Preeclampsia: Maternal Systemic Vascular Disorder Caused by Generalized Endothelial Dysfunction Due to Placental Antiangiogenic Factors


 ,
, 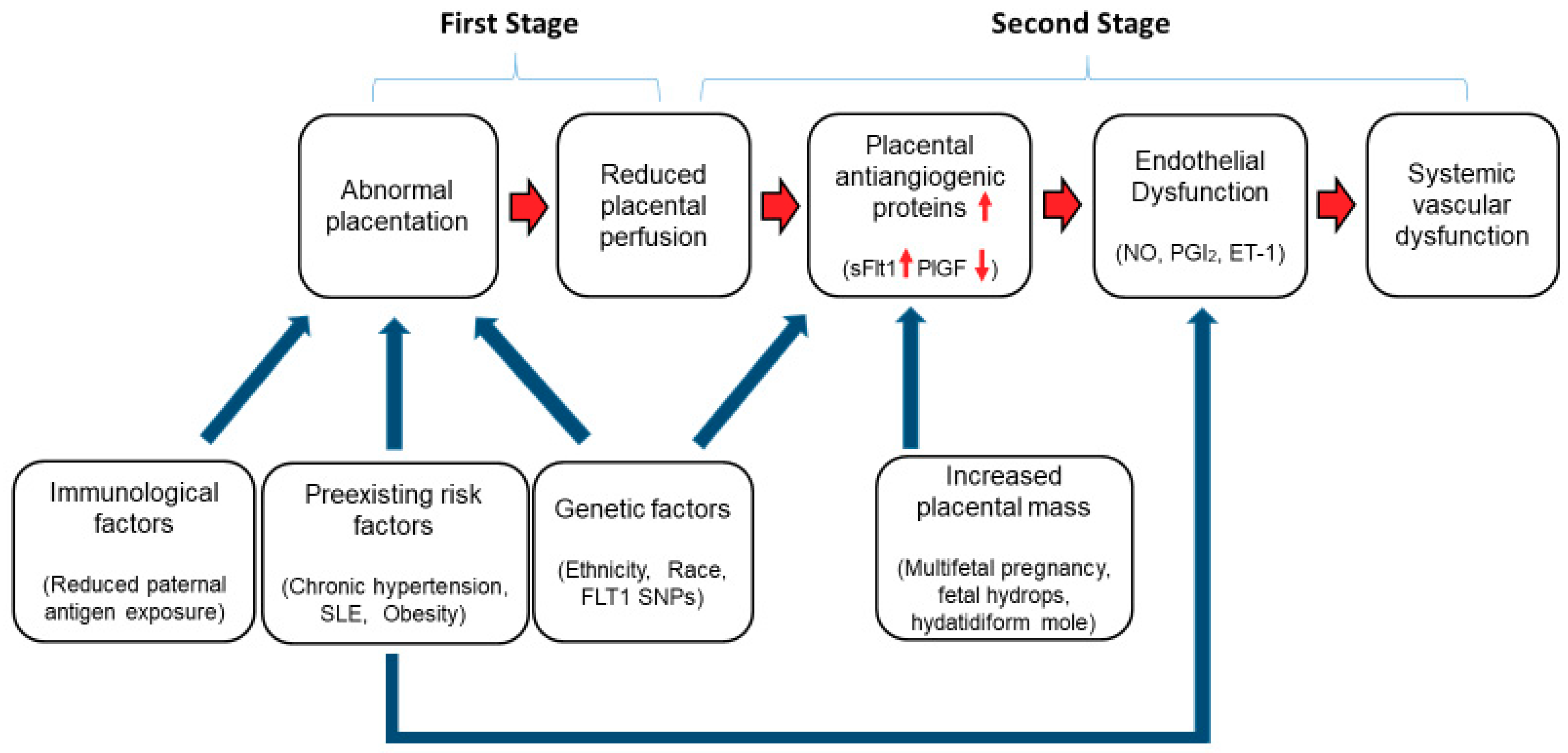{kind=link}
{kind=link}
Abstract
1. Introduction
2. Diagnosis and Risk Factors of Preeclampsia
3. Maternal Antiangiogenic State in Preeclampsia
4. Mechanisms of Endothelial Dysfunction by Inhibiting the VEGF Signal Pathway
5. Mechanism of Renal Injury by Inhibiting the VEGF Signal Pathway
6. Complement System and Angiogenic Imbalance
7. Regulating sFlt1 Production by Trophoblastic Cells
8. Preeclampsia as a Systemic Vascular Disorder of Pregnancy
9. Novel Clinical and Therapeutic Strategies from the Viewpoints of Maternal Angiogenic Imbalance
9.1. Prediction of Disease
9.2. Prediction of Adverse Maternal and Perinatal Complications
9.3. Assessing Angiogenic Imbalance in Differential Diagnosis
9.4. Therapeutic Potential of Modulating Angiogenic Factors
10. Prevention of Preeclampsia
10.1. Low-Dose Aspirin
10.2. Low-Dose Aspirin plus Heparin
11. Preeclampsia and Future Risk of Cardiovascular Disease
12. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| FMD | Flow-mediated dilation |
| sFlt1 | Soluble fms-like tyrosine kinase 1 |
| VEGF | Vascular endothelial growth factor |
| PlGF | Placental growth factor |
| sVEGFR1 | Soluble vascular endothelial growth factor receptor 1 |
| NO | Nitric oxide |
| NOS | Nitric oxide synthase |
| PGI2 | Prostacyclin |
| TXA2 | Thromboxane A2 |
| COX | Cyclooxygenase |
| sGC | Soluble guanylate cyclase |
| AC | Adenylyl cyclase |
| ET-1 | Endothelin 1 |
| ECE-1 | Endothelin-converting enzyme 1 |
| aHUS | Atypical hemolytic uremic syndrome |
| IUGR | Intrauterine growth restriction |
| APS | Antiphospholipid syndrome |
| CKD | Chronic kidney disease |
| SLE | Systemic lupus erythematosus |
| PWA | Pulse wave analysis |
| PWV | Pulse wave velocity |
| RNAi | RNA interference |
| MAP | Mean arterial pressure, and |
| UtA-PI | Uterine artery pulsatility index |
| MMPs | Matrix metalloproteinases |
| ECM | Extracellular matrix |
| HIF1α | Hypoxia-inducible factor 1α |
References
- Hepner, D.L.; Wilkins-Haug, L.; Marks, P.W. Hematologic disease. In Anesthetic and Obstetric Management of High-Risk Pregnancy; Cunningham, F.G., Leveno, K.J., Eds.; Springer: New York, NY, USA, 2004; pp. 309–332. [Google Scholar]
- Sibai, B.; Dekker, G.; Kupferminc, M. Preeclampsia. Lancet 2005, 365, 785–799. [Google Scholar] [CrossRef]
- Roberts, J.M. Endothelial dysfunction in preclampsia. Semin. Reprod. Endocrinol. 1998, 16, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Stout, M. Flow-mediated dilatation: A review of techniques and applications. Echocardiography 2009, 26, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Cockell, A.P.; Poston, L. Flow-mediated vasodilatation is enhanced in normal pregnancy but reduced in preeclampsia. Hypertension 1997, 30, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.E.; Min, J.Y.; Merchan, J.; Lim, K.H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin. Investig. 2003, 111, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.J.; Maynard, S.E.; Qian, C.; Lim, K.H.; England, L.J.; Yu, K.F.; Schisterman, E.F.; Thadhani, R.; Sachs, B.P.; Epstein, F.H.; et al. Circulating angiogenic factors and the risk of preeclampsia. N. Engl. J. Med. 2004, 350, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Venkatesha, S.; Toporsian, M.; Lam, C.; Hanai, J.; Mammoto, T.; Kim, Y.M.; Bdolah, Y.; Lim, K.H.; Yuan, H.T.; Libermann, T.A.; et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat. Med. 2006, 12, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.J.; Lam, C.; Qian, C.; Yu, K.F.; Maynard, S.E.; Sachs, B.P.; Sibai, B.M.; Epstein, F.H.; Romero, R.; Thadhani, R.; et al. Soluble endoglin andother circulating antiangiogenic factors in preeclampsia. N. Engl. J. Med. 2006, 355, 992–1005. [Google Scholar] [CrossRef] [PubMed]
- National Institute for Health and Clinical Excellence (NICE). Hypertension in Pregnancy; RCOG Press: London, UK, 2011. [Google Scholar]
- Shahul, S.; Tung, A.; Minhaj, M.; Nizamuddin, J.; Wenger, J.; Mahmood, E.; Mueller, A.; Shaefi, S.; Scavone, B.; Kociol, R.D.; et al. Racial disparities in comorbidities, complications, and maternal and fetal outcomes in women with preeclampsia/eclampsia. Hypertens. Pregnancy 2015, 34, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Skjaerven, R.; Vatten, L.J.; Wilcox, A.J.; Rønning, T.; Irgens, L.M.; Lie, R.T. Recurrence of pre-eclampsia across generations: Exploring fetal and maternal genetic components in a population based cohort. BMJ 2005, 331, 877. [Google Scholar] [CrossRef]
- Esplin, M.S.; Fausett, M.B.; Fraser, A.; Kerber, R.; Mineau, G.; Carrillo, J.; Varner, M.W. Paternal and maternal components of the predisposition to preeclampsia. N. Engl. J. Med. 2001, 344, 867–872. [Google Scholar] [CrossRef] [PubMed]
- McGinnis, R.; Steinthorsdottir, V.; Williams, N.O.; Thorleifsson, G.; Shooter, S.; Hjartardottir, S.; Bumpstead, S.; Stefansdottir, L.; Hildyard, L.; Sigurdsson, J.K.; et al. Variants in the fetal genome near FLT1 are associated with risk of preeclampsia. Nat. Genet. 2017, 49, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Gray, K.J.; Saxena, R.; Karumanchi, S.A. Genetic predisposition to preeclampsia is conferred by fetal DNA variants near FLT1, a gene involved in the regulation of angiogenesis. Am. J. Obstet. Gynecol. 2018, 218, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Bdolah, Y.; Palomaki, G.E.; Yaron, Y.; Bdolah-Abram, T.; Goldman, M.; Levine, R.J.; Sachs, B.P.; Haddow, J.E.; Karumanchi, S.A. Circulating angiogenic proteins in trisomy 13. Am. J. Obstet. Gynecol. 2006, 194, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.E.; Crawford, S.L.; Bathgate, S.; Yan, J.; Robidoux, L.; Moore, M.; Moore Simas, T.A. Gestational angiogenic biomarker patterns in high risk preeclampsia groups. Am. J. Obstet. Gynecol. 2013, 209, e1–e9. [Google Scholar] [CrossRef]
- Koga, K.; Osuga, Y.; Tajima, T.; Hirota, Y.; Igarashi, T.; Fujii, T.; Yano, T.; Taketani, Y. Elevated serum soluble fms-like tyrosine kinase 1 (sFlt1) level in women with hydatidiform mole. Fertil. Steril. 2010, 94, 305–308. [Google Scholar] [CrossRef]
- Goa, S.; Mimura, K.; Kakigano, A.; Tomimatsu, T.; Kinugasa-Taniguchi, Y.; Endo, M.; Kanagawa, T.; Kimura, T. Normalisation of angiogenic imbalance after intra-uterine transfusion for mirror syndrome caused by parvovirus B19. Fetal. Diagn. Ther. 2013, 34, 176–179. [Google Scholar] [CrossRef]
- Robillard, P.Y.; Dekker, G.A.; Hulsey, T.C. Revisiting the epidemiological standard of preeclampsia: Primigravidity or primipaternity? Eur. J. Obstet. Gynecol. Reprod. Biol. 1999, 84, 37–41. [Google Scholar] [CrossRef]
- Deen, M.E.; Ruurda, L.G.; Wang, J.; Dekker, G.A. Risk factors for preeclampsia in multiparous women: Primipaternity versus the birth interval hypothesis. J. Matern. Fetal Neonatal Med. 2006, 19, 79–84. [Google Scholar] [CrossRef]
- Zhang, H.N.; Xu, Q.Q.; Thakur, A.; Alfred, M.O.; Chakraborty, M.; Ghosh, A.; Yu, X.B. Endothelial dysfunction in diabetes and hypertension: Role of microRNAs and long non-coding RNAs. Life Sci. 2018, 213, 258–268. [Google Scholar] [CrossRef]
- Roberts, J.M.; Hubel, C.A. The two stage model of preeclampsia: Variations on the theme. Placenta 2009, 30, S32–S37. [Google Scholar] [CrossRef] [PubMed]
- Brosens, I.A.; Robertson, W.B.; Dixon, H.G. The role of the spiral arteries in the pathogenesis of preeclampsia. Obstet. Gynecol. Annu. 1972, 1, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Robertson, W.B.; Brosens, I.; Dixon, H.G. The pathological response of the vessels of the placental bed to hypertensive pregnancy. J. Pathol. Bacteriol. 1967, 93, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Pijnenborg, R.; Anthony, J.; Davey, D.A.; Rees, A.; Tiltman, A.; Vercruysse, L.; van Assche, A. Placental bed spiral arteries in the hypertensive disorders of pregnancy. Br. J. Obstet. Gynaecol. 1991, 98, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Damsky, C.H.; Chiu, K.; Roberts, J.M.; Fisher, S.J. Preeclampsia is associated with abnormal expression of adhesion molecules by invasive cytotrophoblasts. J. Clin. Investig. 1993, 91, 950–960. [Google Scholar] [CrossRef] [PubMed]
- Hecht, J.L.; Zsengeller, Z.K.; Spiel, M.; Karumanchi, S.A.; Rosen, S. Revisiting decidual vasculopathy. Placenta 2016, 42, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Tomimatsu, T.; Mimura, K.; Endo, M.; Kumasawa, K.; Kimura, T. Pathophysiology of preeclampsia: An angiogenic imbalance and long-lasting systemic vascular dysfunction. Hypertens. Res. 2017, 40, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Kendall, R.L.; Thomas, K.A. Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 10705–10709. [Google Scholar] [CrossRef] [PubMed]
- Mustonen, T.; Alitalo, K. Endothelial receptor tyrosine kinases involved in angiogenesis. J. Cell Biol. 1995, 129, 895–898. [Google Scholar] [CrossRef]
- Clark, D.E.; Smith, S.K.; He, Y.; Day, K.A.; Licence, D.R.; Corps, A.N.; Lammoglia, R.; Charnock-Jones, D.S. A vascular endothelial growth factor antagonist is produced by the human placenta and released into the maternal circulation. Biol. Reprod. 1998, 59, 1540–1548. [Google Scholar] [CrossRef]
- Van der Zee, R.; Murohara, T.; Luo, Z.; Zollmann, F.; Passeri, J.; Lekutat, C.; Isner, J.M. Vascular endothelial growth factor/vascular permeability factor augments nitric oxide release from quiescent rabbit and human vascular endothelium. Circulation 1997, 95, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, J.R.; Rivard, A.; van der Zee, R.; Hariawala, M.; Sheriff, D.D.; Esakof, D.D.; Chaudhry, G.M.; Symes, J.F.; Isner, J.M. Vascular endothelial growth factor/vascular permeability factor produces nitric oxide-dependent hypotension. Evidence for a maintenance role in quiescent adult endothelium. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2793–2800. [Google Scholar] [CrossRef] [PubMed]
- Bouloumié, A.; Schini-Kerth, V.B.; Busse, R. Vascular endothelial growth factor up-regulates nitric oxide synthase expression in endothelial cells. Cardiovasc. Res. 1999, 41, 773–780. [Google Scholar] [CrossRef]
- Hood, J.D.; Meininger, C.J.; Ziche, M.; Granger, H.J. VEGF upregulates ecNOS message, protein, and NO production in human endothelial cells. Am. J. Physiol. 1998, 274, H1054–H1058. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ogasawara, A.K.; Yang, R.; Wei, W.; He, G.W.; Zioncheck, T.F.; Bunting, S.; de Vos, A.M.; Jin, H. KDR (VEGF receptor 2) is the major mediator for the hypotensive effect of VEGF. Hypertension 2002, 39, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Neagoe, P.E.; Lemieux, C.; Sirois, M.G. Vascular endothelial growth factor (VEGF)-A165-induced prostacyclin synthesis requires the activation of VEGF receptor-1 and -2 heterodimer. J. Biol. Chem. 2005, 280, 9904–9912. [Google Scholar] [CrossRef] [PubMed]
- Wheeler-Jones, C.; Abu-Ghazaleh, R.; Cospedal, R.; Houliston, R.A.; Martin, J.; Zachary, I. Vascular endothelial growth factor stimulates prostacyclin production and activation of cytosolic phospholipase A2 in endothelial cells via p42/p44 mitogen-activated protein kinase. FEBS Lett. 1997, 420, 28–32. [Google Scholar] [CrossRef]
- Robinson, E.S.; Khankin, E.V.; Karumanchi, S.A.; Humphreys, B.D. Hypertension induced by vascular endothelial growth factor signaling pathway inhibition: Mechanisms and potential use as a biomarker. Semin. Nephrol. 2010, 30, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Eremina, V.; Jefferson, J.A.; Kowalewska, J.; Hochster, H.; Haas, M.; Weisstuch, J.; Richardson, C.; Kopp, J.B.; Kabir, M.G.; Backx, P.H.; et al. VEGF inhibition and renal thrombotic microangiopathy. N. Engl. J. Med. 2008, 358, 1129–1136. [Google Scholar] [CrossRef]
- Patel, T.V.; Morgan, J.A.; Demetri, G.D.; George, S.; Maki, R.G.; Quigley, M.; Humphreys, B.D. A preeclampsia-like syndrome characterized by reversible hypertension and proteinuria induced by the multitargeted kinase inhibitors sunitinib and sorafenib. J. Natl. Cancer Inst. 2008, 100, 282–284. [Google Scholar] [CrossRef]
- Vigneau, C.; Lorcy, N.; Dolley-Hitze, T.; Jouan, F.; Arlot-Bonnemains, Y.; Laguerre, B.; Verhoest, G.; Goujon, J.M.; Belaud-Rotureau, M.A.; Rioux-Leclercq, N. All anti-vascular endothelial growth factor drugs can induce ‘pre- eclampsia-like syndrome’: A RARe study. Nephrol. Dial. Transplant. 2014, 29, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Launay-Vacher, V.; Deray, G. Hypertension and proteinuria: A class-effect of antiangiogenic therapies. Anticancer Drugs 2009, 20, 81–82. [Google Scholar] [CrossRef] [PubMed]
- Thijs, A.M.J.; van Herpen, C.M.L.; Sweep, F.C.G.J.; Geurts-Moespot, A.; Smits, P.; van der Graaf, W.T.A.; Rongen, G.A. Role of endogenous vascular endothelial growth factor in endothelium-dependent vasodilation in humans. Hypertension 2013, 61, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Maitland, M.L.; Bakris, G.L.; Black, H.R.; Chen, H.X.; Durand, J.B.; Elliott, W.J.; Ivy, S.P.; Leier, C.V.; Lindenfeld, J.; Liu, G.; et al. Initial assessment, surveillance, and management of blood pressure in patients receiving vascular endothelial growth factor signaling pathway inhibitors. J. Natl. Cancer Inst. 2010, 102, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Lankhorst, S.; Kappers, M.H.W.; van Esch, J.H.M.; Danser, A.H.J.; van den Meiracker, A.H. Mechanism of hypertension and proteinuria during angiogenesis inhibition: Evolving role of endothelin-1. J. Hypertens. 2013, 31, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Lankhorst, S.; Danser, A.H.; van den Meiracker, A.H. Endothelin-1 and antiangiogenesis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R230–R234. [Google Scholar] [CrossRef] [PubMed]
- Kappers, M.H.W.; de Beer, V.J.; Zhou, Z.; Danser, A.H.J.; Sleijfer, S.; Duncker, D.J.; van den Meiracker, A.H.; Merkus, D. Sunitinib-induced systemic vasoconstriction in swine is endothelin mediated and does not involve nitric oxide or oxidative stress. Hypertension 2012, 59, 151–157. [Google Scholar] [CrossRef] [PubMed]
- George, E.M.; Palei, A.C.; Granger, J.P. Endothelin as a final common pathway in the pathophysiology of preeclampsia: Therapeutic implications. Curr. Opin. Nephrol. Hypertens. 2012, 21, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, K.; Saleh, L.; Lankhorst, S.; Smilde, J.E.; van Ingen, M.M.; Garrelds, I.M.; Friesema, E.C.; Russcher, H.; van den Meiracker, A.H.; Visser, W.; et al. Association studies suggest a key role for endothelin-1 in the pathogenesis of preeclampsia and the accompanying renin-angiotensin-aldosterone system suppression. Hypertension 2015, 65, 1316–1323. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, A.; Kawashima, S.; Yamochi, W.; Hirata, K.; Yamaguchi, T.; Emoto, N.; Yokoyama, M. Vascular endothelial growth factor increases endothelin-converting enzyme expression in vascular endothelial cells. Biochem. Biophys. Res. Commun. 1997, 235, 713–716. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, A.; Yamochi, W.; Hirata, K.I.; Kawashima, S.; Yokoyama, M. Stimulatory interaction between vascular endothelial growth factor and endothelin-1 on each gene expression. Hypertension 1998, 32, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Eremina, V.; Quaggin, S.E. The role of VEGF-A in glomerular development and function. Curr. Opin. Nephrol. Hypertens. 2004, 13, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Mattot, V.; Moons, L.; Lupu, F.; Chernavvsky, D.; Gómez, R.A.; Collen, D.; Carmeliet, P. Loss of the VEGF(164) and VEGF(188) isoforms impairs postnatal glomerular angiogenesis and renal arteriogenesis in mice. J. Am. Soc. Nephrol. 2002, 13, 1548–1560. [Google Scholar] [CrossRef] [PubMed]
- Eremina, V.; Sood, M.; Haigh, J.; Nagy, A.; Lajoie, G.; Ferrara, N.; Gerber, H.P.; Kikkawa, Y.; Miner, J.H.; Quaggin, S.E. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J. Clin. Investig. 2003, 111, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Zou, A.P.; Cowley, A.W. Role of nitric oxide in the control of renal function and salt sensitivity. Curr. Hypertens. Rep. 1999, 1, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Facemire, C.S.; Nixon, A.B.; Griffiths, R.; Hurwitz, H.; Coffman, T.M. Vascular endothelial growth factor receptor 2 controls blood pressure by regulating nitric oxide synthase expression. Hypertension 2009, 54, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Lankhorst, S.; Kappers, M.H.; van Esch, J.H.; Smedts, F.M.; Sleijfer, S.; Mathijssen, R.H.; Baelde, H.J.; Danser, A.H.; van den Meiracker, A.H. Treatment of hypertension and renal injury induced by the angiogenesis inhibitor sunitinib: Preclinical study. Hypertension 2014, 64, 1289–1290. [Google Scholar] [CrossRef] [PubMed]
- Derzsy, Z.; Prohaszka, Z.; Rigo, J.J.; Fust, G.; Molvarec, A. Activation of the complement system in normal pregnancy and preeclampsia. Mol. Immunol. 2010, 47, 1500–1506. [Google Scholar] [CrossRef] [PubMed]
- Jean, F.R.; Richard, M.B.; Sherry, D.F. The complement system and preeclampsia. Curr. Hypertens. Rep. 2017, 19, 2–12. [Google Scholar]
- Vaught, A.J.; Gavriilaki, E.; Hueppchen, N.; Blakemore, K.; Yuan, X.; Seifert, S.M.; York, S.; Brodsky, R.A. Direct evidence of complement activation in HELLP syndrome: A link to atypical hemolytic uremic syndrome. Exp. Hematol. 2016, 44, 390–398. [Google Scholar] [CrossRef]
- Vaught, A.J.; Braunstein, E.M.; Jasem, J.; Yuan, X.; Makhlin, I.; Eloundou, S.; Baines, A.C.; Merrill, S.A.; Chaturvedi, S.; Blakemore, K.; et al. Germline mutations in the alternative pathway of complement predispose to HELLP syndrome. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Burwick, R.M.; Fichorova, R.N.; Dawood, H.Y.; Yamamoto, H.S.; Feinberg, B.B. Urinary excretion of c5b-9 in severe preeclampsia: Tipping the balance of complement activation in pregnancy. Hypertension 2013, 62, 1040–1045. [Google Scholar] [CrossRef] [PubMed]
- Burwick, R.M.; Velásquez, J.A.; Valencia, C.M.; Gutiérrez-Marín, J.; Edna-Estrada, F.; Silva, J.L.; Trujillo-Otálvaro, J.; Vargas-Rodríguez, J.; Bernal, Y.; Quintero, A.; et al. Terminal complement activation in preeclampsia. Obstet. Gynecol. 2018, 132, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Penning, M.; Chua, J.S.; van Kooten, C.; Zandbergen, M.; Buurma, A.; Schutte, J.; Bruijn, J.A.; Khankin, E.V.; Bloemenkamp, K.; Karumanchi, S.A.; et al. Classical complement pathway activation in the kidneys of women with preeclampsia. Hypertension 2015, 66, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Vakiti, A.; Singh, D.; Pilla, R.; Alhaj-Moustafa, M.; Fitzpatrick, K.W. Bevacizumab-induced atypical hemolytic uremic syndrome and treatment with eculizumab. J. Oncol. Pharm. Pract. 2019, 25, 1011–1015. [Google Scholar] [CrossRef] [PubMed]
- Keir, L.S.; Firth, R.; Aponik, L.; Feitelberg, D.; Sakimoto, S.; Aguilar, E.; Welsh, G.I.; Richards, A.; Usui, Y.; Satchell, S.C.; et al. VEGF regulates local inhibitory complement proteins in the eye and kidney. J. Clin. Investig. 2017, 127, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Kong, L.R.; Ge, Q.; Lu, Y.Y.; Hong, M.N.; Zhang, Y.; Ruan, C.C.; Gao, P.J. Complement 5a-mediated trophoblasts dysfunction is involved in the development of pre-eclampsia. J. Cell Mol. Med. 2018, 22, 1034–1046. [Google Scholar] [CrossRef]
- Nagamatsu, T.; Fujii, T.; Kusumi, M.; Zou, L.; Yamashita, T.; Osuga, Y.; Momoeda, M.; Kozuma, S.; Taketani, Y. Cytotrophoblasts up-regulate soluble fms-like tyrosine kinase-1 expression under reduced oxygen: An implication for the placental vascular development and the pathophysiology of preeclampsia. Endocrinology 2004, 145, 4838–4845. [Google Scholar] [CrossRef]
- Nevo, O.; Soleymanlou, N.; Wu, Y.; Xu, J.; Kingdom, J.; Many, A.; Zamudio, S.; Caniggia, I. Increased expression of sFlt-1 in in vivo and in vitro models of human placental hypoxia is mediated by HIF-1. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, R1085–R1093. [Google Scholar] [CrossRef]
- Girardi, G.; Yarilin, D.; Thurman, J.M.; Holers, V.M.; Salmon, J.E. Complement activation induces dysregulation of angiogenic factors and causes fetal rejection and growth restriction. J. Exp. Med. 2006, 203, 2165–2175. [Google Scholar] [CrossRef]
- Vaka, V.R.; McMaster, K.M.; Cunningham, M.W., Jr.; Ibrahim, T.; Hazlewood, R.; Usry, N.; Cornelius, D.C.; Amaral, L.M.; LaMarca, B. Role of mitochondrial dysfunction and reactive oxygen species in mediating hypertension in the reduced uterine perfusion pressure rat model of preeclampsia. Hypertension 2018, 72, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Rai, A.; Kambham, N.; Sung, J.F.; Singh, N.; Petitt, M.; Dhal, S.; Agrawal, R.; Sutton, R.E.; Druzin, M.L.; et al. Endometrial VEGF induces placental sFLT1 and leads to pregnancy complications. J. Clin. Investig. 2014, 124, 4941–4952. [Google Scholar] [CrossRef] [PubMed]
- Mashini, I.S.; Albazzaz, S.J.; Fadel, H.E.; Abdulla, A.M.; Hadi, H.A.; Harp, R.; Devoe, L.D. Serial noninvasive evaluation of cardiovascular hemodynamics during pregnancy. Am. J. Obstet. Gynecol. 1987, 156, 1208–1213. [Google Scholar] [CrossRef]
- Robson, S.C.; Hunter, S.; Boys, R.J.; Dunlop, W. Serial study of factors influencing changes in cardiac output during human pregnancy. Am. J. Physiol. 1989, 256, H1060–H1065. [Google Scholar] [CrossRef] [PubMed]
- Duvekot, J.J.; Cheriex, E.C.; Pieters, F.A.; Menheere, P.P.; Peeters, L.H. Early pregnancy changes in hemodynamics and volume homeostasis are consecutive adjustments triggered by a primary fall in systemic vascular tone. Am. J. Obstet. Gynecol. 1993, 169, 1382–1392. [Google Scholar] [CrossRef]
- Mabie, W.C.; DiSessa, T.G.; Crocker, L.G.; Sibai, B.M.; Arheart, K.L. A longitudinal study of cardiac output in normal human pregnancy. Am. J. Obstet. Gynecol. 1994, 170, 849–856. [Google Scholar] [CrossRef]
- Christianson, R.E. Studies on blood pressure during pregnancy. I. Influence of parity and age. Am. J. Obstet. Gynecol. 1976, 125, 509–513. [Google Scholar] [CrossRef]
- Moutquin, J.M.; Rainville, C.; Giroux, L.; Raynauld, P.; Amyot, G.; Bilodeau, R.; Pelland, N. A prospective study of blood pressure in pregnancy: Prediction of preeclampsia. Am. J. Obstet. Gynecol. 1985, 151, 191–196. [Google Scholar] [CrossRef]
- Yu, W.; Gao, W.; Rong, D.; Wu, Z.; Khalil, R.A. Molecular determinants of microvascular dysfunction in hypertensive pregnancy and preeclampsia. Microcirculation 2019, 26, e12508. [Google Scholar] [CrossRef]
- Tomiyama, H.; Yamashina, A. Non-invasive vascular function tests: Their pathophysiological background and clinical application. Circ. J. 2010, 74, 24–33. [Google Scholar] [CrossRef]
- Dørup, I.; Skajaa, K.; Sørensen, K.E. Normal pregnancy is associated with enhanced endothelium-dependent flow-mediated vasodilation. Am. J. Physiol. 1999, 276, H821–H825. [Google Scholar] [CrossRef] [PubMed]
- Weissgerber, T.L.; Milic, N.M.; Milin-Lazovic, J.S.; Garovic, V.D. Impaired flow-mediated dilation before, during, and after preeclampsia: A systematic review and meta-analysis. Hypertension 2016, 67, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.; Hayward, C.S.; Avolio, A.; O’Rourke, M.F. Non-invasive registration of the arterial pressure pulse waveform using high-fidelity applanation tonometry. J. Vasc. Med. Biol. 1989, 1, 142–149. [Google Scholar]
- Nichols, W.W. Clinical measurement of arterial stiffness obtained from noninvasive pressure waveforms. Am. J. Hypertens. 2005, 18, 3S–10S. [Google Scholar] [CrossRef] [PubMed]
- Nichols, W.W.; Singh, B.M. Augmentation index as a measure of peripheral vascular disease state. Curr. Opin. Cardiol. 2002, 17, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Fujime, M.; Tomimatsu, T.; Okaue, Y.; Koyama, S.; Kanagawa, T.; Taniguchi, T.; Kimura, T. Central aortic blood pressure and augmentation index during normal pregnancy. Hypertens. Res. 2012, 35, 633–638. [Google Scholar] [CrossRef]
- Macedo, M.L.; Luminoso, D.; Savvidou, M.D.; McEniery, C.M.; Nicolaides, K.H. Maternal wave reflections and arterial stiffness in normal pregnancy as assessed by applanation tonometry. Hypertension 2008, 51, 1047–1051. [Google Scholar] [CrossRef]
- Khalil, A.; Jauniaux, E.; Cooper, D.; Harrington, K. Pulse wave analysis in normal pregnancy: A prospective longitudinal study. PLoS ONE 2009, 4, e6134. [Google Scholar] [CrossRef]
- Spasojevic, M.; Smith, S.A.; Morris, J.M.; Gallery, E.D. Peripheral arterial pulse wave analysis in women with pre-eclampsia and gestational hypertension. BJOG 2005, 112, 1475–1478. [Google Scholar] [CrossRef]
- Khalil, A.; Jauniaux, E.; Harrington, K. Antihypertensive therapy and central hemodynamics in women with hypertensive disorders in pregnancy. Obstet. Gynecol. 2009, 113, 646–654. [Google Scholar] [CrossRef]
- Khalil, A.A.; Cooper, D.J.; Harrington, K.F. Pulse wave analysis: A preliminary study of a novel technique for the prediction of pre-eclampsia. BJOG 2009, 116, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.; Akolekar, R.; Syngelaki, A.; Elkhouli, M.; Nicolaides, K.H. Maternal hemodynamics at 11–13 weeks’ gestation and risk of pre-eclampsia. Ultrasound Obstet. Gynecol. 2012, 40, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Yinon, Y.; Kingdom, J.C.; Odutayo, A.; Moineddin, R.; Drewlo, S.; Lai, V.; Cherney, D.Z.; Hladunewich, M.A. Vascular dysfunction in women with a history of preeclampsia and intrauterine growth restriction: Insights into future vascular risk. Circulation 2010, 122, 1846–1853. [Google Scholar] [CrossRef] [PubMed]
- Tomimatsu, T.; Fujime, M.; Kanayama, T.; Mimura, K.; Koyama, S.; Kanagawa, T.; Kimura, T. Maternal arterial stiffness in normotensive pregnant women who subsequently deliver babies that are small for gestational age. Eur. J. Obstet. Gynecol. Reprod. Biol. 2013, 169, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Tomimatsu, T.; Fujime, M.; Kanayama, T.; Mimura, K.; Koyama, S.; Kanagawa, T.; Endo, M.; Shimoya, K.; Kimura, T. Abnormal pressure-wave reflection in pregnant women with chronic hypertension: Association with maternal and fetal outcomes. Hypertens. Res. 2014, 37, 989–992. [Google Scholar] [CrossRef]
- Asmar, R. Pulse wave velocity: Principle and measurement. In Arterial Stiffness and Pulse Wave Velocity; Asmar, R., Ed.; Elsevier: Amsterdam, The Netherlands, 1999; pp. 25–56. [Google Scholar]
- McEniery, C.M.; Yasmin; Hall, I.R.; Qasem, A.; Wilkinson, I.B.; Cockcroft, J.R. Normal vascular aging: Differential effects on wave reflection and aortic pulse wave velocity: The Anglo-Cardiff Collaborative Trial (ACCT). J. Am. Coll. Cardiol. 2005, 46, 1753–1760. [Google Scholar] [CrossRef]
- Oyama-Kato, M.; Ohmichi, M.; Takahashi, K.; Suzuki, S.; Henmi, N.; Yokoyama, Y.; Kurachi, H. Change in pulse wave velocity throughout normal pregnancy and its value in predicting pregnancy-induced hypertension: A longitudinal study. Am. J. Obstet. Gynecol. 2006, 195, 464–469. [Google Scholar] [CrossRef]
- Kaihura, C.; Savvidou, M.D.; Anderson, J.M.; McEniery, C.M.; Nicolaides, K.H. Maternal arterial stiffness in pregnancies affected by preeclampsia. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H759–H764. [Google Scholar] [CrossRef]
- Bujold, E.; Roberge, S.; Lacasse, Y.; Bureau, M.; Audibert, F.; Marcoux, S.; Forest, J.C.; Giguère, Y. Prevention of preeclampsia and intrauterine growth restriction with aspirin started in early pregnancy: A meta-analysis. Obstet. Gynecol. 2010, 116, 402–414. [Google Scholar] [CrossRef]
- Jacobs, M.; Nassar, N.; Roberts, C.L.; Hadfield, R.; Morris, J.M.; Ashton, A.W. Levels of soluble fms-like tyrosine kinase one in first trimester and outcomes of pregnancy: A systematic review. Reprod. Biol. Endocrinol. 2011, 9, 77. [Google Scholar] [CrossRef]
- Poon, L.C.; Nicolaides, K.H. First-trimester maternal factors and biomarker screening for preeclampsia. Prenat. Diagn. 2014, 34, 618–627. [Google Scholar] [CrossRef]
- O’Gorman, N.; Wright, D.; Poon, L.C.; Rolnik, D.L.; Syngelaki, A.; de Alvarado, M.; Carbone, I.F.; Dutemeyer, V.; Fiolna, M.; Frick, A.; et al. Multicenter screening for pre-eclampsia by maternal factors and biomarkers at 11–13 weeks’ gestation: Comparison with NICE guidelines and ACOG recommendations. Ultrasound Obstet. Gynecol. 2017, 49, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Rolnik, D.L.; Wright, D.; Poon, L.C.; O’Gorman, N.; Syngelaki, A.; de Paco Matallana, C.; Akolekar, R.; Cicero, S.; Janga, D.; Singh, M.; et al. Aspirin versus placebo in pregnancies at high risk for preterm preeclampsia. N. Engl. J. Med. 2017, 377, 613–622. [Google Scholar] [CrossRef]
- Sibai, B.M.; Stella, C.L. Diagnosis and management of atypical preeclampsia-eclampsia. Am. J. Obstet. Gynecol. 2009, 200, e1–e7. [Google Scholar] [CrossRef] [PubMed]
- Stella, C.L.; Malik, K.M.; Sibai, B.M. HELLP syndrome: An atypical presentation. Am. J. Obstet. Gynecol. 2008, 198, e6–e8. [Google Scholar] [CrossRef] [PubMed]
- Ohno, Y.; Terauchi, M.; Tamakoshi, K.; Shiozaki, A.; Saito, S. The risk factors for labor onset hypertension. Hypertens. Res. 2016, 39, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Powe, C.E.; Salahuddin, S.; Verlohren, S.; Perschel, F.H.; Levine, R.J.; Lim, K.H.; Wenger, J.B.; Thadhani, R.; Karumanchi, S.A. Angiogenic factors and the risk of adverse outcomes in women with suspected preeclampsia. Circulation 2012, 125, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Chaiworapongsa, T.; Romero, R.; Savasan, Z.A.; Kusanovic, J.P.; Ogge, G.; Soto, E.; Dong, Z.; Tarca, A.; Gaurav, B.; Hassan, S.S. Maternal plasma concentrations of angiogenic/antiangiogenic factors are of prognostic value in patients presenting to the obstetrical triage area with the suspicion of preeclampsia. J. Matern. Fetal. Neonatal. Med. 2011, 24, 1187–1207. [Google Scholar] [CrossRef]
- Rana, S.; Schnettler, W.T.; Powe, C.; Wenger, J.; Salahuddin, S.; Cerdeira, A.S.; Verlohren, S.; Perschel, F.H.; Arany, Z.; Lim, K.H.; et al. Clinical characterization and outcomes of preeclampsia with normal angiogenic profile. Hypertens. Pregnancy 2013, 32, 189–201. [Google Scholar] [CrossRef]
- Zeisler, H.; Llurba, E.; Chantraine, F.; Vatish, M.; Staff, A.C.; Sennström, M.; Olovsson, M.; Brennecke, S.P.; Stepan, H.; Allegranza, D.; et al. Predictive value of the sFlt-1:PlGF ratio in women with suspected preeclampsia. N. Engl. J. Med. 2016, 374, 13–22. [Google Scholar] [CrossRef]
- Zeisler, H.; Llurba, E.; Chantraine, F.; Vatish, M.; Staff, A.C.; Sennström, M.; Olovsson, M.; Brennecke, S.P.; Stepan, H.; Allegranza, D.; et al. Soluble fms-like tyrosine kinase-1-to-placental growth factor ratio and time to delivery in women with suspected preeclampsia. Obstet. Gynecol. 2016, 128, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Saleh, L.; van den Meiracker, A.H.; Geensen, R.; Kaya, A.; Roeters van Lennep, J.E.; Duvekot, J.J.; Verdonk, K.; Steegers, E.A.P.; Russcher, H.; Danser, A.H.J.; et al. Soluble fms-like tyrosine kinase-1 and placental growth factor kinetics during and after pregnancy in women with suspected or confirmed pre-eclampsia. Ultrasound Obstet. Gynecol. 2018, 51, 751–757. [Google Scholar] [CrossRef]
- Duhig, K.E.; Myers, J.; Seed, P.T.; Sparkes, J.; Lowe, J.; Hunter, R.M.; Shennan, A.H.; Chappell, L.C.; PARROT trial group. Placental growth factor testing to assess women with suspected pre-eclampsia: A multicentre, pragmatic, stepped-wedge cluster-randomised controlled trial. Lancet 2019, 393, 1807–1818. [Google Scholar] [CrossRef]
- Rolfo, A.; Attini, R.; Nuzzo, A.M.; Piazzese, A.; Parisi, S.; Ferraresi, M.; Todros, T.; Piccoli, G.B. Chronic kidney disease may be differentially diagnosed from preeclampsia by serum biomarkers. Kidney Int. 2013, 83, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Young, B.; Levine, R.J.; Salahuddin, S.; Qian, C.; Lim, K.H.; Karumanchi, S.A.; Rana, S. The use of angiogenic biomarkers to differentiate non-HELLP related thrombocytopenia from HELLP syndrome. J. Matern. Fetal Neonat. Med. 2010, 23, 366–370. [Google Scholar] [CrossRef]
- Perni, U.; Sison, C.; Sharma, V.; Helseth, G.; Hawfield, A.; Suthanthiran, M.; August, P. Angiogenic factors in superimposed preeclampsia: A longitudinal study of women with chronic hypertension during pregnancy. Hypertension 2012, 59, 740–746. [Google Scholar] [CrossRef]
- Leaños-Miranda, A.; Campos-Galicia, I.; Berumen-Lechuga, M.G.; Molina-Pérez, C.J.; García-Paleta, Y.; Isordia-Salas, I.; Ramírez-Valenzuela, K.L. Circulating angiogenic factors and the risk of preeclampsia in systemic lupus erythematosus pregnancies. J. Rheumatol. 2015, 42, 1141–1149. [Google Scholar] [CrossRef]
- Qazi, U.; Lam, C.; Karumanchi, S.A.; Petri, M. Soluble Fms-like tyrosine kinase associated with preeclampsia in pregnancy in systemic lupus erythematosus. J. Rheumatol. 2008, 35, 631–634. [Google Scholar]
- Kim, M.Y.; Buyon, J.P.; Guerra, M.M.; Rana, S.; Zhang, D.; Laskin, C.A.; Petri, M.; Lockshin, M.D.; Sammaritano, L.R.; Branch, D.W.; et al. Angiogenic factor imbalance early in pregnancy predicts adverse outcomes in patients with lupus and antiphospholipid antibodies: Results of the PROMISSE study. Am. J. Obstet. Gynecol. 2016, 214, 108. [Google Scholar] [CrossRef]
- Karumanchi, S.A. Angiogenic factors in preeclampsia: From diagnosis to therapy. Hypertension 2016, 67, 1072–1079. [Google Scholar] [CrossRef]
- Phipps, E.A.; Thadhani, R.; Benzing, T.; Karumanchi, S.A. Pre-eclampsia: Pathogenesis, novel diagnostics and therapies. Nat. Rev. Nephrol. 2019, 15, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Kumasawa, K.; Ikawa, M.; Kidoya, H.; Hasuwa, H.; Saito-Fujita, T.; Morioka, Y.; Takakura, N.; Kimura, T.; Okabe, M. Pravastatin induces placental growth factor (PGF) and ameliorates preeclampsia in a mouse model. Proc. Natl. Acad. Sci. USA 2011, 108, 1451–1455. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.S.; Verzwyvelt, J.; Colson, D.; Arany, M.; Karumanchi, S.A.; Granger, J.P. Recombinant vascular endothelial growth factor 121 infusion lowers blood pressure and improves renal function in rats with placental ischemia-induced hypertension. Hypertension 2010, 55, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Ohkuchi, A.; Matsubara, S.; Takei, Y.; Murakami, M.; Shibuya, M.; Suzuki, M.; Sato, Y. Effect of recombinant placental growth factor 2 on hypertension induced by full-length mouse soluble fms-like tyrosine kinase 1 adenoviral vector in pregnant mice. Hypertension 2009, 54, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Mimura, K.; Tomimatsu, T.; Sharentuya, N.; Tskitishvili, E.; Kinugasa-Taniguchi, Y.; Kanagawa, T.; Kimura, T. Nicotine restores endothelial dysfunction caused by excess sFlt1 and sEng in an in vitro model of preeclamptic vascular endothelium: A possible therapeutic role of nicotinic acetylcholine receptor (nAChR) agonists for preeclampsia. Am. J. Obstet. Gynecol. 2010, 202, e1–e6. [Google Scholar] [CrossRef] [PubMed]
- Kakigano, A.; Tomimatsu, T.; Mimura, K.; Kanayama, T.; Fujita, S.; Minato, K.; Kumasawa, K.; Taniguchi, Y.; Kanagawa, T.; Endo, M.; et al. Drug repositioning for preeclampsia therapeutics by in vitro screening: phosphodiesterase-5 inhibitor vardenafil restores endothelial dysfunction via induction of placental growth factor. Reprod. Sci. 2015, 22, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Turanov, A.A.; Lo, A.; Hassler, M.R.; Makris, A.; Ashar-Patel, A.; Alterman, J.F.; Coles, A.H.; Haraszti, R.A.; Roux, L.; Godinho, B.M.D.C.; et al. RNAi modulation of placental sFLT1 for the treatment of preeclampsia. Nat. Biotechnol. 2018, 36, 1164–1173. [Google Scholar] [CrossRef] [PubMed]
- Thadhani, R.; Kisner, T.; Hagmann, H.; Bossung, V.; Noack, S.; Schaarschmidt, W.; Jank, A.; Kribs, A.; Cornely, O.A.; Kreyssig, C.; et al. Pilot study of extracorporeal removal of soluble fms-like tyrosine kinase 1 in preeclampsia. Circulation 2011, 124, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.W. Low-dose aspirin: treatment for the imbalance of increased thromboxane and decreased prostacyclin in preeclampsia. Am. J. Perinatol. 1989, 6, 124–132. [Google Scholar] [CrossRef]
- Smith, W.L. The eicosanoids and their biochemical mechanisms of action. Biochem. J. 1989, 259, 315. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. COX-2 inhibitors and metabolism of essential fatty acids. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2005, 11, RA233–RA237. [Google Scholar]
- Sibai, B.M.; Mirro, R.; Chesney, C.M.; Leffler, C. Low-dose aspirin in pregnancy. Obstet. Gynecol. 1989, 74, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.S.; Joyce, R.M.; McLeod, L.J.; Vial, J.H.; Seville, P.R. Slow release aspirin and prostaglandin inhibition. Lancet Lond. Engl. 1986, 1, 1153–1154. [Google Scholar] [CrossRef]
- Perneby, C.; Vahter, M.; Akesson, A.; Bremme, K.; Hjemdahl, P. Thromboxane metabolite excretion during pregnancy—influence of preeclampsia and aspirin treatment. Thromb. Res. 2011, 127, 605–606. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Raikwar, N.S.; Santillan, M.K.; Santillan, D.A.; Thomas, C.P. Aspirin inhibits expression of sFLT1 from human cytotrophoblasts induced by hypoxia, via cyclo-oxygenase 1. Placenta 2015, 36, 446–453. [Google Scholar] [CrossRef]
- Panagodage, S.; Yong, H.E.; Da Silva Costa, F.; Borg, A.J.; Kalionis, B.; Brennecke, S.P.; Murthi, P. Low-dose acetylsalicylic acid treatment modulates the production of cytokines and improves trophoblast function in an in vitro model of early-onset preeclampsia. Am. J. Pathol. 2016, 186, 3217–3224. [Google Scholar] [CrossRef]
- Beaufils, M.; Donsimoni, R.; Uzan, S.; Colau, J.C. Prevention of preeclampsia by early antiplatelet therapy. Lancet 1985, 325, 840–842. [Google Scholar] [CrossRef]
- Beroyz, G.; Casale, R.; Farreiros, A. CLASP: A randomised trial of low-dose aspirin for the prevention and treatment of pre-eclampsia among 9364 pregnant women. Lancet 1994, 343, 619–629. [Google Scholar]
- Caritis, S.; Sibai, B.; Hauth, J.; Lindheimer, M.D.; Klebanoff, M.; Thom, E.; Van Dorsten, P.; Landon, M.; Paul, R.; Miodovnik, M.; et al. Low-dose aspirin to prevent preeclampsia in women at high risk. N. Engl. J. Med. 1998, 338, 701–705. [Google Scholar] [CrossRef]
- Duley, L.; Henderson-Smart, D.; Knight, M.; King, J. Antiplatelet drugs for prevention of pre-eclampsia and its consequences: Systematic review. BMJ 2001, 322, 329–333. [Google Scholar] [CrossRef]
- Kemp, M.; Thomas, W. Antiphospholipid syndrome in obstetrics. Lupus 2018, 27, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Girardi, G.; Redecha, P.; Salmon, J.E. Heparin prevents antiphospholipid antibody-induced fetal loss by inhibiting complement activation. Nat. Med. 2004, 10, 1222–1226. [Google Scholar] [CrossRef] [PubMed]
- Roberge, S.; Demers, S.; Nicolaides, K.H.; Bureau, M.; Côté, S.; Bujold, E. Prevention of pre-eclampsia by low-molecular-weight heparin in addition to aspirin: A meta-analysis. Ultrasound Obstet. Gynecol. 2016, 47, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Groom, K.M.; McCowan, L.M.; Mackay, L.K.; Lee, A.C.; Said, J.M.; Kane, S.C.; Walker, S.P.; van Mens, T.E.; Hannan, N.J.; Tong, S.; et al. Enoxaparin for the prevention of preeclampsia and intrauterine growth restriction in women with a history: A randomized trial. Am. J. Obstet. Gynecol. 2017, 216, e1–e14. [Google Scholar] [CrossRef][Green Version]
- Haddad, B.; Winer, N.; Chitrit, Y.; Houfflin-Debarge, V.; Chauleur, C.; Bages, K.; Tsatsaris, V.; Benachi, A.; Bretelle, F.; Gris, J.-C.; et al. Enoxaparin and aspirin compared with aspirin alone to prevent placenta-mediated pregnancy complications: A randomized controlled trial. Obstet. Gynecol. 2016, 128, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Searle, J.; Mockel, M.; Gwosc, S.; Datwyler, S.A.; Qadri, F.; Albert, G.I.; Holert, F.; Isbruch, A.; Klug, L.; Muller, D.N.; et al. Heparin strongly induces soluble fms-like tyrosine kinase 1 release in vivo and in vitro—Brief report. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2972–2974. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, V.A.; Buhimschi, I.A.; Lockwood, C.J.; Paidas, M.J.; Dulay, A.T.; Ramma, W.; Abdel-Razeq, S.S.; Zhao, G.; Ahmad, S.; Ahmed, A.; et al. Heparin elevates circulating soluble fms-like tyrosine kinase-1 immunoreactivity in pregnant women receiving anticoagulation therapy. Circulation 2011, 124, 2543–2553. [Google Scholar] [CrossRef]
- Yinon, Y.; Ben Meir, E.; Margolis, L.; Lipitz, S.; Schiff, E.; Mazaki-Tovi, S.; Simchen, M.J. Low molecular weight heparin therapy during pregnancy is associated with elevated circulatory levels of placental growth factor. Placenta 2015, 36, 121–124. [Google Scholar] [CrossRef]
- Wu, P.; Haththotuwa, R.; Kwok, C.S.; Babu, A.; Kotronias, R.A.; Rushton, C.; Zaman, A.; Fryer, A.A.; Kadam, U.; Chew-Graham, C.A.; et al. Preeclampsia and future cardiovascular health: A systematic review and meta-analysis. Circ. Cardiovasc. Qual. Outcomes 2017, 10. [Google Scholar] [CrossRef]
- Bellamy, L.; Casas, J.P.; Hingorani, A.D.; Williams, D.J. Pre-eclampsia and risk of cardiovascular disease and cancer in later life: Systematic review and meta-analysis. BMJ 2007, 335, 974. [Google Scholar] [CrossRef]
- McDonald, S.D.; Malinowski, A.; Zhou, Q.; Yusuf, S.; Devereaux, P.J. Cardiovascular sequelae of preeclampsia/eclampsia: A systematic review and meta-analyses. Am. Heart J. 2008, 56, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, T.; Lamai, O.; Nerenberg, K. Hypertensive disorders of pregnancy and cardiovascular diseases: Current knowledge and future directions. Curr. Treat. Opt. Cardiovasc. Med. 2018, 20, 56. [Google Scholar] [CrossRef] [PubMed]
- Orabona, R.; Sciatti, E.; Vizzardi, E.; Bonadei, I.; Valcamonico, A.; Metra, M.; Frusca, T. Endothelial dysfunction and vascular stiffness in women with previous pregnancy complicated by early or late pre-eclampsia. Ultrasound Obstet. Gynecol. 2017, 49, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Orabona, R.; Vizzardi, E.; Sciatti, E.; Bonadei, I.; Valcamonico, A.; Metra, M.; Frusca, T. Insights into cardiac alterations after pre-eclampsia: An echocardiographic study. Ultrasound Obstet. Gynecol. 2017, 49, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Akhter, T.; Wikström, A.K.; Larsson, M.; Larsson, A.; Wikström, G.; Naessen, T. Association between angiogenic factors and signs of arterial aging in women with pre-eclampsia. Ultrasound Obstet. Gynecol. 2017, 50, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Kvehaugen, A.S.; Dechend, R.; Ramstad, H.B.; Troisi, R.; Fugelseth, D.; Staff, A.C. Endothelial function and circulating biomarkers are disturbed in women and children after preeclampsia. Hypertension 2011, 58, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Onoue, K.; Uemura, S.; Takeda, Y.; Somekawa, S.; Iwama, H.; Nishida, T.; Morikawa, Y.; Nakagawa, H.; Tsutsumi, T.; Sung, J.H.; et al. Usefulness of soluble Fms-like tyrosine kinase-1 as a biomarker of acute severe heart failure in patients with acute myocardial infarction. Am. J. Cardiol. 2009, 104, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- Patten, I.S.; Rana, S.; Shahul, S.; Rowe, G.C.; Jang, C.; Liu, L.; Hacker, M.R.; Rhee, J.S.; Mitchell, J.; Mahmood, F.; et al. Cardiac angiogenic imbalance leads to peripartum cardiomyopathy. Nature 2012, 485, 333–338. [Google Scholar] [CrossRef]
- Gamble, D.T.; Brikinns, B.; Myint, P.K.; Bhattacharya, S. Hypertensive disorders of pregnancy and subsequent cardiovascular disease: Current national and international guidelines and the need for future research. Front. Cardiovasc. Med. 2019, 6, 55. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomimatsu, T.; Mimura, K.; Matsuzaki, S.; Endo, M.; Kumasawa, K.; Kimura, T. Preeclampsia: Maternal Systemic Vascular Disorder Caused by Generalized Endothelial Dysfunction Due to Placental Antiangiogenic Factors. Int. J. Mol. Sci. 2019, 20, 4246. https://doi.org/10.3390/ijms20174246
Tomimatsu T, Mimura K, Matsuzaki S, Endo M, Kumasawa K, Kimura T. Preeclampsia: Maternal Systemic Vascular Disorder Caused by Generalized Endothelial Dysfunction Due to Placental Antiangiogenic Factors. International Journal of Molecular Sciences. 2019; 20(17):4246. https://doi.org/10.3390/ijms20174246
Chicago/Turabian StyleTomimatsu, Takuji, Kazuya Mimura, Shinya Matsuzaki, Masayuki Endo, Keiichi Kumasawa, and Tadashi Kimura. 2019. "Preeclampsia: Maternal Systemic Vascular Disorder Caused by Generalized Endothelial Dysfunction Due to Placental Antiangiogenic Factors" International Journal of Molecular Sciences 20, no. 17: 4246. https://doi.org/10.3390/ijms20174246
APA StyleTomimatsu, T., Mimura, K., Matsuzaki, S., Endo, M., Kumasawa, K., & Kimura, T. (2019). Preeclampsia: Maternal Systemic Vascular Disorder Caused by Generalized Endothelial Dysfunction Due to Placental Antiangiogenic Factors. International Journal of Molecular Sciences, 20(17), 4246. https://doi.org/10.3390/ijms20174246