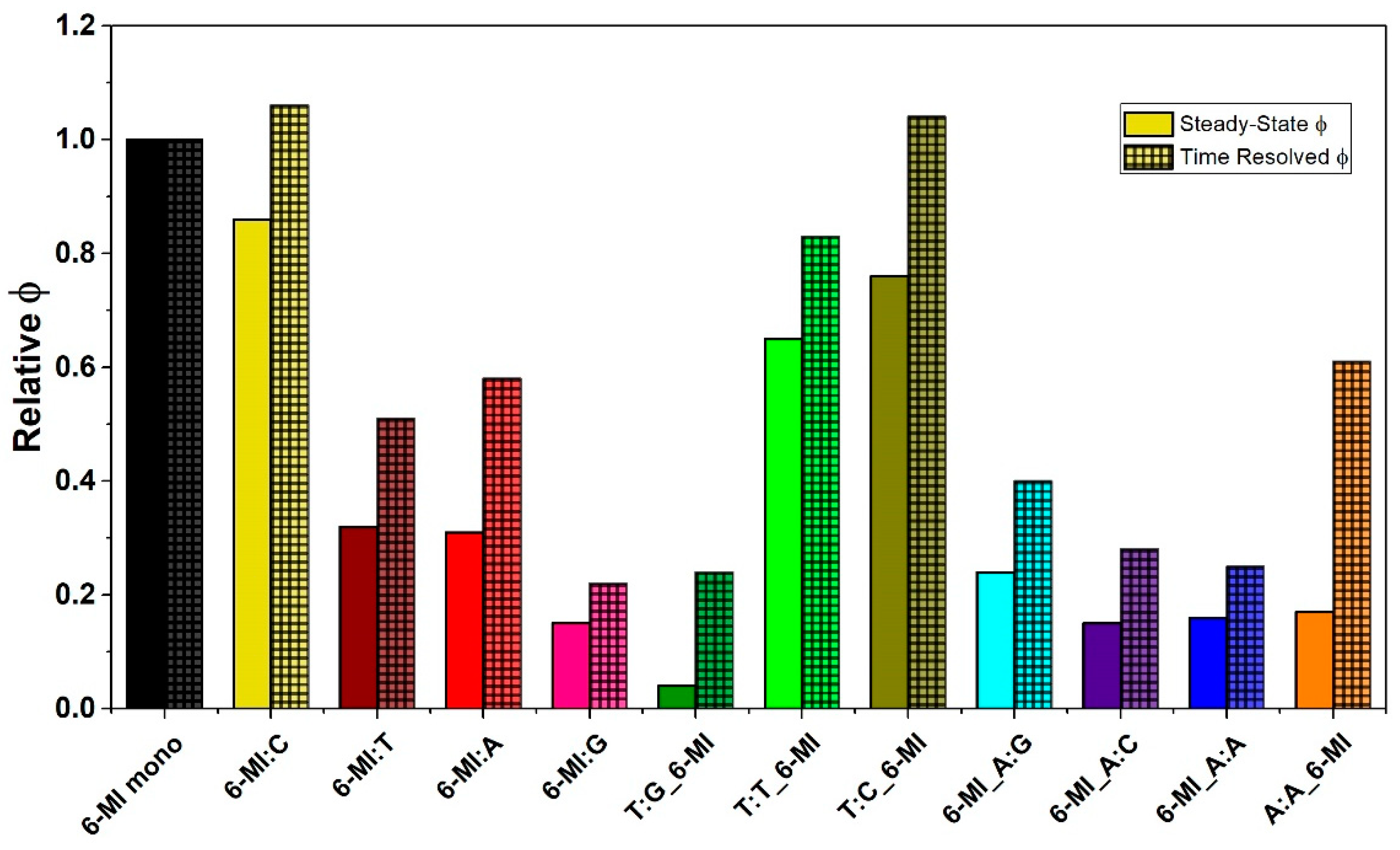
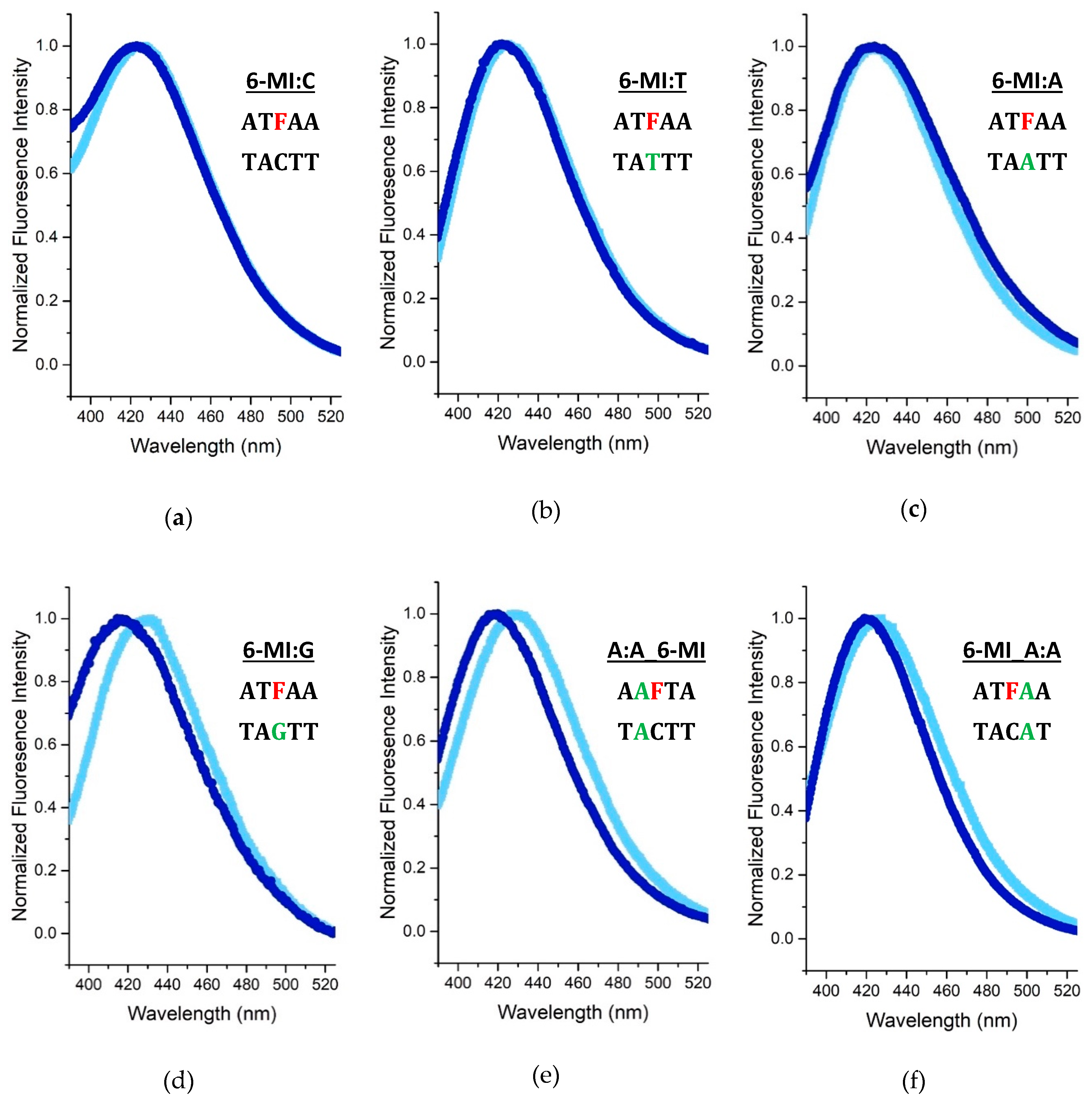
2.2. 6-MI Fluorescence Reveals Significant Variations among Mismatched Base Pairs
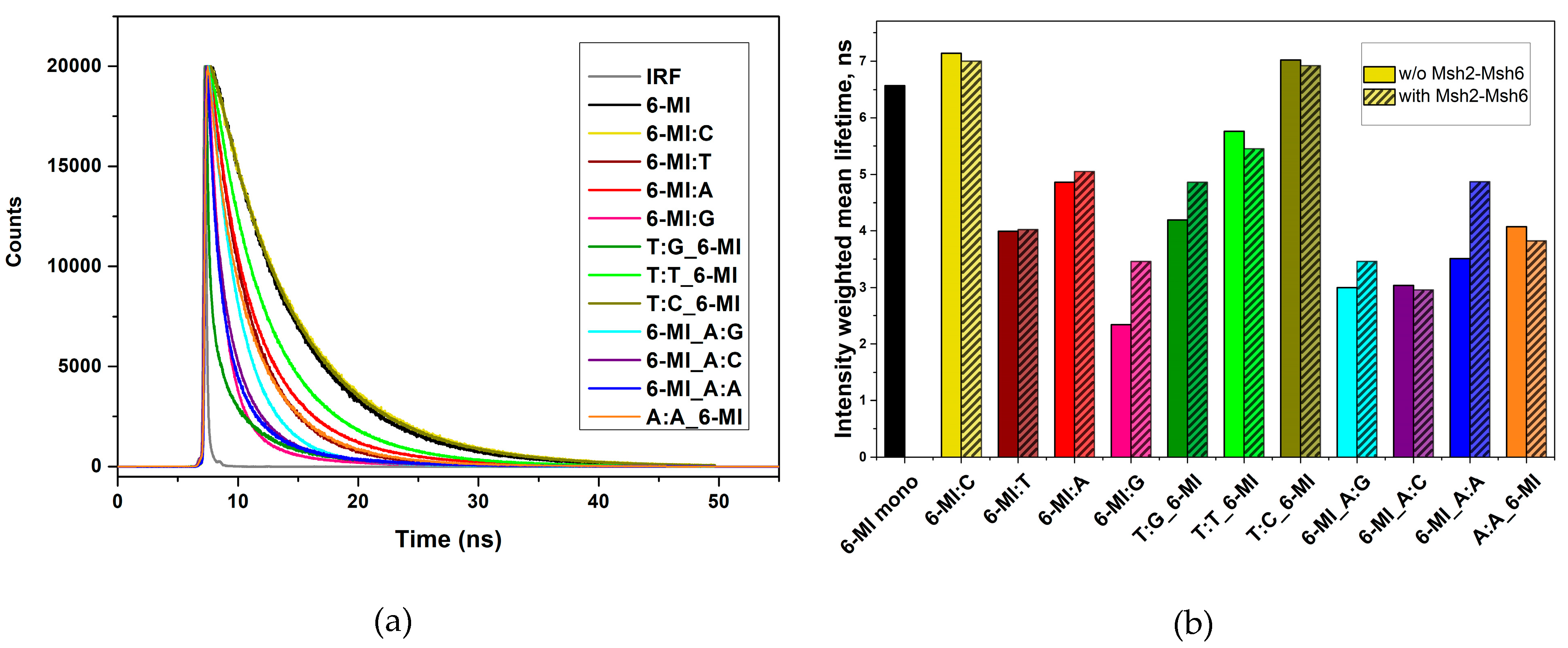
Local Structure and Dynamics of Mismatched Base Pairs
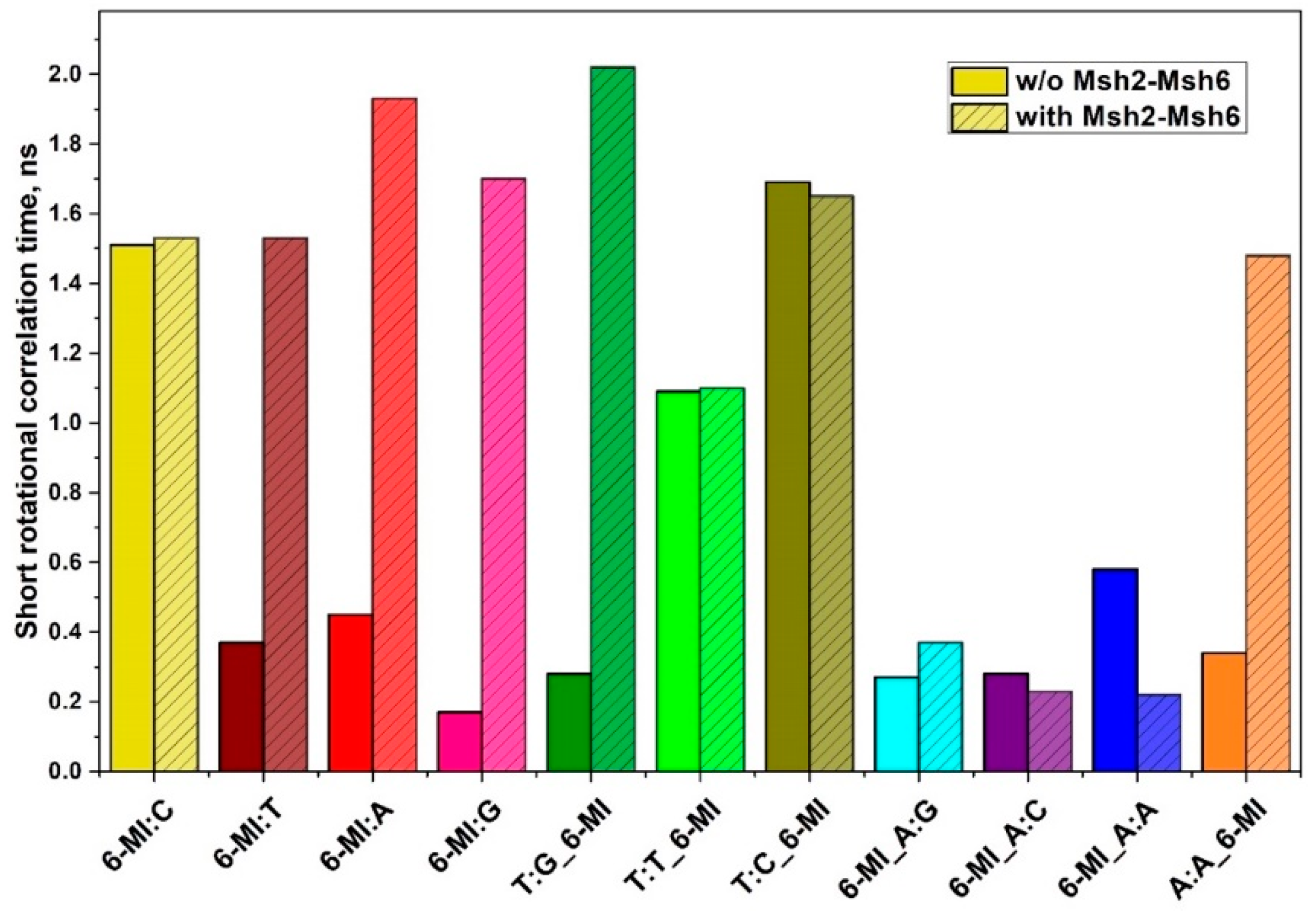
2.3. Msh2-Msh6 Binding Affects Mismatched Base Pair Dynamics
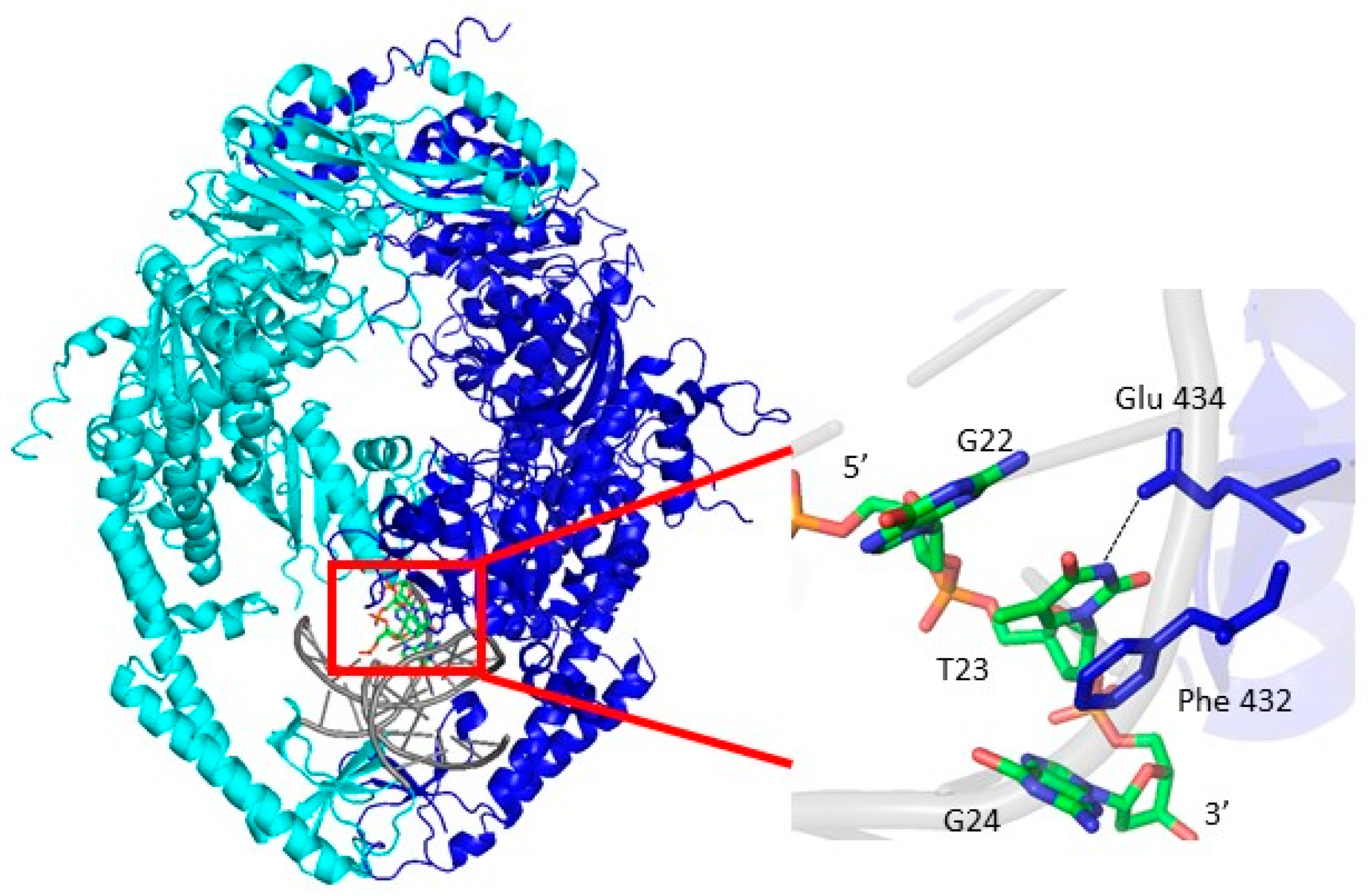
2.4. Msh2-Msh6 Preferred Binding Orientation and Phe Intercalation Reported by 6-MI Fluorescence
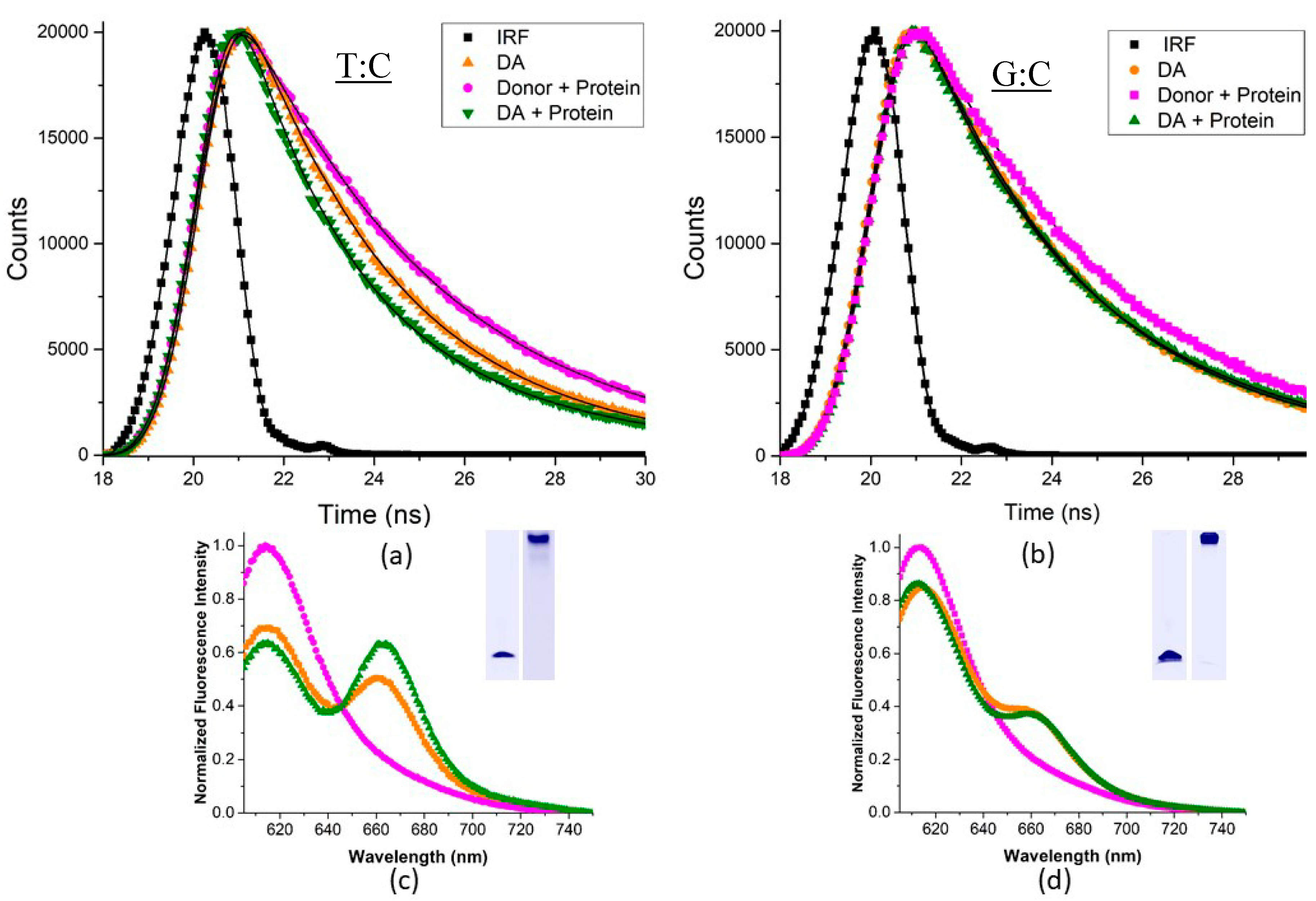
2.5. Msh2-Msh6-Induced DNA Bending Measured by Förster Resonance Energy Transfer
3. Discussion
3.1. Msh2–Msh6 Induces the Same Amount of Bending in Different Mismatched Duplexes but Not Homoduplex DNA
3.2. Msh2–Msh6 Binding Affinity Depends on Mismatch Type
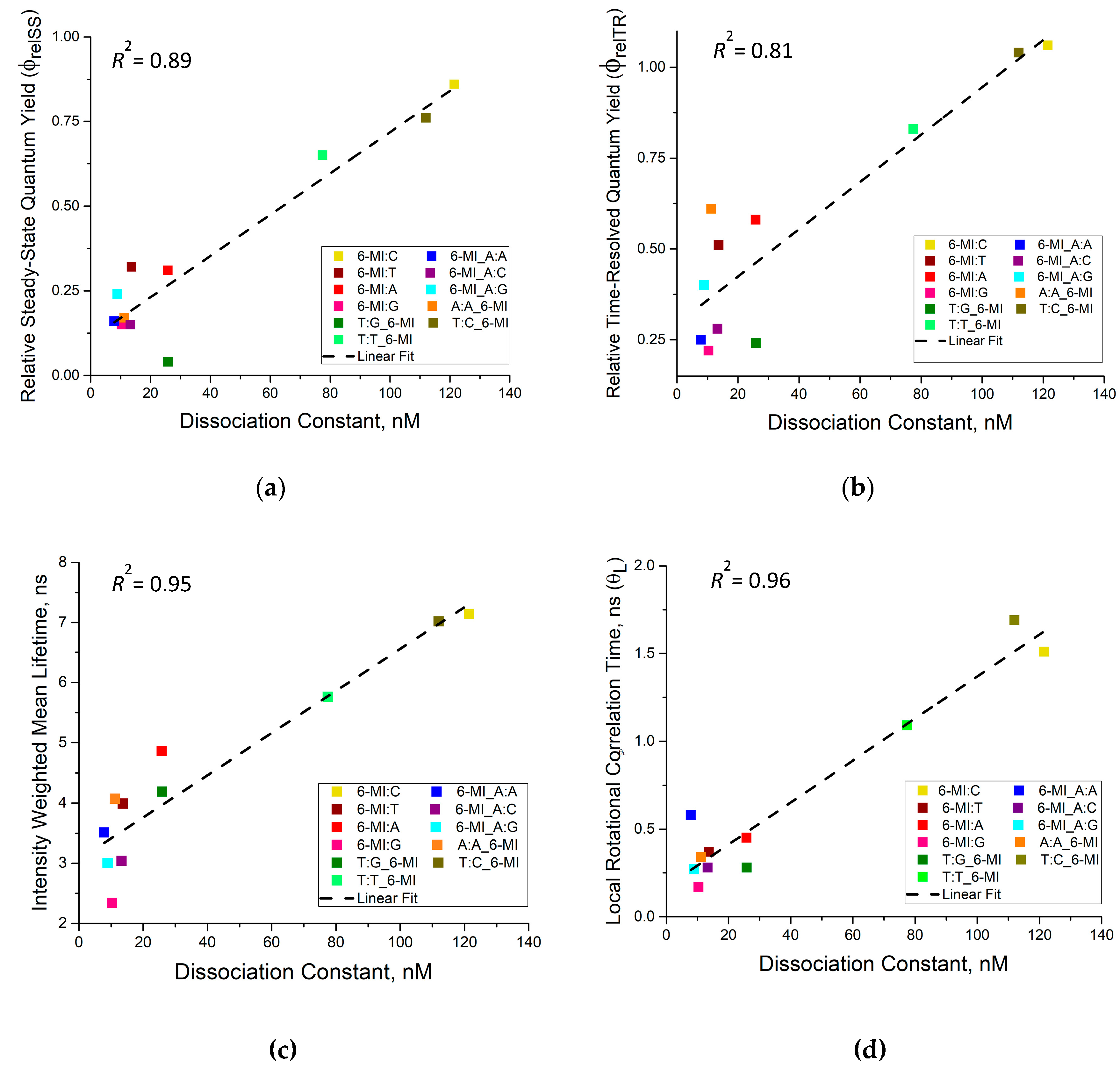
3.3. Msh2-Msh6 Binding Affinity Correlates with Enhanced Dynamics at or next to a Mismatch Site
3.4. Msh2–Msh6 Stabilizes the Mismatch Upon Binding
4. Materials and Methods
4.1. S. cerevisiae Msh2–Msh6 Purification from E. coli Cells
4.2. Oligonucleotides
4.3. Steady-State Quantum Yield
4.4. Fluorescence Emission Spectra
4.5. Electrophoretic Mobility Shift Assay
4.6. Fluorescence Anisotropy Equilibrium Binding Experiments
4.7. Fluorescence Lifetime and Anisotropy Decays
4.8. Steady-State and Time-Resolved Förster Resonance Energy Transfer (FRET) Assays and Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MMR | Mismatch repair |
| 6-MI | 6-Methylisoxanthopterin |
| Msh | MutS Homolog |
| IDL | Insertion/deletion loop |
| AFM | Atomic force microscopy |
| FRET | Förster resonance energy transfer |
| sm-FRET | Single-molecule Förster resonance energy transfer |
| MD | Molecular dynamics |
| bp | Base pair |
| EMSA | Electrophoretic mobility shift assay |
| 𝜙𝑟𝑒𝑙SS | Relative steady-state fluorescence quantum yield |
| 𝜙𝑟𝑒𝑙TR | Relative time-resolved fluorescence quantum yield |
| IRF | Instrument response function |
| 2-AP | 2-Aminopurine |
| SPR | Surface plasmon resonance |
| TCSPC | Time-correlated single-photon counting |
| FWHM | Full width at half-maximum |
| FAM | 5-Carboxyfluorescein succinimidyl ester |
| TAMRA | 5-Carboxytetramethylrhodamine succinimidyl ester |
| LED | Light-emitting diode |
| MWCO | Molecular weight cut off |
References
- Kunkel, T.A.; Erie, D.A. DNA mismatch repair. Annu. Rev. Biochem. 2005, 74, 681–710. [Google Scholar] [CrossRef] [PubMed]
- Jiricny, J. MutLalpha: at the cutting edge of mismatch repair. Cell 2006, 126, 239–241. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Spies, M.; Fishel, R. Mismatch repair during homologous and homeologous recombination. Cold Spring Harbor Perspect. Biol. 2015, 7, a022657. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, T.A.; Erie, D.A. Eukaryotic mismatch repair in relation to DNA replication. Annu. Rev. Genet. 2015, 49, 291–313. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R.; Lynch, H.T. The history of Lynch syndrome. Fam. Cancer 2013, 12, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; Snyder, C.L.; Shaw, T.G.; Heinen, C.D.; Hitchins, M.P. Milestones of Lynch syndrome: 1895–2015. Nat. Rev. Cancer 2015, 15, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Lebbink, J.H.; Drost, M.; de Wind, N. DNA mismatch repair: from biophysics to bedside. DNA Repair 2016, 38, 1–2. [Google Scholar] [CrossRef]
- Heinen, C.D. Mismatch repair defects and Lynch syndrome: The role of the basic scientist in the battle against cancer. DNA Repair 2016, 38, 127–134. [Google Scholar] [CrossRef]
- Fishel, R. Mismatch repair. J. Biol. Chem. 2015, 290, 26395–26403. [Google Scholar] [CrossRef]
- Modrich, P. Mechanisms in eukaryotic mismatch repair. J. Biol. Chem. 2006, 281, 30305–30309. [Google Scholar] [CrossRef]
- Groothuizen, F.S.; Sixma, T.K. The conserved molecular machinery in DNA mismatch repair enzyme structures. DNA Repair 2016, 38, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, M.M. Mismatch binding, ADP-ATP exchange and intramolecular signaling during mismatch repair. DNA Repair 2016, 38, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Reyes, G.X.; Schmidt, T.T.; Kolodner, R.D.; Hombauer, H. New insights into the mechanism of DNA mismatch repair. Chromosoma 2015, 124, 443–462. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, A.A.; McMurray, C.T. Close encounters: Moving along bumps, breaks, and bubbles on expanded trinucleotide tracts. DNA Repair 2017, 56, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.H.; Pearson, C.E. Disease-associated repeat instability and mismatch repair. DNA Repair 2016, 38, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Lamers, M.H.; Perrakis, A.; Enzlin, J.H.; Winterwerp, H.H.; de Wind, N.; Sixma, T.K. The crystal structure of DNA mismatch repair protein MutS binding to a G x T mismatch. Nature 2000, 407, 711–717. [Google Scholar] [CrossRef]
- Obmolova, G.; Ban, C.; Hsieh, P.; Yang, W. Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature 2000, 407, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Natrajan, G.; Lamers, M.H.; Enzlin, J.H.; Winterwerp, H.H.; Perrakis, A.; Sixma, T.K. Structures of Escherichia coli DNA mismatch repair enzyme MutS in complex with different mismatches: a common recognition mode for diverse substrates. Nucleic Acids Res. 2003, 31, 4814–4821. [Google Scholar] [CrossRef]
- Warren, J.J.; Pohlhaus, T.J.; Changela, A.; Iyer, R.R.; Modrich, P.L.; Beese, L.S. Structure of the human MutSalpha DNA lesion recognition complex. Mol. Cell 2007, 26, 579–592. [Google Scholar] [CrossRef]
- Groothuizen, F.S.; Winkler, I.; Cristóvão, M.; Fish, A.; Winterwerp, H.H.K.; Reumer, A.; Marx, A.D.; Hermans, N.; Nicholls, R.A.; Murshudov, G.N.; et al. MutS/MutL crystal structure reveals that the MutS sliding clamp loads MutL onto DNA. eLife 2015, 4, e06744. [Google Scholar] [CrossRef]
- Wang, H.; Yang, Y.; Schofield, M.J.; Du, C.; Fridman, Y.; Lee, S.D.; Larson, E.D.; Drummond, J.T.; Alani, E.; Hsieh, P.; et al. DNA bending and unbending by MutS govern mismatch recognition and specificity. Proc. Natl. Acad. Sci. USA 2003, 100, 14822–14827. [Google Scholar] [CrossRef] [PubMed]
- Tessmer, I.; Yang, Y.; Zhai, J.; Du, C.; Hsieh, P.; Hingorani, M.M.; Erie, D.A. Mechanism of MutS searching for DNA mismatches and signaling repair. J. Biol. Chem. 2008, 283, 36646–36654. [Google Scholar] [CrossRef] [PubMed]
- DeRocco, V.C.; Sass, L.E.; Qiu, R.; Weninger, K.R.; Erie, D.A. Dynamics of MutS-mismatched DNA complexes are predictive of their repair phenotypes. Biochemistry 2014, 53, 2043–2052. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.B.; Cho, W.K.; Park, J.; Jeon, Y.; Kim, D.; Lee, S.H.; Fishel, R. Single-molecule views of MutS on mismatched DNA. DNA Repair 2014, 20, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Jeong, C.; Cho, W.K.; Song, K.M.; Cook, C.; Yoon, T.Y.; Ban, C.; Fishel, R.; Lee, J.B. MutS switches between two fundamentally distinct clamps during mismatch repair. Nat. Struct. Mol. Biol. 2011, 18, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Gorman, J.; Wang, F.; Redding, S.; Plys, A.J.; Fazio, T.; Wind, S.; Alani, E.E.; Greene, E.C. Single-molecule imaging reveals target-search mechanisms during DNA mismatch repair. Proc. Natl. Acad. Sci. USA 2012, 109, E3074–E3083. [Google Scholar] [CrossRef]
- Sharma, M.; Predeus, A.V.; Mukherjee, S.; Feig, M. DNA bending propensity in the presence of base mismatches: implications for DNA repair. J. Phys. Chem. B 2013, 117, 6194–6205. [Google Scholar] [CrossRef]
- Marsischky, G.T.; Kolodner, R.D. Biochemical characterization of the interaction between the Saccharomyces cerevisiae MSH2-MSH6 complex and mispaired bases in DNA. J. Biol. Chem. 1999, 274, 26668–26682. [Google Scholar] [CrossRef]
- Brown, J.; Brown, T.; Fox, K.R. Affinity of mismatch-binding protein MutS for heteroduplexes containing different mismatches. Biochem. J. 2001, 354, 627–633. [Google Scholar] [CrossRef]
- Mazurek, A.; Johnson, C.N.; Germann, M.W.; Fishel, R. Sequence context effect for hMSH2-hMSH6 mismatch-dependent activation. Proc. Natl. Acad. Sci. USA 2009, 106, 4177–4182. [Google Scholar] [CrossRef]
- Kramer, B.; Kramer, W.; Fritz, H.J. Different base/base mismatches are corrected with different efficiencies by the methyl-directed DNA mismatch-repair system of E. coli. Cell 1984, 38, 879–887. [Google Scholar] [CrossRef]
- Dohet, C.; Wagner, R.; Radman, M. Repair of defined single base-pair mismatches in Escherichia coli. Proc. Natl. Acad. Sci. USA 1985, 82, 503–505. [Google Scholar] [CrossRef] [PubMed]
- David, P.; Efrati, E.; Tocco, G.; Krauss, S.W.; Goodman, M.F. DNA replication and postreplication mismatch repair in cell-free extracts from cultured human neuroblastoma and fibroblast cells. J. Neurosci. 1997, 17, 8711–8720. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, R.J.; Rayens, W.S.; Spielmann, H.P. Structural differences in the NOE-derived structure of G-T mismatched DNA relative to normal DNA are correlated with differences in (13)C relaxation-based internal dynamics. J. Mol. Biol. 2002, 319, 191–207. [Google Scholar] [CrossRef]
- Nag, N.; Rao, B.J.; Krishnamoorthy, G. Altered dynamics of DNA bases adjacent to a mismatch: a cue for mismatch recognition by MutS. J. Mol. Biol. 2007, 374, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, G.; Dans, P.D.; Gomez-Pinto, I.; Ivani, I.; Gonzalez, C.; Orozco, M. The structural impact of DNA mismatches. Nucleic Acids Res. 2015, 43, 4309–4321. [Google Scholar] [CrossRef] [PubMed]
- Imhof, P.; Zahran, M. The effect of a G:T mispair on the dynamics of DNA. PLoS ONE 2013, 8, e53305. [Google Scholar] [CrossRef]
- Isaacs, R.J.; Spielmann, H.P. A model for initial DNA lesion recognition by NER and MMR based on local conformational flexibility. DNA Repair 2004, 3, 455–464. [Google Scholar] [CrossRef]
- Hawkins, M.E. Fluorescent pteridine nucleoside analogs: A window on DNA interactions. Cell Biochem. Biophys. 2001, 34, 257–281. [Google Scholar] [CrossRef]
- Moreno, A.; Knee, J.; Mukerji, I. Applying 6-methylisoxanthopterin-enhanced fluorescence to examine protein-DNA interactions in the picomolar range. Biochemistry 2012, 51, 6847–6859. [Google Scholar] [CrossRef]
- Fried, M.; Liu, G. Molecular sequestration stabilizes CAP-DNA complexes during polyacrylamide gel electrophoresis. Nucleic Acids Res. 1994, 23, 5054–5059. [Google Scholar] [CrossRef] [PubMed]
- Hellman, L.M.; Fried, M.G. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat. Protoc. 2007, 2, 1849–1861. [Google Scholar] [CrossRef] [PubMed]
- Cristovao, M.; Sisamakis, E.; Hingorani, M.M.; Marx, A.D.; Jung, C.P.; Rothwell, P.J.; Seidel, C.A.; Friedhoff, P. Single-molecule multiparameter fluorescence spectroscopy reveals directional MutS binding to mismatched bases in DNA. Nucleic Acids Res. 2012, 40, 5448–5464. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hawkins, M.E.; Pfleiderer, W.; Balis, F.M.; Porter, D.; Knutson, J.R. Fluorescence properties of pteridine nucleoside analogs as monomers and incorporated into oligonucleotides. Anal. Biochem. 1997, 244, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, M.E.; Pfleiderer, W.; Mazumder, A.; Pommier, Y.G.; Balis, F.M. Incorporation of a fluorescent guanosine analog into oligonucleotides and its application to a real time assay for the HIV-1 integrase 3′-processing reaction. Nucleic Acids Res. 1995, 23, 2872–2880. [Google Scholar] [CrossRef] [PubMed]
- Moreno, A.; Knee, J.L.; Mukerji, I. Photophysical characterization of enhanced 6-methylisoxanthopterin fluorescence in duplex DNA. J. Phys. Chem. B 2016, 120, 12232–12248. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Plenum Press: New York, NY, USA, 2006; p. 954. [Google Scholar]
- Wojtuszewski Poulin, K.; Smirnov, A.V.; Hawkins, M.E.; Balis, F.M.; Knutson, J.R. Conformational heterogeneity and quasi-static self-quenching in DNA containing a fluorescent guanine analogue, 3MI or 6MI. Biochemistry 2009, 48, 8861–8868. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hawkins, M.E. Fluorescent pteridine probes for nucleic acid analysis. Methods Enzymol. 2008, 450, 201–231. [Google Scholar]
- Ivanov, V.; Li, M.; Mizuuchi, K. Impact of emission anisotropy on fluorescence spectroscopy and FRET distance measurements. Biophys. J. 2009, 97, 922–929. [Google Scholar] [CrossRef][Green Version]
- Thermo Fisher Scientific-US, Fluorescence Quantum Yields (QY) and Lifetimes (τ) for Alexa Fluor Dyes-Table 1.5. Available online: www.thermofisher.com/us/en/home/references/molecular-probes-the-handbook/tables/fluorescence-quantum-yields-and-lifetimes-for-alexa-fluor-dyes.html (accessed on 6 July 2019).
- Sass, L.E.; Lanyi, C.; Weninger, K.; Erie, D.A. Single-molecule FRET TACKLE reveals highly dynamic mismatched DNA-MutS complexes. Biochemistry 2010, 49, 3174–3190. [Google Scholar] [CrossRef][Green Version]
- Litke, J.L.; Li, Y.; Nocka, L.M.; Mukerji, I. Probing the ion binding site in a DNA holliday junction using forster resonance energy transfer (FRET). Int. J. Mol. Sci. 2016, 17, 366. [Google Scholar] [CrossRef] [PubMed]
- Bishop, D.K.; Andersen, J.; Kolodner, R.D. Specificity of mismatch repair following transformation of Saccharomyces cerevisiae with heteroduplex plasmid DNA. Proc. Natl. Acad. Sci. USA 1989, 86, 3713–3717. [Google Scholar] [CrossRef] [PubMed]
- Kramer, B.; Kramer, W.; Williamson, M.S.; Fogel, S. Heteroduplex DNA correction in Saccharomyces cerevisiae is mismatch specific and requires functional PMS genes. Mol. Cell. Biol. 1989, 9, 4432–4440. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.; Wagner, R.; Radman, M. Repair of a mismatch is influenced by the base composition of the surrounding nucleotide sequence. Genetics 1987, 115, 605–610. [Google Scholar] [PubMed]
- Su, S.S.; Lahue, R.S.; Au, K.G.; Modrich, P. Mispair specificity of methyl-directed DNA mismatch correction in vitro. J. Biol. Chem. 1988, 263, 6829–6835. [Google Scholar]
- Allawi, H.T.; SantaLucia, J. Thermodynamics and NMR of internal G.T mismatches in DNA. Biochemistry 1997, 36, 10581–10594. [Google Scholar] [CrossRef] [PubMed]
- Allawi, H.T.; SantaLucia, J. Nearest neighbor thermodynamic parameters for internal G.A mismatches in DNA. Biochemistry 1998, 37, 2170–2179. [Google Scholar] [CrossRef]
- Allawi, H.T.; SantaLucia, J. Nearest-neighbor thermodynamics of internal A.C mismatches in DNA: sequence dependence and pH effects. Biochemistry 1998, 37, 9435–9444. [Google Scholar] [CrossRef]
- Li, G.M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef]
- Shweta, H.; Singh, M.K.; Yadav, K.; Verma, S.D.; Pal, N.; Sen, S. Effect of T.T mismatch on DNA dynamics probed by minor groove binders: Comparison of dynamic stokes shifts of hoechst and DAPI. J. Phys. Chem. B 2017, 121, 10735–10748. [Google Scholar] [CrossRef]
- Brown, T.; Hunter, W.N.; Kneale, G.; Kennard, O. Molecular structure of the G.A base pair in DNA and its implications for the mechanism of transversion mutations. Proc. Natl. Acad. Sci. USA 1986, 83, 2402–2406. [Google Scholar] [CrossRef] [PubMed]
- Boulard, Y.; Cognet, J.A.; Fazakerley, G.V. Solution structure as a function of pH of two central mismatches, C . T and C . C, in the 29 to 39 K-ras gene sequence, by nuclear magnetic resonance and molecular dynamics. J. Mol. Biol. 1997, 268, 331–347. [Google Scholar] [CrossRef]
- Gorman, J.; Chowdhury, A.; Surtees, J.A.; Shimada, J.; Reichman, D.R.; Alani, E.; Greene, E.C. Dynamic basis for one-dimensional DNA scanning by the mismatch repair complex Msh2-Msh6. Mol. Cell. 2007, 28, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, E.S.; Kimsey, I.J.; Al-Hashimi, H.M. Direct NMR evidence that transient tautomeric and anionic states in dG.dT form watson-crick-like base pairs. J. Am. Chem. Soc. 2017, 139, 4326–4329. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M.F. Smoking gun for a rare mutation mechanism. Nature 2018, 554, 180–181. [Google Scholar] [CrossRef] [PubMed]
- Kimsey, I.J.; Szymanski, E.S.; Zahurancik, W.J.; Shakya, A.; Xue, Y.; Chu, C.C.; Sathyamoorthy, B.; Suo, Z.; Al-Hashimi, H.M. Dynamic basis for dG*dT misincorporation via tautomerization and ionization. Nature 2018, 554, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Kouchakdjian, M.; Li, B.F.; Swann, P.F.; Patel, D.J. Pyrimidine.pyrimidine base-pair mismatches in DNA. A nuclear magnetic resonance study of T.T pairing at neutral pH and C.C pairing at acidic pH in dodecanucleotide duplexes. J. Mol. Biol. 1988, 202, 139–155. [Google Scholar] [CrossRef]
- Cornelis, A.G.; Haasnoot, J.H.; den Hartog, J.F.; de Rooij, M.; van Boom, J.H.; Cornelis, A. Local destabilisation of a DNA double helix by a T--T wobble pair. Nature 1979, 281, 235–236. [Google Scholar] [CrossRef]
- Yamamoto, A.; Schofield, M.J.; Biswas, I.; Hsieh, P. Requirement for Phe36 for DNA binding and mismatch repair by Escherichia coli MutS protein. Nucleic Acids Res. 2000, 28, 3564–3569. [Google Scholar] [CrossRef] [PubMed]
- Bowers, J.; Sokolsky, T.; Quach, T.; Alani, E. A mutation in the MSH6 subunit of the saccharomyces cerevisiae MSH2-MSH6 complex disrupts mismatch recognition. J. Biol. Chem. 1999, 274, 16115–16125. [Google Scholar] [CrossRef]
- Biswas, I.; Hsieh, P. Interaction of MutS protein with the major and minor grooves of a heteroduplex DNA. J. Biol. Chem. 1997, 272, 13355–13364. [Google Scholar] [CrossRef] [PubMed]
- Antony, E.; Hingorani, M.M. Mismatch recognition-coupled stabilization of Msh2-Msh6 in an ATP-bound state at the initiation of DNA repair. Biochemistry 2003, 42, 7682–7693. [Google Scholar] [CrossRef] [PubMed]
- Thermo Fisher Scientific. Amine Reactive Probe Labeling Protocol. Available online: https://www.thermofisher.com/us/en/home/references/protocols/cell-and-tissue-analysis/labeling-chemistry-protocols/amine-reactive-probe-labeling-protocol.html (accessed on 11 June 2019).
- Horiba Scientific. A Guide to Recording Fluorescence Quantum Yields. Available online: http://www.horiba.com/fileadmin/uploads/Scientific/Documents/Fluorescence/quantumyieldstrad.pdf (accessed on 11 June 2019).
- Umberger, J.Q.; LaMer, V.K. The kinetics of diffusion controlled molecular and ionic reactions in solution as determined by measurements of quenching of fluorescence. J. Am. Chem. Soc. 1945, 67, 1099–1109. [Google Scholar] [CrossRef]
- Wojtuszewski, K.; Mukerji, I. HU binding to bent DNA: A fluorescence resonance energy transfer and anisotropy study. Biochemistry 2003, 42, 3096–3104. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNA Substrate | KD1 (nM) | KD2 (nM) | KD3 (nM) |
|---|---|---|---|
| 6-MI:C | 121.5 ± 12.1 | - | 106.5 ± 13.9 |
| 6-MI:T | 13.7 ± 3.4 | - | 13.3 ± 3.9 |
| 6-MI:A | 25.8 ± 12.3 | - | - |
| 6-MI:G | 10.4 ± 5.8 | - | 15.6 ± 3.4 |
| T:G_6-MI | 25.9 ± 1.2 | - | - |
| T:T_6-MI | 77.5 ± 20.0 | - | - |
| T:C_6-MI | 112.6 ± 18.7 | - | - |
| A:A_6-MI | 11.3 ± 3.1 | 10.4 ± 2.4 | - |
| 6-MI_A:A | 7.9 ± 2.0 | 19.1 ± 3.5 | - |
| 6-MI_A:G | 9.0 ± 2.8 | 11.8 ± 2.8 | - |
| 6-MI_A:C | 13.3 ± 3.6 | 17.1 ± 4.3 | - |
| +T_6-MI | 16.0 ± 2.9 | 18.1 ± 2.6 | - |
| Mismatch Type | Sample 3 | α1 | τ1 | α2 | τ2 | χ2 | TR EFRET 4 ±0.013 | TR ΔEFRET | TR R 5 (Å) ±15 Å | TR Angle 6 (°) ±10° | SS EFRET 4 | SS ΔEFRET | SS R 5 (Å) ±15Å | SS Angle 6 (°) ±10° |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T:T | D | 1.00 | 4.28 | 1.25 | 0.47 | 0.14 | 77 | 70 | 0.42 | 0.27 | 80 | 79 | ||
| DA | 0.82 | 4.28 | 0.18 | 2.26 | 1.18 | |||||||||
| T:T with Msh2–Msh6 | D | 1.00 | 4.36 | 1.29 | 0.62 | 70 | 0.69 | 66 | ||||||
| DA | 0.80 | 4.36 | 0.20 | 1.66 | 1.24 | |||||||||
| G:T | D | 1.00 | 4.28 | 1.19 | 0.37 | 0.15 | 82 | 59 | 0.43 | 0.21 | 79 | 73 | ||
| DA | 0.84 | 4.28 | 0.16 | 2.69 | 0.98 | |||||||||
| G:T with Msh2–Msh6 | D | 1.00 | 4.33 | 1.17 | 0.53 | 74 | 0.64 | 69 | ||||||
| DA | 0.90 | 4.33 | 0.10 | 2.05 | 1.01 | |||||||||
| A:A | D | 1.00 | 4.25 | 1.23 | 0.42 | 0.18 | 80 | 68 | 0.42 | 0.28 | 80 | 78 | ||
| DA | 0.82 | 4.25 | 0.18 | 2.47 | 1.03 | |||||||||
| A:A with Msh2–Msh6 | D | 1.00 | 4.31 | 1.38 | 0.60 | 71 | 0.69 | 66 | ||||||
| DA | 0.90 | 4.31 | 0.10 | 1.72 | 1.12 | |||||||||
| T:C | D | 1.00 | 4.28 | 1.25 | 0.38 | 0.22 | 80 | 68 | 0.37 | 0.20 | 82 | 64 | ||
| DA | 0.52 | 4.28 | 0.48 | 2.62 | 0.85 | |||||||||
| T:C with Msh2–Msh6 | D | 1.00 | 4.29 | 1.13 | 0.60 | 71 | 0.57 | 72 | ||||||
| DA | 0.64 | 4.28 | 0.36 | 1.72 | 1.95 | |||||||||
| G:C | D | 1.00 | 4.29 | 1.20 | 0.45 | 0.00 | 78 | 0.43 | −0.02 | 79 | ||||
| DA | 0.78 | 4.29 | 0.22 | 2.37 | 1.21 | |||||||||
| G:C with Msh2–Msh6 | D | 1.00 | 4.41 | 1.42 | 0.45 | 78 | 0.41 | 80 | ||||||
| DA | 0.82 | 4.41 | 0.18 | 2.42 | 0.97 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Lombardo, Z.; Joshi, M.; Hingorani, M.M.; Mukerji, I. Mismatch Recognition by Saccharomyces cerevisiae Msh2-Msh6: Role of Structure and Dynamics. Int. J. Mol. Sci. 2019, 20, 4271. https://doi.org/10.3390/ijms20174271
Li Y, Lombardo Z, Joshi M, Hingorani MM, Mukerji I. Mismatch Recognition by Saccharomyces cerevisiae Msh2-Msh6: Role of Structure and Dynamics. International Journal of Molecular Sciences. 2019; 20(17):4271. https://doi.org/10.3390/ijms20174271
Chicago/Turabian StyleLi, Yan, Zane Lombardo, Meera Joshi, Manju M. Hingorani, and Ishita Mukerji. 2019. "Mismatch Recognition by Saccharomyces cerevisiae Msh2-Msh6: Role of Structure and Dynamics" International Journal of Molecular Sciences 20, no. 17: 4271. https://doi.org/10.3390/ijms20174271
APA StyleLi, Y., Lombardo, Z., Joshi, M., Hingorani, M. M., & Mukerji, I. (2019). Mismatch Recognition by Saccharomyces cerevisiae Msh2-Msh6: Role of Structure and Dynamics. International Journal of Molecular Sciences, 20(17), 4271. https://doi.org/10.3390/ijms20174271