Structural Insights into the Interaction of Cytochrome P450 3A4 with Suicide Substrates: Mibefradil, Azamulin and 6′,7′-Dihydroxybergamottin


Abstract
1. Introduction
2. Results and Discussion
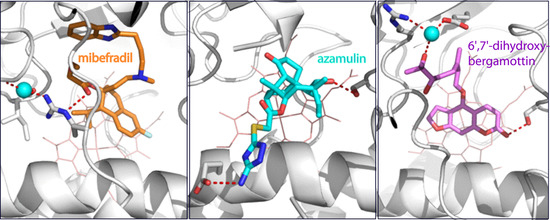
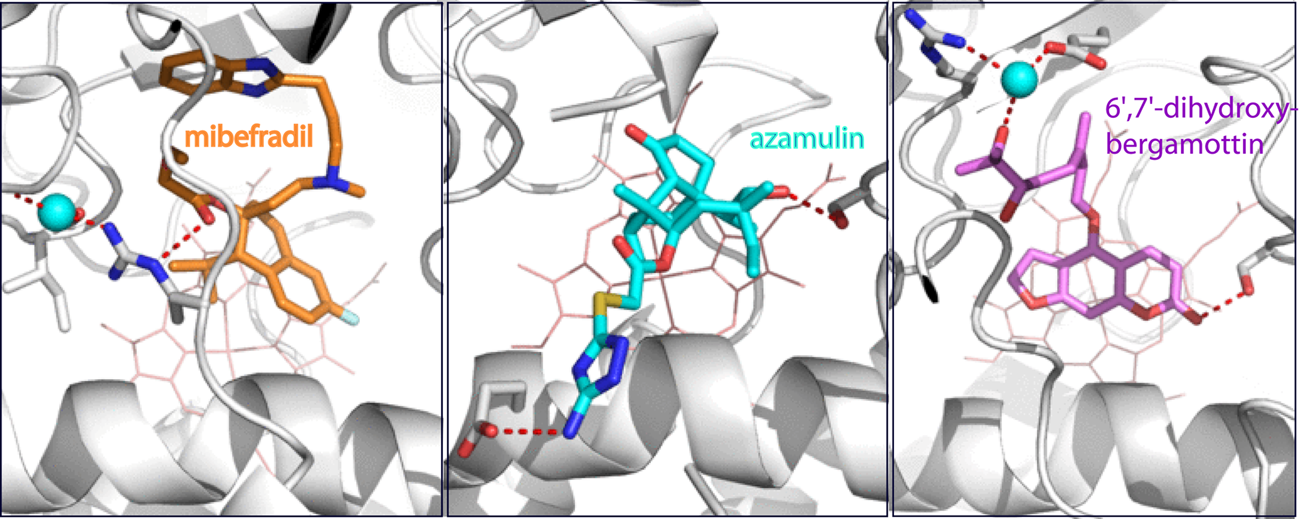
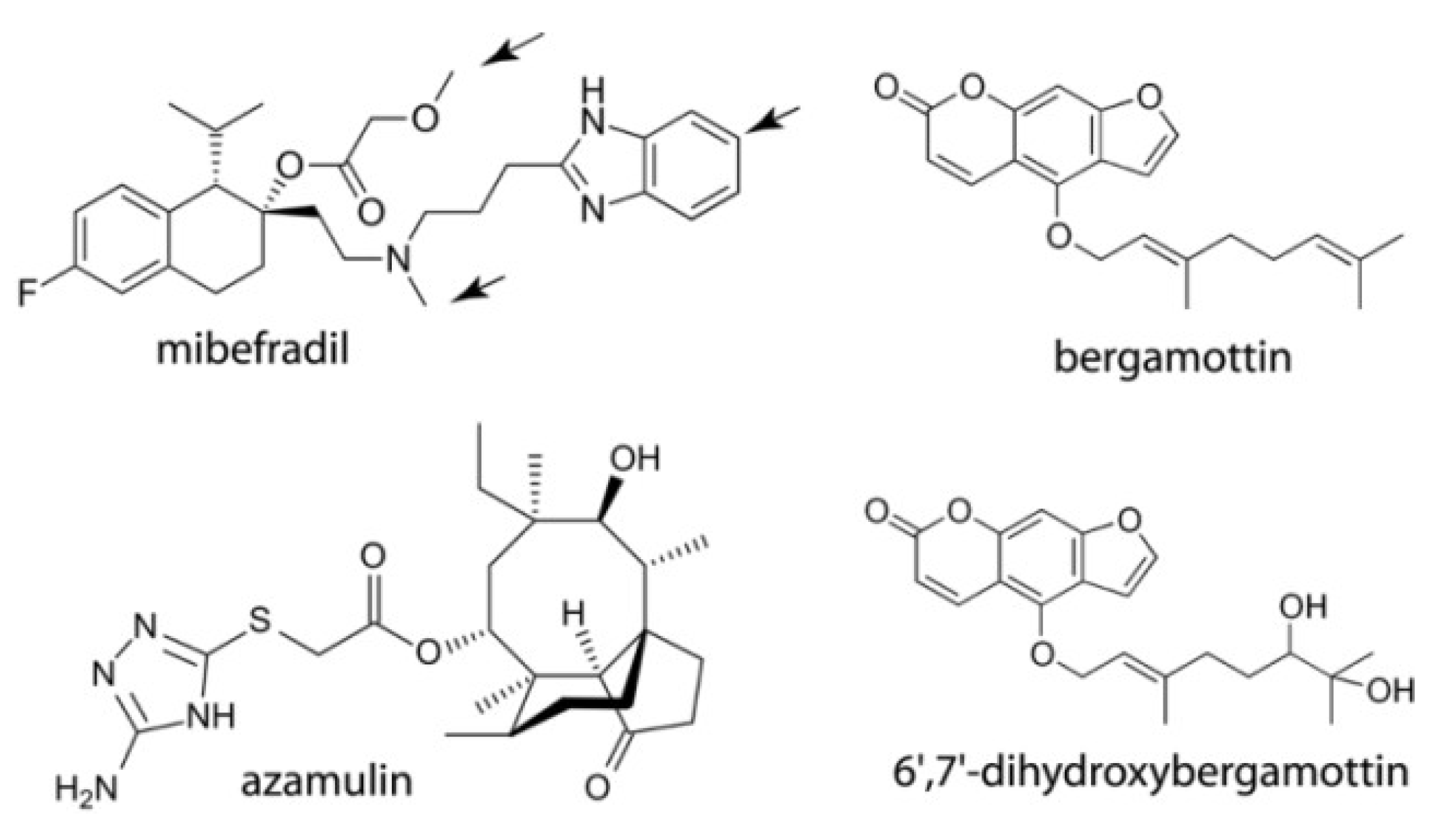
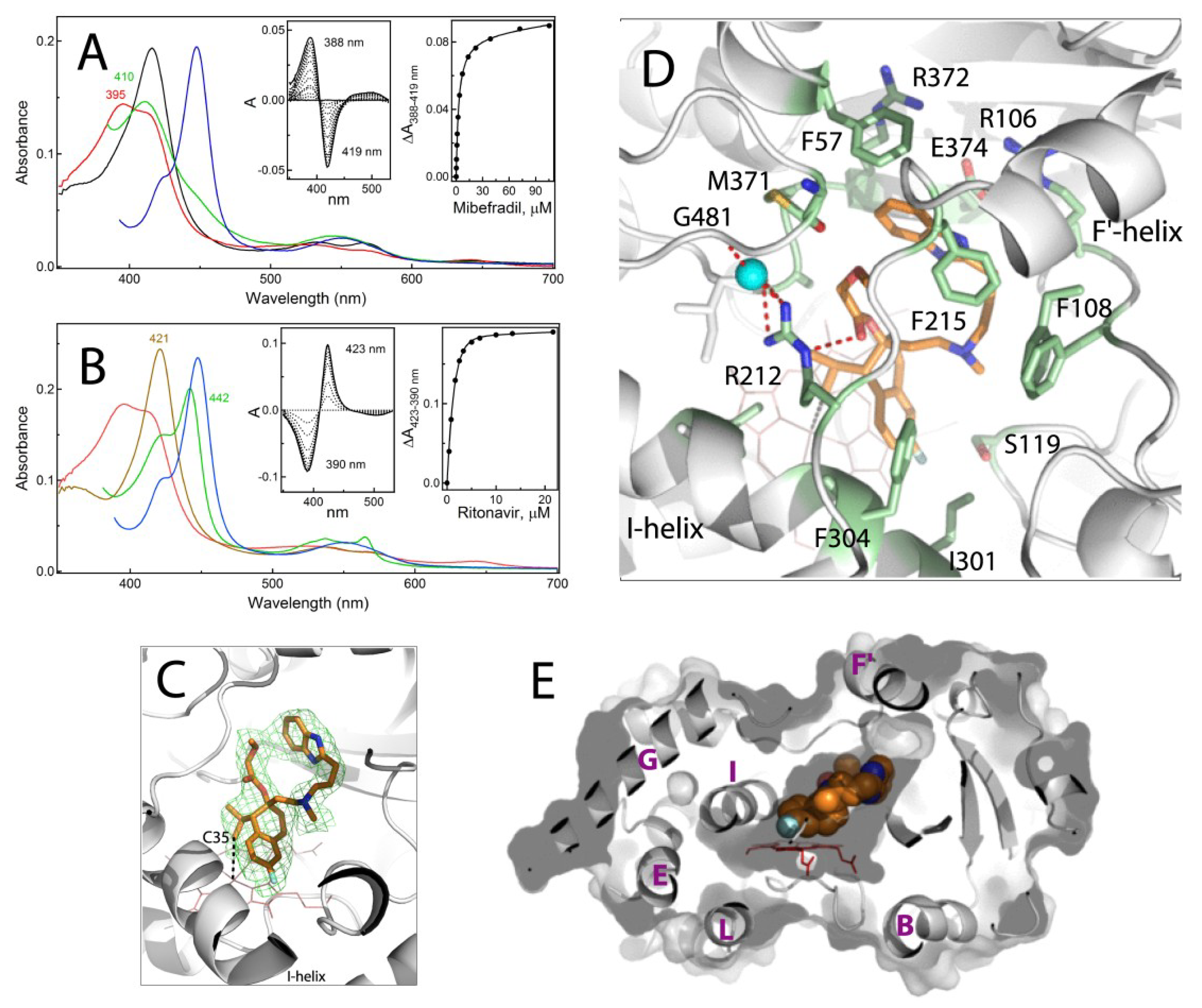
2.1. Interaction of CYP3A4 with Mibefradil
2.1.1. Crystal Structure of the CYP3A4-Mibefradil Complex
2.1.2. Possible Inhibitory Mechanism of Mibefradil
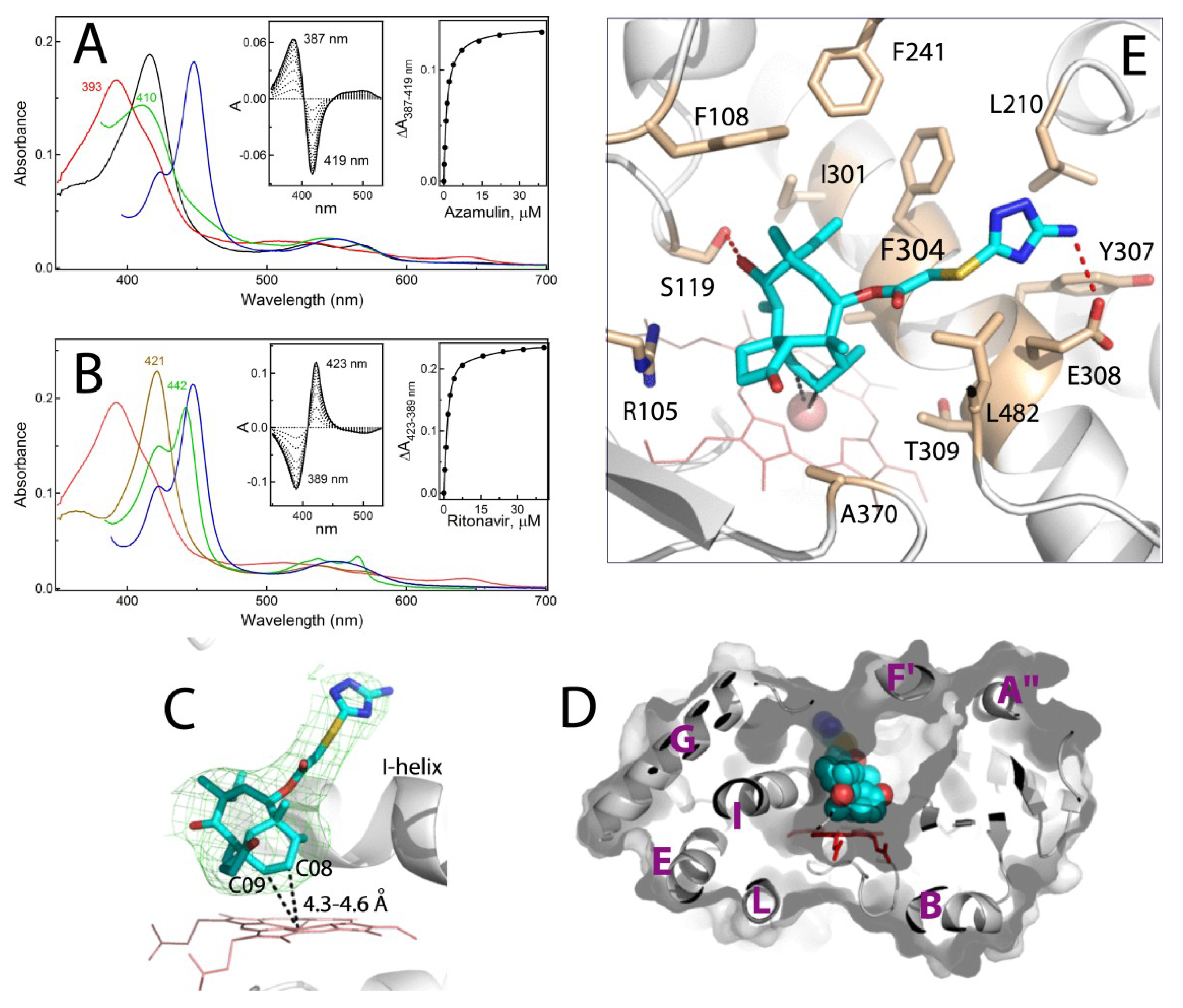
2.2. Interaction of CYP3A4 with Azamulin
Crystal Structure of CYP3A4 Bound to Azamulin
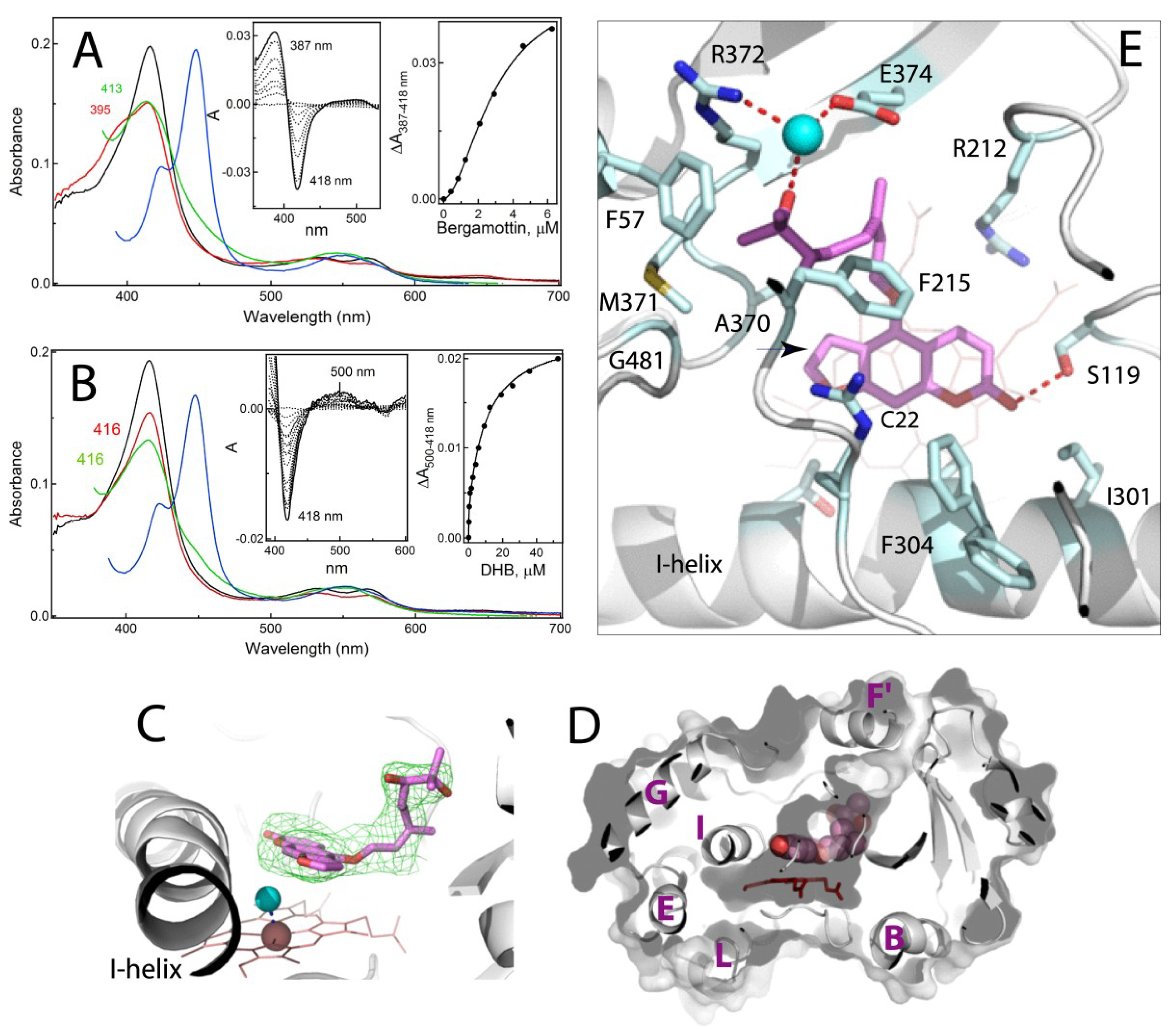
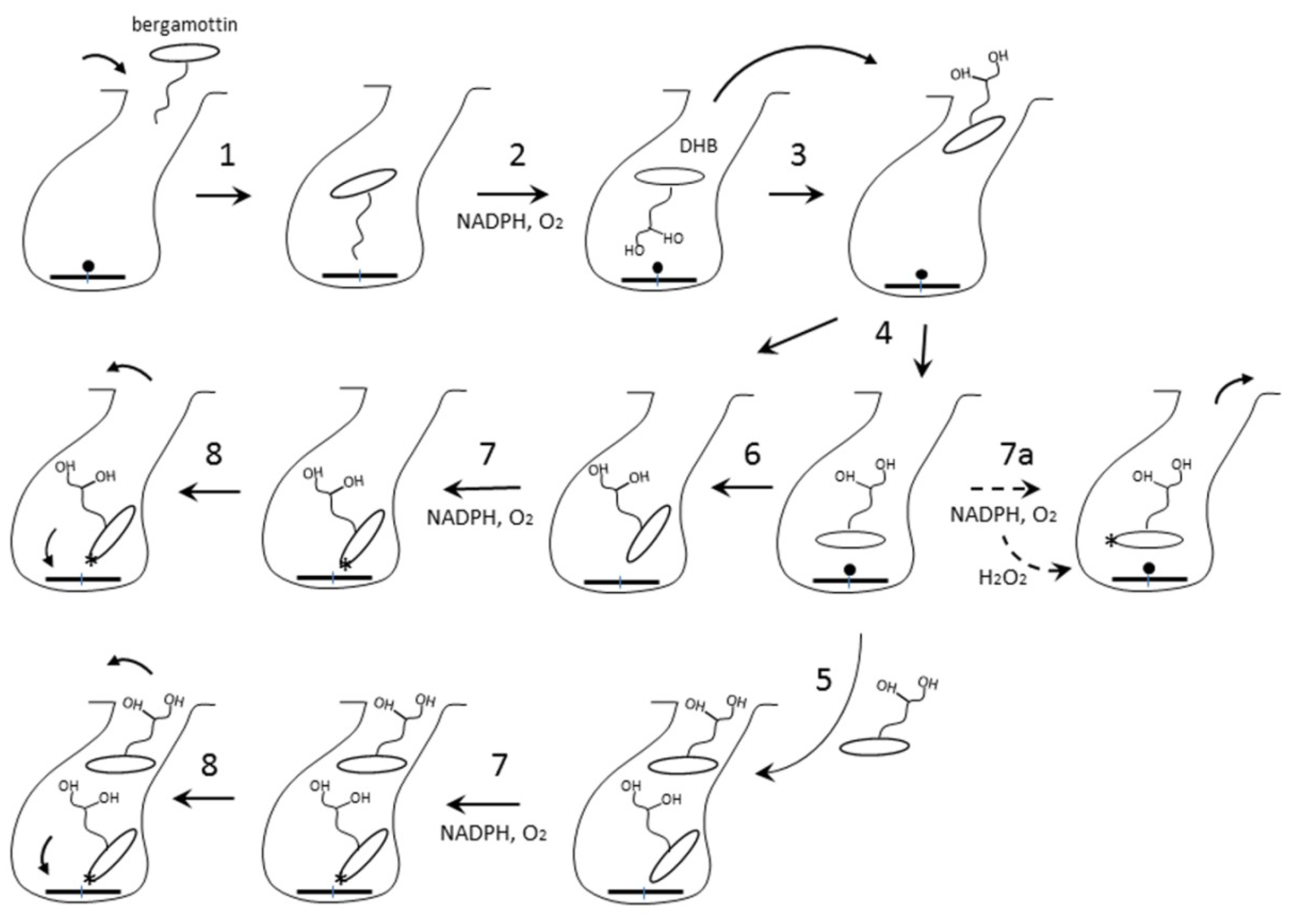
2.3. Interaction of CYP3A4 with Bergamottin and DHB
2.3.1. Crystal Structure of the CYP3A4-DHB Complex
2.3.2. Possible Mechanism for DHB Bioactivation
3. Materials and Methods
3.1. Protein Expression and Purification
3.2. Spectral Binding Titrations
3.3. Determination of the X-ray Structures
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CYP3A4 | cytochrome P450 3A4 |
| DDI | drug-drug interaction |
| DHB | 6′,7′-dihydroxybergamottin |
| MBI | mechanism-based inactivation |
References
- Guengerich, F.P. Cytochrome P-450 3A4: Regulation and role in drug metabolism. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F. Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4. Curr. Drug Metab. 2008, 9, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Yung Chan, S.; Cher Goh, B.; Chan, E.; Duan, W.; Huang, M.; McLeod, H.L. Mechanism-based inhibition of cytochrome P450 3A4 by therapeutic drugs. Clin. Pharmacokinet. 2005, 44, 279–304. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F. Potential strategies for minimizing mechanism-based inhibition of cytochrome P450 3A4. Curr. Pharm. Des. 2008, 14, 990–1000. [Google Scholar] [CrossRef] [PubMed]
- Li, A.P.; Kaminski, D.L.; Rasmussen, A. Substrates of human hepatic cytochrome P450 3A4. Toxicology 1995, 104, 1–8. [Google Scholar] [CrossRef]
- Ekroos, M.; Sjogren, T. Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc. Natl. Acad. Sci. USA 2006, 103, 13682–13687. [Google Scholar] [CrossRef] [PubMed]
- Sevrioukova, I.F.; Poulos, T.L. Structural basis for regiospecific midazolam oxidation by human cytochrome P450 3A4. Proc. Natl. Acad. Sci. USA 2017, 114, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Skopalik, J.; Anzenbacher, P.; Otyepka, M. Flexibility of human cytochromes P450: Molecular dynamics reveals differences between CYPs 3A4, 2C9, and 2A6, which correlate with their substrate preferences. J. Phys. Chem. B. 2008, 112, 8165–8173. [Google Scholar] [CrossRef]
- Sevrioukova, I.F.; Poulos, T.L. Structural and mechanistic insights into the interaction of cytochrome P4503A4 with bromoergocryptine, a type I ligand. J. Biol. Chem. 2012, 287, 3510–3517. [Google Scholar] [CrossRef]
- Welker, H.A.; Wiltshire, H.; Bullingham, R. Clinical pharmacokinetics of mibefradil. Clin. Pharmacokinet. 1998, 35, 405–423. [Google Scholar] [CrossRef]
- Stresser, D.M.; Broudy, M.I.; Ho, T.; Cargill, C.E.; Blanchard, A.P.; Sharma, R.; Dandeneau, A.A.; Goodwin, J.J.; Turner, S.D.; Erve, J.C.; et al. Highly selective inhibition of human CYP3Aa in vitro by azamulin and evidence that inhibition is irreversible. Drug Metab. Dispos. 2004, 32, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Khojasteh, S.C.; Prabhu, S.; Kenny, J.R.; Halladay, J.S.; Lu, A.Y. Chemical inhibitors of cytochrome P450 isoforms in human liver microsomes: A re-evaluation of P450 isoform selectivity. Eur. J. Drug Metab. Pharmacokinet. 2011, 36, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hung, W.L.; Suh, J.H.; Wang, Y. Chemistry and health effects of furanocoumarins in grapefruit. J. Food Drug Anal. 2017, 25, 71–83. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Iyer, K.R.; Hayes, R.N.; Sinz, M.W.; Woolf, T.F.; Hollenberg, P.F. Inactivation of cytochrome P450 3A4 by bergamottin, a component of grapefruit juice. Chem. Res. Toxicol. 1998, 11, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Foti, R.S.; Rock, D.A.; Pearson, J.T.; Wahlstrom, J.L.; Wienkers, L.C. Mechanism-based inactivation of cytochrome P450 3A4 by mibefradil through heme destruction. Drug Metab. Dispos. 2011, 39, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Samuels, E.R.; Sevrioukova, I. Structure-activity relationships of rationally designed ritonavir analogs: Impact of side-group stereochemistry, head-group spacing, and backbone composition on the interaction with CYP3A4. Biochemistry 2019, 58, 2077–2087. [Google Scholar] [CrossRef] [PubMed]
- Prueksaritanont, T.; Ma, B.; Tang, C.; Meng, Y.; Assang, C.; Lu, P.; Reider, P.J.; Lin, J.H.; Baillie, T.A. Metabolic interactions between mibefradil and HMG-CoA reductase inhibitors: An in vitro investigation with human liver preparations. Br. J. Clin. Pharmacol. 1999, 47, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Bui, P.H.; Quesada, A.; Handforth, A.; Hankinson, O. The mibefradil derivative NNC55-0396, a specific T-type calcium channel antagonist, exhibits less CYP3A4 inhibition than mibefradil. Drug Metab. Dispos. 2008, 36, 1291–1299. [Google Scholar] [CrossRef]
- Tassaneeyakul, W.; Guo, L.Q.; Fukuda, K.; Ohta, T.; Yamazoe, Y. Inhibition selectivity of grapefruit juice components on human cytochromes P450. Arch. Biochem. Biophys. 2000, 378, 356–363. [Google Scholar] [CrossRef]
- Edwards, D.J.; Bellevue, F.H., 3rd; Woster, P.M. Identification of 6′,7′-dihydroxybergamottin, a cytochrome P450 inhibitor, in grapefruit juice. Drug Metab. Dispos. 1996, 24, 1287–1290. [Google Scholar]
- Guo, L.Q.; Taniguchi, M.; Xiao, Y.Q.; Baba, K.; Ohta, T.; Yamazoe, Y. Inhibitory effect of natural furanocoumarins on human microsomal cytochrome P450 3A activity. Jpn. J. Pharmacol. 2000, 82, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.L.; Kenaan, C.; Hollenberg, P.F. Identification of the residue in human CYP3A4 that is covalently modified by bergamottin and the reactive intermediate that contributes to the grapefruit juice effect. Drug Metab. Dispos. 2012, 40, 998–1006. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.L.; Kent, U.M.; Hollenberg, P.F. The grapefruit juice effect is not limited to cytochrome P450 (P450) 3A4: Evidence for bergamottin-dependent inactivation, heme destruction, and covalent binding to protein in P450s 2B6 and 3A5. J. Pharmacol. Exp. Ther. 2005, 313, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Bart, A.G.; Scott, E.E. Structures of human cytochrome P450 1A1 with bergamottin and erlotinib reveal active-site modifications for binding of diverse ligands. J. Biol. Chem. 2018, 293, 19201–19210. [Google Scholar] [CrossRef] [PubMed]
- Row, E.; Brown, S.A.; Stachulski, A.V.; Lennard, M.S. Development of novel furanocoumarin dimers as potent and selective inhibitors of CYP3A4. Drug Metab. Dispos. 2006, 34, 324–330. [Google Scholar] [CrossRef]
- Oda, K.; Yamaguchi, Y.; Yoshimura, T.; Wada, K.; Nishizono, N. Synthetic models related to furanocoumarin-CYP 3A4 interactions. comparison of furanocoumarin, coumarin, and benzofuran dimers as potent inhibitors of CYP3A4 activity. Chem. Pharm. Bull. 2007, 55, 1419–1421. [Google Scholar] [CrossRef] [PubMed]
- Grinkova, Y.V.; Denisov, I.G.; McLean, M.A.; Sligar, S.G. Oxidase uncoupling in heme monooxygenases: Human cytochrome P450 CYP3A4 in Nanodiscs. Biochem. Biophys. Res. Commun. 2013, 430, 1223–1227. [Google Scholar] [CrossRef]
- Paine, M.F.; Criss, A.B.; Watkins, P.B. Two major grapefruit juice components differ in time to onset of intestinal CYP3A4 inhibition. J. Pharmacol. Exp. Ther. 2005, 312, 1151–1160. [Google Scholar] [CrossRef]
- Sevrioukova, I.F. High-level production and properties of the cysteine-depleted cytochrome P450 3A4. Biochemistry 2017, 56, 3058–3067. [Google Scholar] [CrossRef]
- Karplus, P.A.; Diederichs, K. Linking crystallographic model and data quality. Science 2012, 336, 1030–1033. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Section D 2010, 66, 213–321. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Section D 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Ks a μM | High-Spin Shift | KsRIT b μM |
|---|---|---|---|
| mibefradil | 3.3 ± 0.4 | 65% | 1.42 ± 0.03 |
| azamulin | 1.7 ± 0.3 | 100% | 0.88 ± 0.09 |
| bergamottin | 3.0 ± 0.3 c (n = 1.8) | 48% | 0.035 ± 0.008 |
| DHB | 0.22 ± 0.03 d | <3% | 0.032 ± 0.002 |
| 10.3 ± 1.4 d | |||
| bromocryptine | 0.43 ± 0.08 | 90% | 0.070 ± 0.005 |
| midazolam | 24 ± 3 | 85% | 0.071 ± 0.006 |
| none | 0.019 ± 0.002 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sevrioukova, I.F. Structural Insights into the Interaction of Cytochrome P450 3A4 with Suicide Substrates: Mibefradil, Azamulin and 6′,7′-Dihydroxybergamottin. Int. J. Mol. Sci. 2019, 20, 4245. https://doi.org/10.3390/ijms20174245
Sevrioukova IF. Structural Insights into the Interaction of Cytochrome P450 3A4 with Suicide Substrates: Mibefradil, Azamulin and 6′,7′-Dihydroxybergamottin. International Journal of Molecular Sciences. 2019; 20(17):4245. https://doi.org/10.3390/ijms20174245
Chicago/Turabian StyleSevrioukova, Irina F. 2019. "Structural Insights into the Interaction of Cytochrome P450 3A4 with Suicide Substrates: Mibefradil, Azamulin and 6′,7′-Dihydroxybergamottin" International Journal of Molecular Sciences 20, no. 17: 4245. https://doi.org/10.3390/ijms20174245
APA StyleSevrioukova, I. F. (2019). Structural Insights into the Interaction of Cytochrome P450 3A4 with Suicide Substrates: Mibefradil, Azamulin and 6′,7′-Dihydroxybergamottin. International Journal of Molecular Sciences, 20(17), 4245. https://doi.org/10.3390/ijms20174245