Fluoxetine Suppresses Glutamate- and GABA-Mediated Neurotransmission by Altering SNARE Complex



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
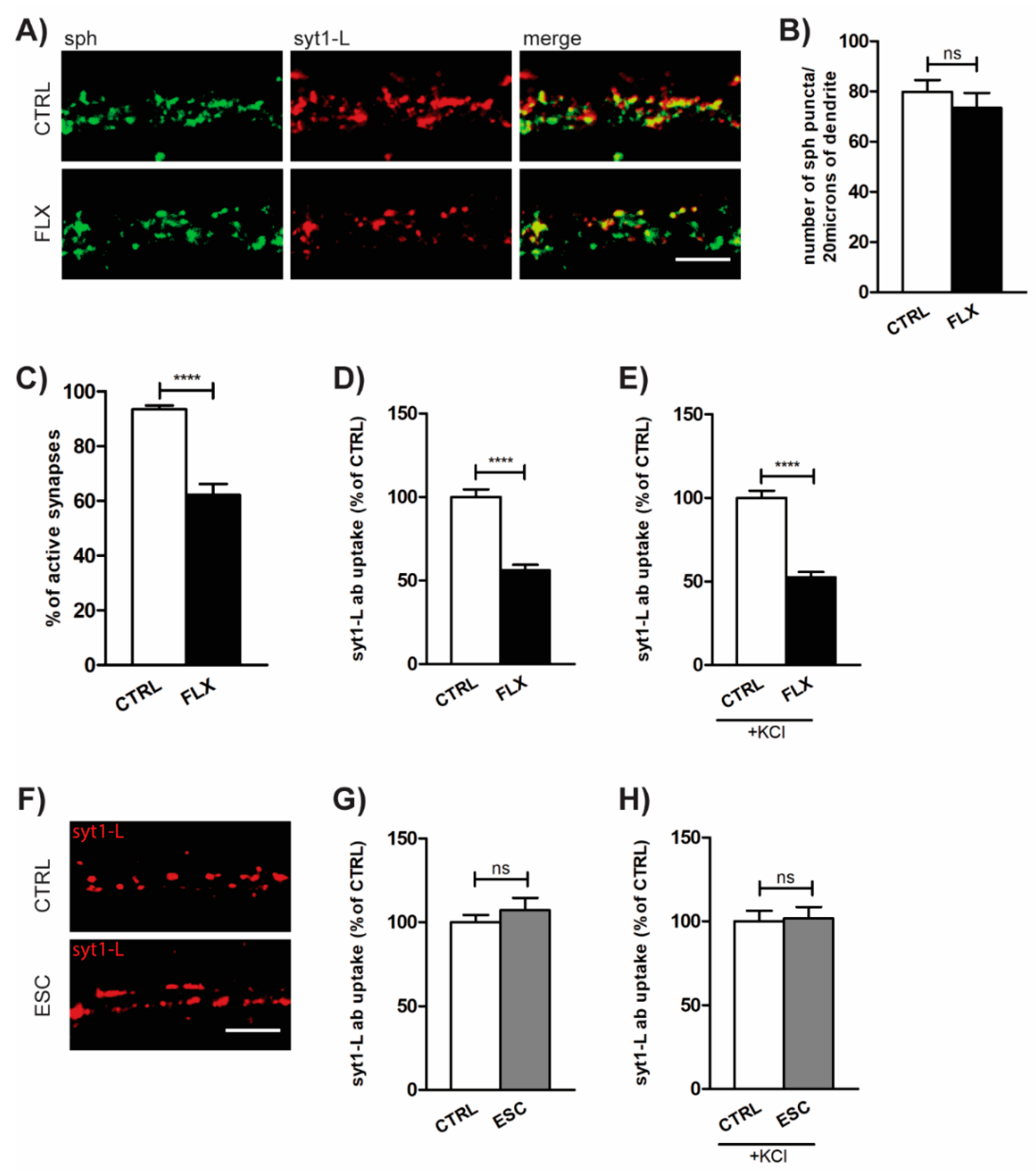
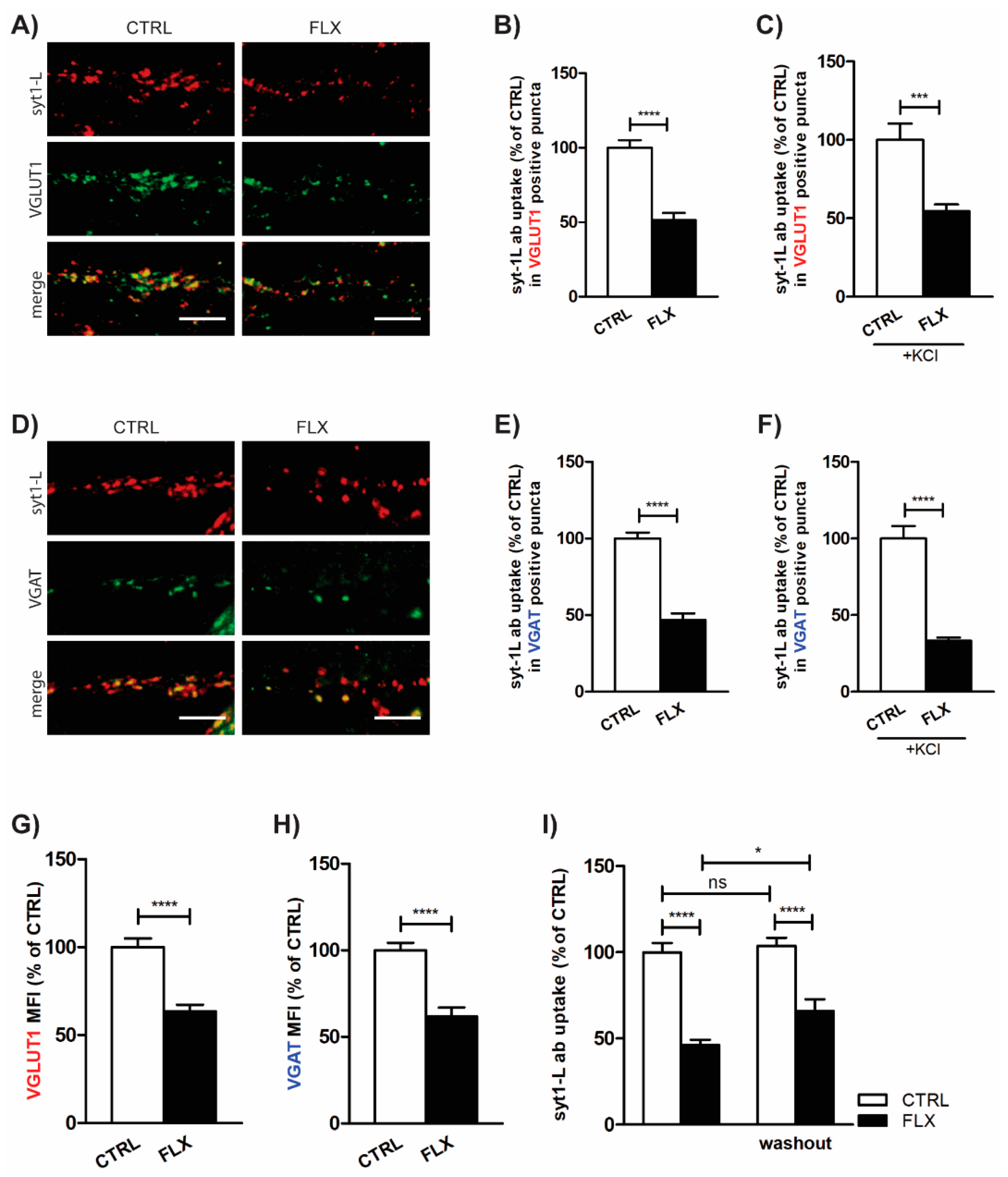
2.1. FLX Reduces Presynaptic Activity
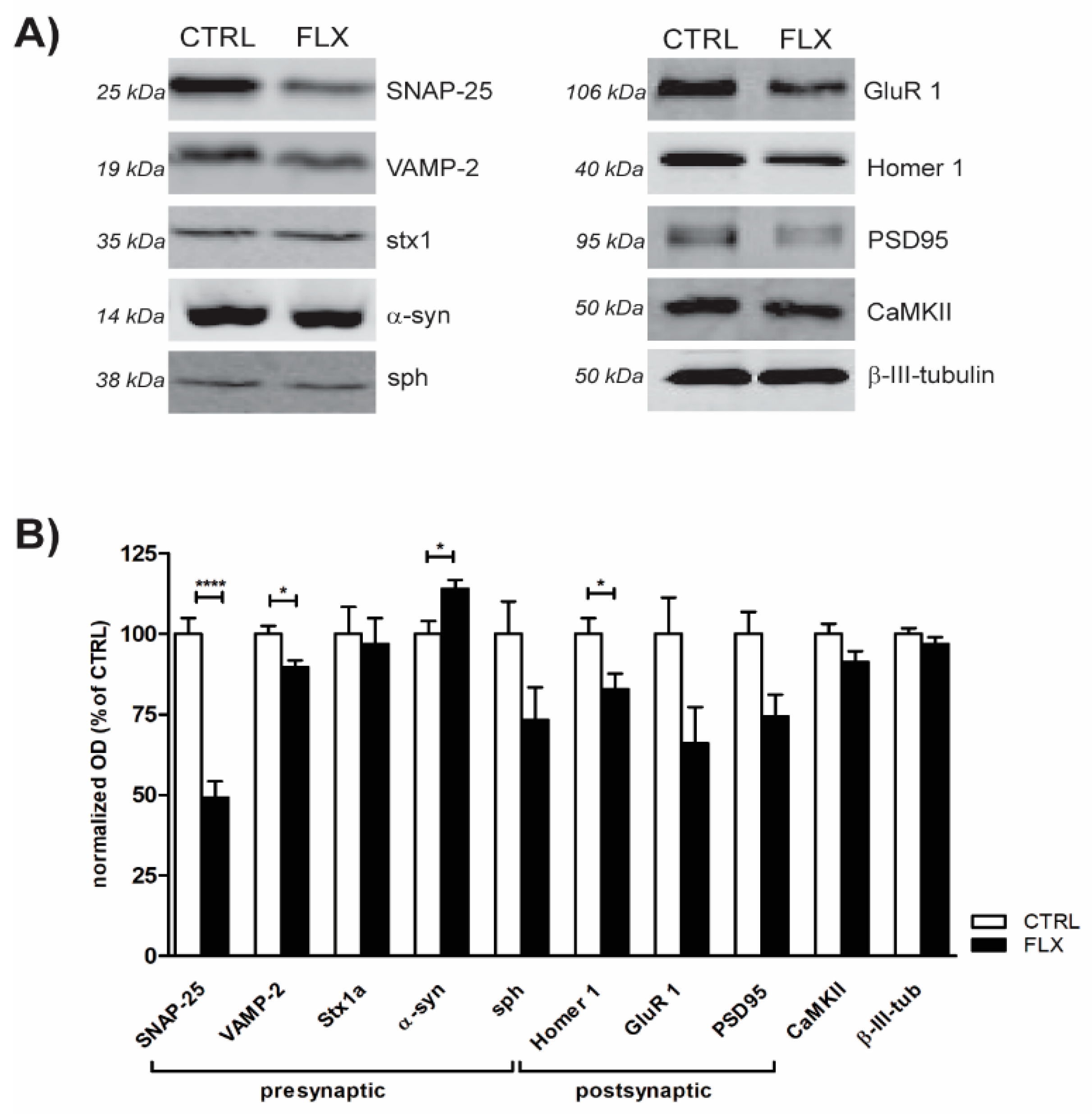
2.2. FLX Treatment Impairs Exocytic Machinery Components
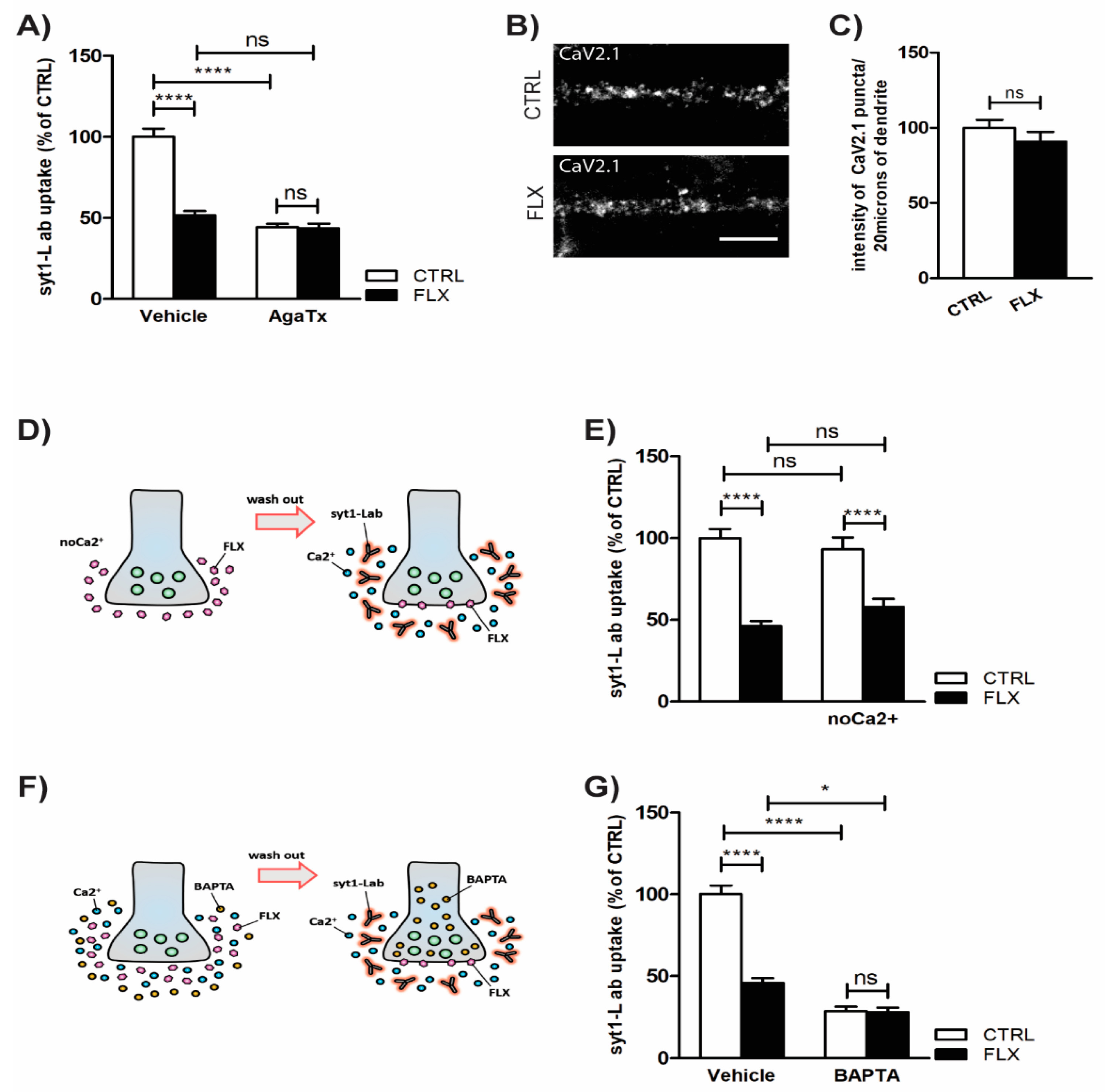
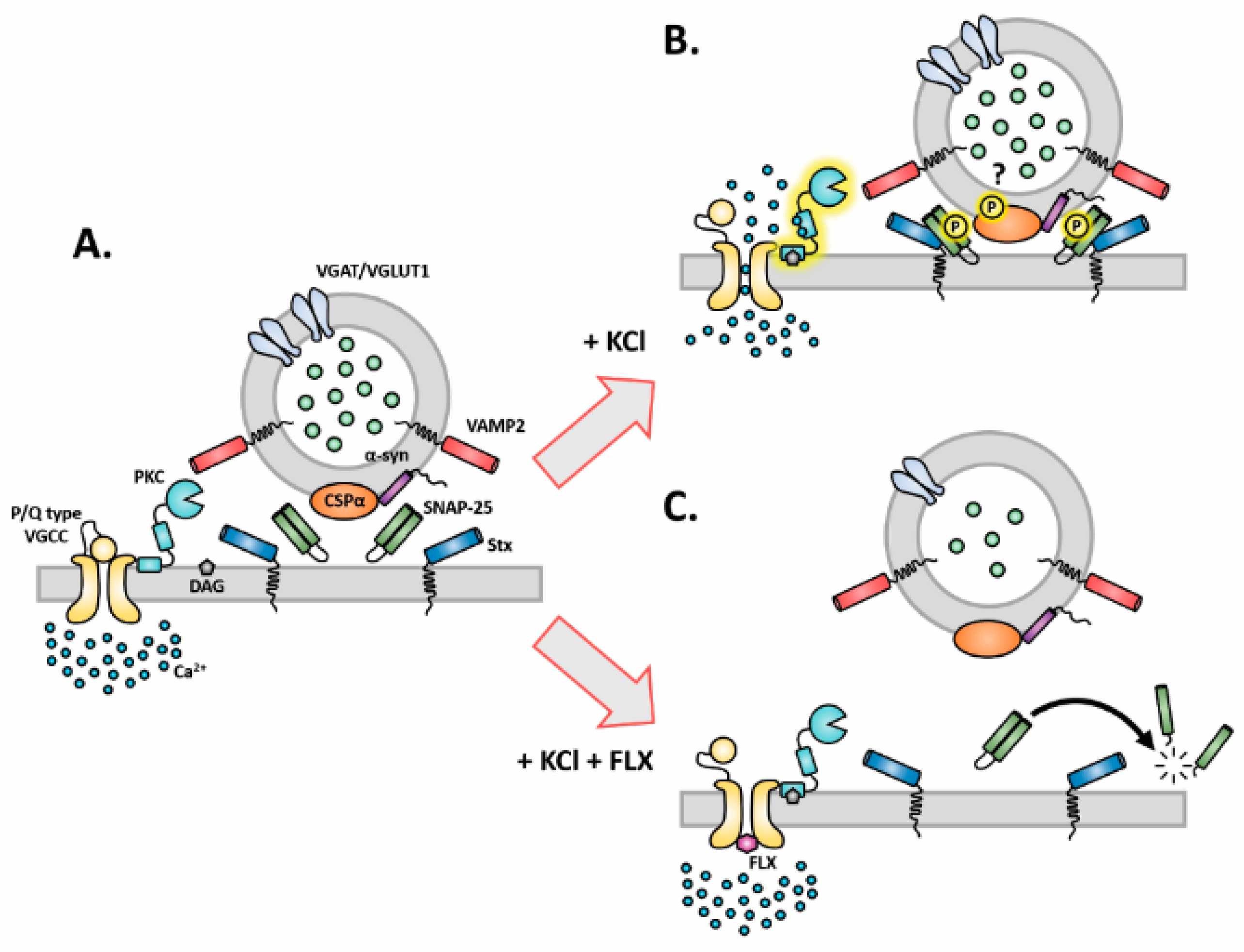
2.3. Inhibition of P/Q Type Ca2+ Channels Mediates FLX-Induced Suppression of Presynaptic Activity
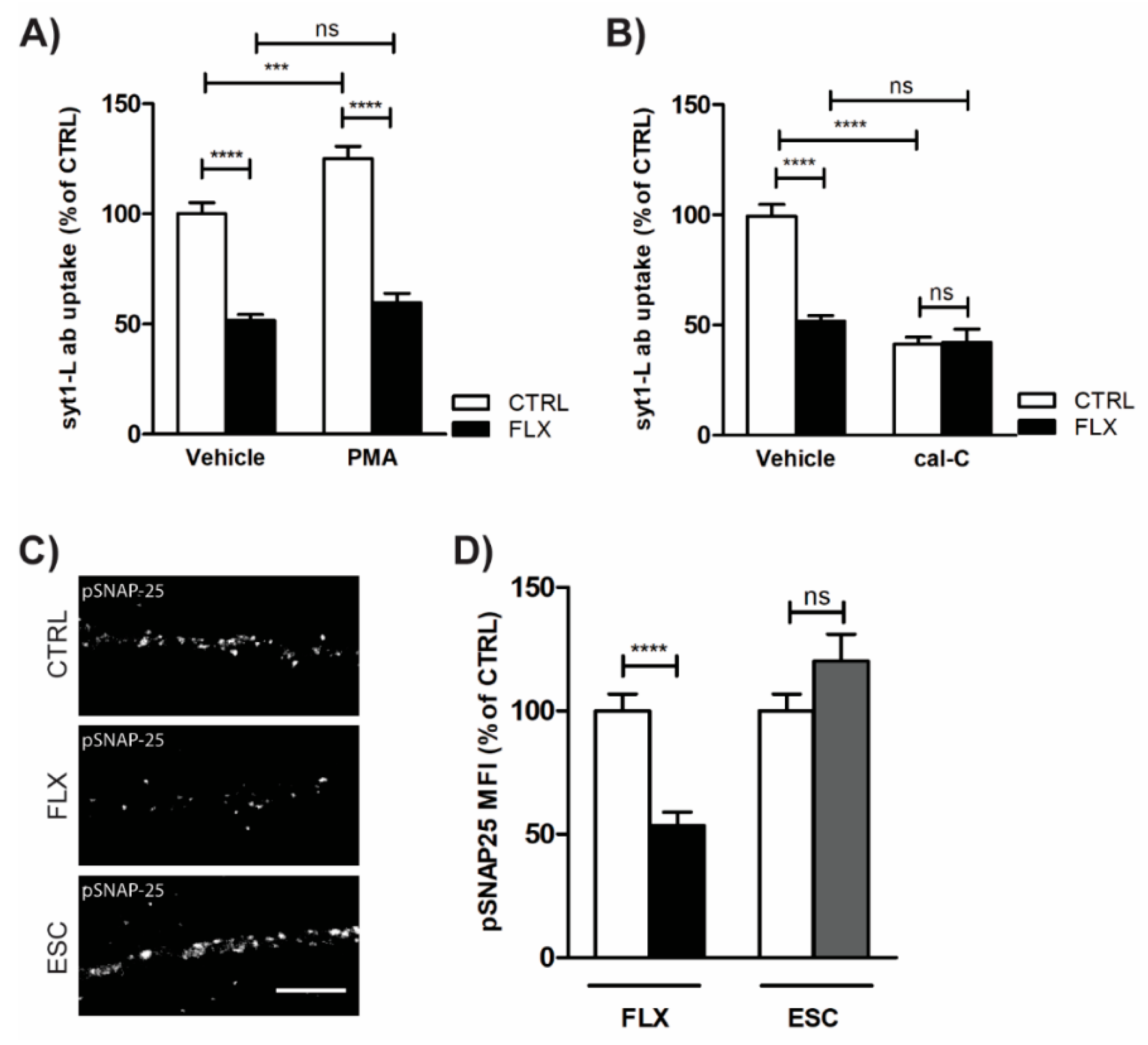
2.4. Impaired Protein Kinase C (PKC) Activity May Account for FLX-Induced Aberrant Neurotransmission
3. Discussion
3.1. FLX Decreases Synaptic Activity through a SERT-Independent Mechanism
3.2. FLX Decreases the Level of Synaptic Machinery Proteins, Especially Core SNARE Molecules
3.3. FLX Inhibits Ca2+-Induced Activation of PKC through Blocking the P/Q Type VGCCs
4. Materials and Methods
4.1. Animals
4.2. Chemical Reagents
4.3. Preparation of Primary Neurons
4.4. Immunocytochemistry and Syt1-L ab Uptake Assay
4.5. Western Blot
4.6. Antibodies
4.7. Image Acquisition and Analysis
4.8. Statistics
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| SSRIs | selective serotonin reuptake inhibitors |
| SERT | serotonin transporter |
| FLX | fluoxetine |
| GABA | gamma-Aminobutyric acid |
| SNAP-25 | synaptosomal nerve-associated protein 25 |
| PKC | protein kinase C |
| VGCCs | voltage-gated calcium channels |
| NSF | N-ethylmaleimide-sensitive factor |
| SNARE | soluble NSF attachment protein receptor |
| MDD | major depressive disorder |
| HPA | hypothalamic–pituitary axis |
| ESC | escitalopram |
| 5HTR | 5-hydroxytryptamine receptor |
| BDNF | brain-derived neurotrophic factor |
| PFC | prefrontal cortex |
| HPC | hippocampal complex |
| SV | synaptic vesicle |
| Sph | synaptophysin |
| syt1-L ab | synaptotagmin 1 luminal domain antibody |
| VGLUT1 | vesicular glutamate transporter 1 |
| VGAT | vesicular GABA transporter |
| VAMP-2 | Vesicle-associated membrane protein 2 |
| AMPA | α-amino-3-hydroxy-5-methyl-4- isoxazole-propanoic acid |
| PSD95 | postsynaptic density protein 95 |
| BAPTA-AM | 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) |
| PMA | phorbol myristate acetate |
| cal-C | Calphostin C |
| SNAP-25 | Synaptosomal nerve-associated protein 25 |
| DR | dorsal raphe |
| TPH2 | Tryptophan hydroxylase 2 |
| CSPα | cysteine string protein α |
| PD | Parkinson’s disease |
References
- Otte, C.; Gold, S.M.; Penninx, B.W.; Pariante, C.M.; Etkin, A.; Fava, M.; Mohr, D.C.; Schatzberg, A.F. Major depressive disorder. Nat. Rev. Dis. Primers 2016, 2, 16065. [Google Scholar] [CrossRef] [PubMed]
- Belmaker, R.H.; Agam, G. Major depressive disorder. New Engl. J. Med. 2008, 358, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.H.; Raison, C.L. The role of inflammation in depression: From evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 2016, 16, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Wohleb, E.S.; Franklin, T.; Iwata, M.; Duman, R.S. Integrating neuroimmune systems in the neurobiology of depression. Nat. Rev. Neurosci. 2016, 17, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Hillhouse, T.M.; Porter, J.H. A brief history of the development of antidepressant drugs: From monoamines to glutamate. Exp. Clin. Psychopharmacol. 2015, 23, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.T.; Perry, K.W.; Bymaster, F.P. Case history: The discovery of fluoxetine hydrochloride (Prozac). Nat. Rev. Drug Discov. 2005, 4, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Marsh, L. Depression and Parkinson’s disease: Current knowledge. Curr. Neurol. Neurosci. Rep. 2013, 13, 409. [Google Scholar] [CrossRef] [PubMed]
- Hurley, L.L.; Tizabi, Y. Neuroinflammation, neurodegeneration, and depression. Neurotox. Res. 2013, 23, 131–144. [Google Scholar] [CrossRef]
- Dupuy, J.M.; Ostacher, M.J.; Huffman, J.; Perlis, R.H.; Nierenberg, A.A. A critical review of pharmacotherapy for major depressive disorder. Int. J. Neuropsychopharmacol. 2011, 14, 1417–1431. [Google Scholar] [CrossRef]
- Gutknecht, L.; Kriegebaum, C.; Waider, J.; Schmitt, A.; Lesch, K.P. Spatio-temporal expression of tryptophan hydroxylase isoforms in murine and human brain: Convergent data from Tph2 knockout mice. Eur. Neuropsychopharmacol. 2009, 19, 266–282. [Google Scholar] [CrossRef]
- Mathews, D.C.; Henter, I.D.; Zarate, C.A. Targeting the glutamatergic system to treat major depressive disorder: Rationale and progress to date. Drugs 2012, 72, 1313–1333. [Google Scholar] [CrossRef] [PubMed]
- Sanacora, G.; Treccani, G.; Popoli, M. Towards a glutamate hypothesis of depression: An emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology 2012, 62, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Murrough, J.W.; Abdallah, C.G.; Mathew, S.J. Targeting glutamate signalling in depression: Progress and prospects. Nat. Rev. Drug Discov. 2017, 16, 472–486. [Google Scholar] [CrossRef] [PubMed]
- Musazzi, L.; Treccani, G.; Mallei, A.; Popoli, M. The action of antidepressants on the glutamate system: Regulation of glutamate release and glutamate receptors. Biol. Psychiatry 2013, 73, 1180–1188. [Google Scholar] [CrossRef] [PubMed]
- Haroon, E.; Miller, A.H.; Sanacora, G. Inflammation, Glutamate, and Glia: A Trio of Trouble in Mood Disorders. Neuropsychopharmacology 2017, 42, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Nautiyal, K.M.; Hen, R. Serotonin receptors in depression: From A to B. F1000Research 2017, 6, 123. [Google Scholar] [CrossRef] [PubMed]
- Svenningsson, P.; Chergui, K.; Rachleff, I.; Flajolet, M.; Zhang, X.; El Yacoubi, M.; Vaugeois, J.M.; Nomikos, G.G.; Greengard, P. Alterations in 5-HT1B receptor function by p11 in depression-like states. Science 2006, 311, 77–80. [Google Scholar] [CrossRef]
- Varnas, K.; Hurd, Y.L.; Hall, H. Regional expression of 5-HT1B receptor mRNA in the human brain. Synapse 2005, 56, 21–28. [Google Scholar] [CrossRef]
- Owens, M.J.; Knight, D.L.; Nemeroff, C.B. Second-generation SSRIs: Human monoamine transporter binding profile of escitalopram and R-fluoxetine. Biol. Psychiatry 2001, 50, 345–350. [Google Scholar] [CrossRef]
- Robinson, R.T.; Drafts, B.C.; Fisher, J.L. Fluoxetine increases GABA(A) receptor activity through a novel modulatory site. J. Pharmacol. Exp. Ther. 2003, 304, 978–984. [Google Scholar] [CrossRef]
- Poulin, H.; Bruhova, I.; Timour, Q.; Theriault, O.; Beaulieu, J.M.; Frassati, D.; Chahine, M. Fluoxetine blocks Nav1.5 channels via a mechanism similar to that of class 1 antiarrhythmics. Mol. Pharmacol. 2014, 86, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Lazarevic, V.; Schone, C.; Heine, M.; Gundelfinger, E.D.; Fejtova, A. Extensive remodeling of the presynaptic cytomatrix upon homeostatic adaptation to network activity silencing. J. Neurosci. 2011, 31, 10189–10200. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.J.; Su, C.F.; Kuo, Y.H. Fluoxetine depresses glutamate exocytosis in the rat cerebrocortical nerve terminals (synaptosomes) via inhibition of P/Q-type Ca2+ channels. Synapse 2003, 48, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Levy, J.M.; Hou, A.; Winters, C.; Azzam, R.; Sousa, A.A.; Leapman, R.D.; Nicoll, R.A.; Reese, T.S. PSD-95 family MAGUKs are essential for anchoring AMPA and NMDA receptor complexes at the postsynaptic density. Proc. Natl. Acad. Sci. USA 2015, 112, E6983–E6992. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Few, A.P. Calcium channel regulation and presynaptic plasticity. Neuron 2008, 59, 882–901. [Google Scholar] [CrossRef] [PubMed]
- Deak, F.; Lasztoczi, B.; Pacher, P.; Petheo, G.L.; Valeria, K.; Spat, A. Inhibition of voltage-gated calcium channels by fluoxetine in rat hippocampal pyramidal cells. Neuropharmacology 2000, 39, 1029–1036. [Google Scholar] [CrossRef]
- Fischer, A.G.; Jocham, G.; Ullsperger, M. Dual serotonergic signals: A key to understanding paradoxical effects? Trends Cogn. Sci. 2014. [Google Scholar] [CrossRef] [PubMed]
- Waselus, M.; Valentino, R.J.; Van Bockstaele, E.J. Collateralized dorsal raphe nucleus projections: A mechanism for the integration of diverse functions during stress. J. Chem. Neuroanat. 2011, 41, 266–280. [Google Scholar] [CrossRef]
- Beaudoin, G.M., 3rd; Lee, S.H.; Singh, D.; Yuan, Y.; Ng, Y.G.; Reichardt, L.F.; Arikkath, J. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat. Protoc. 2012, 7, 1741–1754. [Google Scholar] [CrossRef]
- Narboux-Neme, N.; Pavone, L.M.; Avallone, L.; Zhuang, X.; Gaspar, P. Serotonin transporter transgenic (SERTcre) mouse line reveals developmental targets of serotonin specific reuptake inhibitors (SSRIs). Neuropharmacology 2008, 55, 994–1005. [Google Scholar] [CrossRef]
- Soiza-Reilly, M.; Meye, F.J.; Olusakin, J.; Telley, L.; Petit, E.; Chen, X.; Mameli, M.; Jabaudon, D.; Sze, J.Y.; Gaspar, P. Correction: SSRIs target prefrontal to raphe circuits during development modulating synaptic connectivity and emotional behavior. Mol. Psychiatry 2019, 24, 773. [Google Scholar] [CrossRef] [PubMed]
- Bonanno, G.; Giambelli, R.; Raiteri, L.; Tiraboschi, E.; Zappettini, S.; Musazzi, L.; Raiteri, M.; Racagni, G.; Popoli, M. Chronic antidepressants reduce depolarization-evoked glutamate release and protein interactions favoring formation of SNARE complex in hippocampus. J. Neurosci. 2005, 25, 3270–3279. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Burre, J.; Sudhof, T.C. CSPalpha promotes SNARE-complex assembly by chaperoning SNAP-25 during synaptic activity. Nat. Cell Biol. 2011, 13, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Burre, J.; Bronk, P.; Zhang, Y.; Xu, W.; Sudhof, T.C. CSPalpha knockout causes neurodegeneration by impairing SNAP-25 function. Embo J. 2012, 31, 829–841. [Google Scholar] [CrossRef]
- Fernandez-Chacon, R.; Wolfel, M.; Nishimune, H.; Tabares, L.; Schmitz, F.; Castellano-Munoz, M.; Rosenmund, C.; Montesinos, M.L.; Sanes, J.R.; Schneggenburger, R.; et al. The synaptic vesicle protein CSP alpha prevents presynaptic degeneration. Neuron 2004, 42, 237–251. [Google Scholar] [CrossRef]
- Chandra, S.; Gallardo, G.; Fernandez-Chacon, R.; Schluter, O.M.; Sudhof, T.C. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 2005, 123, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Leenders, A.G.; Sheng, Z.H. Modulation of neurotransmitter release by the second messenger-activated protein kinases: Implications for presynaptic plasticity. Pharmacol. Ther. 2005, 105, 69–84. [Google Scholar] [CrossRef]
- Shirafuji, T.; Ueyama, T.; Adachi, N.; Yoshino, K.I.; Sotomaru, Y.; Uwada, J.; Kaneoka, A.; Ueda, T.; Tanaka, S.; Hide, I.; et al. The Role of Cysteine String Protein alpha Phosphorylation at Serine 10 and 34 by Protein Kinase Cgamma for Presynaptic Maintenance. J. Neurosci. 2018, 38, 278–290. [Google Scholar] [CrossRef]
- Nagy, G.; Matti, U.; Nehring, R.B.; Binz, T.; Rettig, J.; Neher, E.; Sorensen, J.B. Protein kinase C-dependent phosphorylation of synaptosome-associated protein of 25 kDa at Ser187 potentiates vesicle recruitment. J. Neurosci. 2002, 22, 9278–9286. [Google Scholar] [CrossRef]
- Virmani, T.; Ertunc, M.; Sara, Y.; Mozhayeva, M.; Kavalali, E.T. Phorbol esters target the activity-dependent recycling pool and spare spontaneous vesicle recycling. J. Neurosci. 2005, 25, 10922–10999. [Google Scholar] [CrossRef]
- Newton, A.C. Protein kinase C: Structure, function, and regulation. J. Biol. Chem. 1995, 270, 28495–28498. [Google Scholar] [CrossRef] [PubMed]
- Steele, E.C., Jr.; Chen, X.; MacLeish, P.R. Fluoxetine inhibits calcium-activated currents of salamander rod photoreceptor somata and presynaptic terminals via modulation of intracellular calcium dynamics. Mol. Vis. 2005, 11, 1200–1210. [Google Scholar] [PubMed]
- Xu, F.; Luk, C.; Richard, M.P.; Zaidi, W.; Farkas, S.; Getz, A.; Lee, A.; van Minnen, J.; Syed, N.I. Antidepressant fluoxetine suppresses neuronal growth from both vertebrate and invertebrate neurons and perturbs synapse formation between Lymnaea neurons. Eur. J. Neurosci. 2010, 31, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Getz, A.; Xu, F.; Zaidi, W.; Syed, N.I. The antidepressant fluoxetine but not citalopram suppresses synapse formation and synaptic transmission between Lymnaea neurons by perturbing presynaptic and postsynaptic machinery. Eur. J. Neurosci. 2011, 34, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Mohler, H. The GABA system in anxiety and depression and its therapeutic potential. Neuropharmacology 2012, 62, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, X.; Yuan, T.F. Somatostatin-positive GABAergic interneuron: New targets for depression. Mol. Psychiatry 2017, 22, 790–791. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.; Jefferson, S.J.; Hooper, A.; Yee, P.H.; Maguire, J.; Luscher, B. Disinhibition of somatostatin-positive GABAergic interneurons results in an anxiolytic and antidepressant-like brain state. Mol. Psychiatry 2017, 22, 920–930. [Google Scholar] [CrossRef]
- Wieronska, J.M.; Pilc, A. Depression and schizophrenia viewed from the perspective of amino acidergic neurotransmission: Antipodes of psychiatric disorders. Pharmacol. Ther. 2019, 193, 75–82. [Google Scholar] [CrossRef]
- Cipriani, A.; Santilli, C.; Furukawa, T.A.; Signoretti, A.; Nakagawa, A.; McGuire, H.; Churchill, R.; Barbui, C. Escitalopram versus other antidepressive agents for depression. Cochrane Database Syst. Rev. 2009, CD006532. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, M.; Shi, J.; Zhou, Y.; Wan, Z.; Wang, Y.; Wan, Y.; Li, J.; Wang, Z.; Fei, J. Brain-Specific SNAP-25 Deletion Leads to Elevated Extracellular Glutamate Level and Schizophrenia-Like Behavior in Mice. Neural Plast. 2017, 2017, 4526417. [Google Scholar] [CrossRef]
- Corradini, I.; Verderio, C.; Sala, M.; Wilson, M.C.; Matteoli, M. SNAP-25 in neuropsychiatric disorders. Ann. N. Y. Acad. Sci. 2009, 1152, 93–99. [Google Scholar] [CrossRef]
- Knable, M.B.; Barci, B.M.; Webster, M.J.; Meador-Woodruff, J.; Torrey, E.F.; Stanley Neuropathology, C. Molecular abnormalities of the hippocampus in severe psychiatric illness: Postmortem findings from the Stanley Neuropathology Consortium. Mol. Psychiatry 2004, 9, 609–620. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Parkinson’s Disease Is Not Simply a Prion Disorder. J. Neurosci. 2017, 37, 9799–9807. [Google Scholar] [CrossRef]
- Pchitskaya, E.; Popugaeva, E.; Bezprozvanny, I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 2018, 70, 87–94. [Google Scholar] [CrossRef]
- Nimmrich, V.; Gross, G. P/Q-type calcium channel modulators. Br. J. Pharmacol. 2012, 167, 741–759. [Google Scholar] [CrossRef]
- Lazarevic, V.; Yang, Y.; Ivanova, D.; Fejtova, A.; Svenningsson, P. Riluzole attenuates the efficacy of glutamatergic transmission by interfering with the size of the readily releasable neurotransmitter pool. Neuropharmacology 2018, 143, 38–48. [Google Scholar] [CrossRef]
- Tsuriel, S.; Fisher, A.; Wittenmayer, N.; Dresbach, T.; Garner, C.C.; Ziv, N.E. Exchange and redistribution dynamics of the cytoskeleton of the active zone molecule bassoon. J. Neurosci. 2009, 29, 351–358. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lazarevic, V.; Mantas, I.; Flais, I.; Svenningsson, P. Fluoxetine Suppresses Glutamate- and GABA-Mediated Neurotransmission by Altering SNARE Complex. Int. J. Mol. Sci. 2019, 20, 4247. https://doi.org/10.3390/ijms20174247
Lazarevic V, Mantas I, Flais I, Svenningsson P. Fluoxetine Suppresses Glutamate- and GABA-Mediated Neurotransmission by Altering SNARE Complex. International Journal of Molecular Sciences. 2019; 20(17):4247. https://doi.org/10.3390/ijms20174247
Chicago/Turabian StyleLazarevic, Vesna, Ioannis Mantas, Ivana Flais, and Per Svenningsson. 2019. "Fluoxetine Suppresses Glutamate- and GABA-Mediated Neurotransmission by Altering SNARE Complex" International Journal of Molecular Sciences 20, no. 17: 4247. https://doi.org/10.3390/ijms20174247
APA StyleLazarevic, V., Mantas, I., Flais, I., & Svenningsson, P. (2019). Fluoxetine Suppresses Glutamate- and GABA-Mediated Neurotransmission by Altering SNARE Complex. International Journal of Molecular Sciences, 20(17), 4247. https://doi.org/10.3390/ijms20174247