ZO-2 Is a Master Regulator of Gene Expression, Cell Proliferation, Cytoarchitecture, and Cell Size

Abstract
1. Introduction
2. ZO-2 During Early Development
3. ZO-2 Presence at TJs Is Crucial in the Mouse Testis and the Human Liver and Inner Ear
3.1. ZO-2 and the Blood–Testis Barrier
3.2. ZO-2 in the Liver
3.3. ZO-2 in the Inner Ear
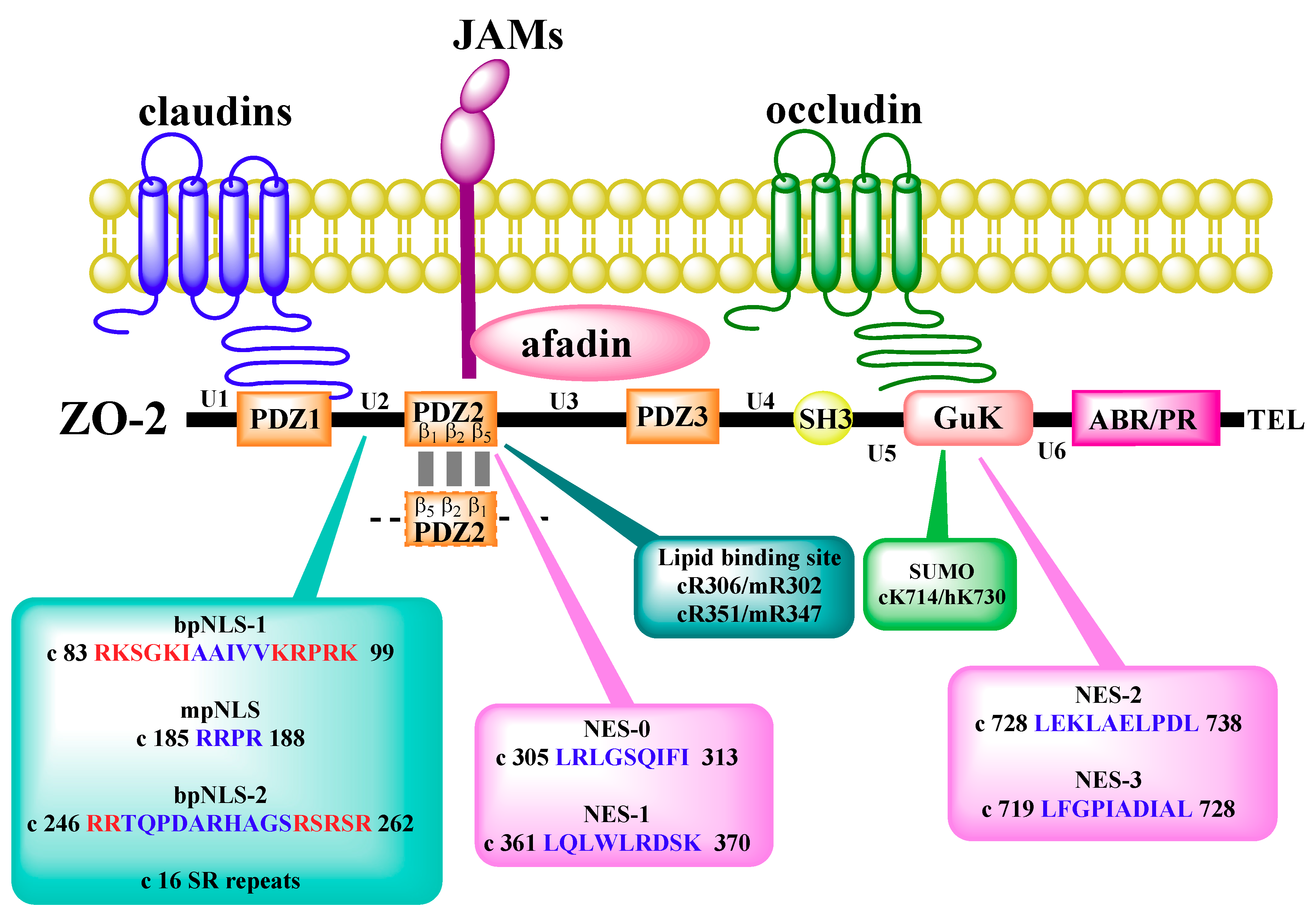
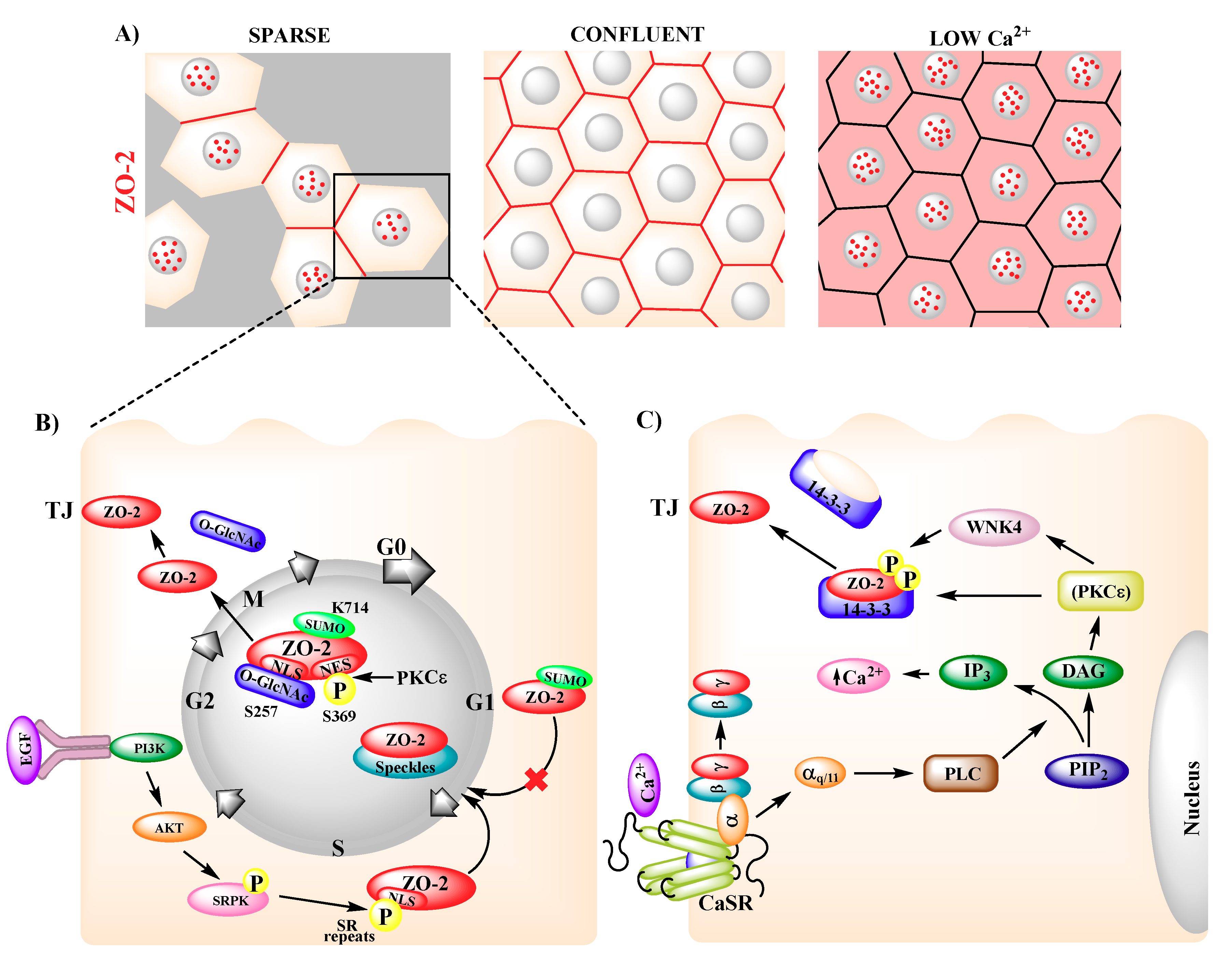
4. Subcellular Localization and Traffic of ZO-2
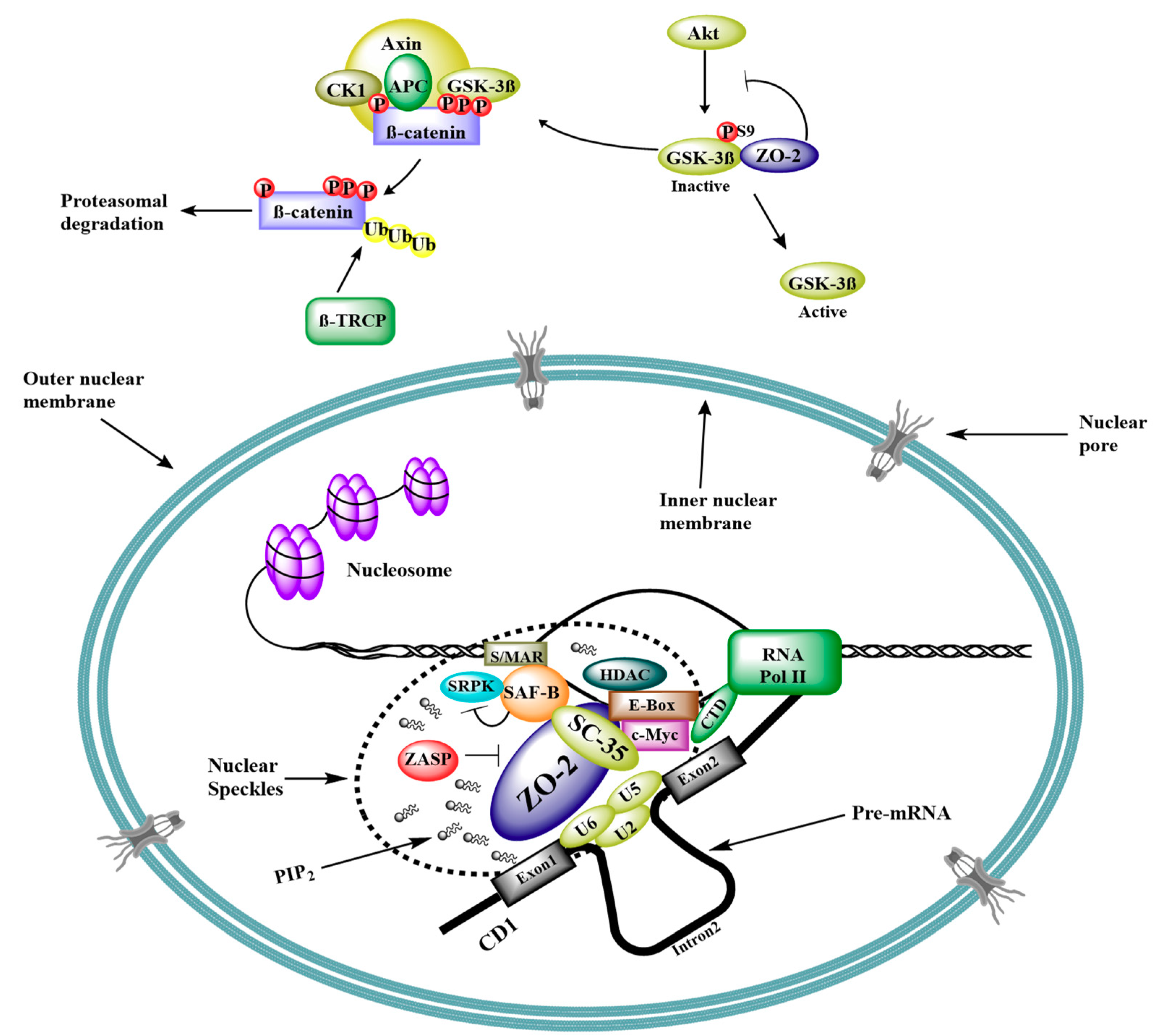
5. Interaction of ZO-2 with Nuclear Proteins
6. ZO-2 as Repressor of Gene Transcription
7. ZO-2 and Cell Proliferation
8. ZO-2 in Apoptosis and Cell Degeneration
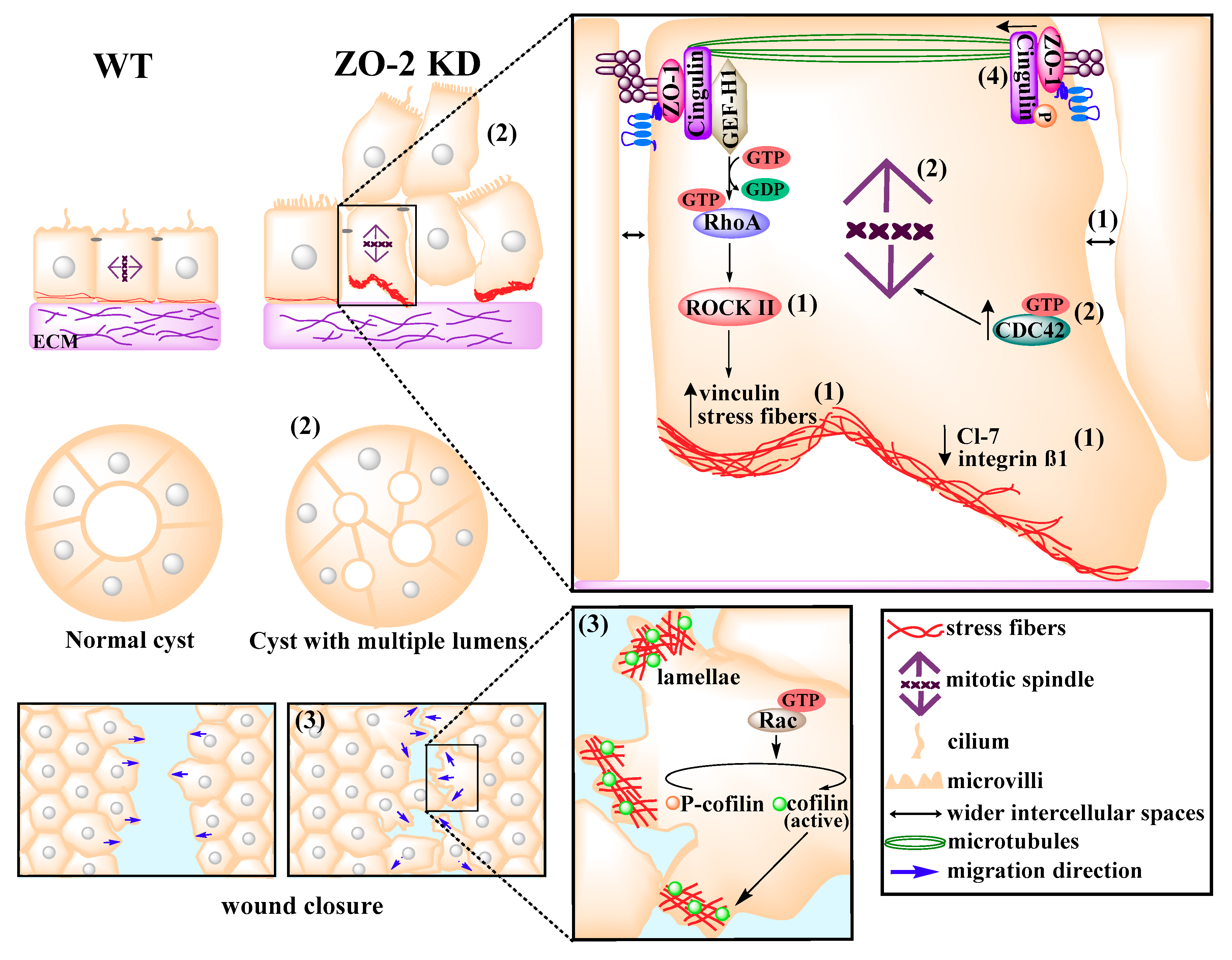
9. ZO-2 Associates with the Cortical Actomyosin Ring and Regulates Cytoarchitecture
10. ZO-2 as a Modulator of Cell Size
11. ZO-2 as a Tumor Regulator Protein
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gonzalez-Mariscal, L.; Quiros, M.; Diaz-Coranguez, M. ZO proteins and redox-dependent processes. Antioxid Redox Signal. 2011, 15, 1235–1253. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Mariscal, L.; Miranda, J.; Ortega-Olvera, J.M.; Gallego-Gutierrez, H.; Raya-Sandino, A.; Vargas-Sierra, O. Zonula Occludens Proteins in Cancer. Curr. Pathobiol. Rep. 2016, 4, 107–116. [Google Scholar] [CrossRef]
- Gumbiner, B.; Lowenkopf, T.; Apatira, D. Identification of a 160-kDa polypeptide that binds to the tight junction protein ZO-1. Proc. Natl. Acad. Sci. USA 1991, 88, 3460–3464. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.; Pierce, S.B.; Lenz, D.R.; Brownstein, Z.; Dagan-Rosenfeld, O.; Shahin, H.; Roeb, W.; McCarthy, S.; Nord, A.S.; Gordon, C.R.; et al. Genomic duplication and overexpression of TJP2/ZO-2 leads to altered expression of apoptosis genes in progressive nonsyndromic hearing loss DFNA51. Am. J. Hum. Genet. 2010, 87, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Morita, K.; Tsukita, S. Characterization of ZO-2 as a MAGUK family member associated with tight as well as adherens junctions with a binding affinity to occludin and alpha catenin. J. Biol. Chem. 1999, 274, 5981–5986. [Google Scholar] [CrossRef] [PubMed]
- Jesaitis, L.A.; Goodenough, D.A. Molecular characterization and tissue distribution of ZO-2, a tight junction protein homologous to ZO-1 and the Drosophila discs-large tumor suppressor protein. J. Cell Biol. 1994, 124, 949–961. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, E.L.; Caputo, M.; Angelini, G.D.; Ghorbel, M.T. Chronic hypoxia down-regulates tight junction protein ZO-2 expression in children with cyanotic congenital heart defect. Esc Heart Fail. 2016, 3, 131–137. [Google Scholar] [CrossRef]
- Gonzalez-Mariscal, L.; Miranda, J.; Raya-Sandino, A.; Dominguez-Calderon, A.; Cuellar-Perez, F. ZO-2, a tight junction protein involved in gene expression, proliferation, apoptosis, and cell size regulation. Ann. New York Acad. Sci. 2017, 1397, 35–53. [Google Scholar] [CrossRef]
- Haskins, J.; Gu, L.; Wittchen, E.S.; Hibbard, J.; Stevenson, B.R. ZO-3, a novel member of the MAGUK protein family found at the tight junction, interacts with ZO-1 and occludin. J. Cell Biol. 1998, 141, 199–208. [Google Scholar] [CrossRef]
- Wittchen, E.S.; Haskins, J.; Stevenson, B.R. Protein interactions at the tight junction. Actin has multiple binding partners, and ZO-1 forms independent complexes with ZO-2 and ZO-3. J. Biol. Chem. 1999, 274, 35179–35185. [Google Scholar] [CrossRef]
- Wu, J.; Yang, Y.; Zhang, J.; Ji, P.; Du, W.; Jiang, P.; Xie, D.; Huang, H.; Wu, M.; Zhang, G.; et al. Domain-swapped dimerization of the second PDZ domain of ZO2 may provide a structural basis for the polymerization of claudins. J. Biol. Chem. 2007, 282, 35988–35999. [Google Scholar] [CrossRef] [PubMed]
- Umeda, K.; Ikenouchi, J.; Katahira-Tayama, S.; Furuse, K.; Sasaki, H.; Nakayama, M.; Matsui, T.; Tsukita, S.; Furuse, M.; Tsukita, S. ZO-1 and ZO-2 independently determine where claudins are polymerized in tight-junction strand formation. Cell 2006, 126, 741–754. [Google Scholar] [CrossRef] [PubMed]
- Sheth, B.; Nowak, R.L.; Anderson, R.; Kwong, W.Y.; Papenbrock, T.; Fleming, T.P. Tight junction protein ZO-2 expression and relative function of ZO-1 and ZO-2 during mouse blastocyst formation. Exp. Cell Res. 2008, 314, 3356–3368. [Google Scholar] [CrossRef] [PubMed]
- Eckert, J.J.; Fleming, T.P. Tight junction biogenesis during early development. Biochim. Et. Biophys. Acta. 2008, 1778, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Kausalya, P.J.; Phua, D.C.; Ali, S.M.; Hossain, Z.; Hunziker, W. Early embryonic lethality of mice lacking ZO-2, but Not ZO-3, reveals critical and nonredundant roles for individual zonula occludens proteins in mammalian development. Mol. Cell Biol. 2008, 28, 1669–1678. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Anuar, F.; Ali, S.M.; Ng, M.Y.; Phua, D.C.; Hunziker, W. Zona occludens-2 is critical for blood-testis barrier integrity and male fertility. Mol. Biol. Cell 2009, 20, 4268–4277. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, S.; Chavez Munguia, B.; Gonzalez-Mariscal, L. ZO-2 silencing in epithelial cells perturbs the gate and fence function of tight junctions and leads to an atypical monolayer architecture. Exp. Cell Res. 2007, 313, 1533–1547. [Google Scholar] [CrossRef] [PubMed]
- Raya-Sandino, A.; Castillo-Kauil, A.; Dominguez-Calderon, A.; Alarcon, L.; Flores-Benitez, D.; Cuellar-Perez, F.; Lopez-Bayghen, B.; Chavez-Munguia, B.; Vazquez-Prado, J.; Gonzalez-Mariscal, L. Zonula occludens-2 regulates Rho proteins activity and the development of epithelial cytoarchitecture and barrier function. Biochim. Biophys. Acta. 2017, 1864, 1714–1733. [Google Scholar] [CrossRef]
- Kim, M.A.; Kim, Y.R.; Sagong, B.; Cho, H.J.; Bae, J.W.; Kim, J.; Lee, J.; Park, H.J.; Choi, J.Y.; Lee, K.Y.; et al. Genetic analysis of genes related to tight junction function in the Korean population with non-syndromic hearing loss. PLoS ONE 2014, 9, e95646. [Google Scholar] [CrossRef]
- Wang, H.Y.; Zhao, Y.L.; Liu, Q.; Yuan, H.; Gao, Y.; Lan, L.; Yu, L.; Wang, D.Y.; Guan, J.; Wang, Q.J. Identification of Two Disease-causing Genes TJP2 and GJB2 in a Chinese Family with Unconditional Autosomal Dominant Nonsyndromic Hereditary Hearing Impairment. Chin. Med. J. 2015, 128, 3345–3351. [Google Scholar] [CrossRef]
- Carlton, V.E.; Harris, B.Z.; Puffenberger, E.G.; Batta, A.K.; Knisely, A.S.; Robinson, D.L.; Strauss, K.A.; Shneider, B.L.; Lim, W.A.; Salen, G.; et al. Complex inheritance of familial hypercholanemia with associated mutations in TJP2 and BAAT. Nat. Genet. 2003, 34, 91–96. [Google Scholar] [CrossRef]
- Vitale, G.; Gitto, S.; Raimondi, F.; Mattiaccio, A.; Mantovani, V.; Vukotic, R.; D'Errico, A.; Seri, M.; Russell, R.B.; Andreone, P. Cryptogenic cholestasis in young and adults: ATP8B1, ABCB11, ABCB4, and TJP2 gene variants analysis by high-throughput sequencing. J. Gastroenterol. 2018, 53, 945–958. [Google Scholar] [CrossRef]
- Sambrotta, M.; Strautnieks, S.; Papouli, E.; Rushton, P.; Clark, B.E.; Parry, D.A.; Logan, C.V.; Newbury, L.J.; Kamath, B.M.; Ling, S.; et al. Mutations in TJP2 cause progressive cholestatic liver disease. Nat. Genet. 2014, 46, 326–328. [Google Scholar] [CrossRef]
- Ge, T.; Zhang, X.; Xiao, Y.; Wang, Y.; Zhang, T. Novel compound heterozygote mutations of TJP2 in a Chinese child with progressive cholestatic liver disease. Bmc Med. Genet. 2019, 20, 18. [Google Scholar] [CrossRef]
- Zhou, S.; Hertel, P.M.; Finegold, M.J.; Wang, L.; Kerkar, N.; Wang, J.; Wong, L.J.; Plon, S.E.; Sambrotta, M.; Foskett, P.; et al. Hepatocellular carcinoma associated with tight-junction protein 2 deficiency. Hepatology 2015, 62, 1914–1916. [Google Scholar] [CrossRef]
- Parsons, D.W.; Roy, A.; Yang, Y.; Wang, T.; Scollon, S.; Bergstrom, K.; Kerstein, R.A.; Gutierrez, S.; Petersen, A.K.; Bavle, A.; et al. Diagnostic Yield of Clinical Tumor and Germline Whole-Exome Sequencing for Children With Solid Tumors. JAMA Oncol. 2016, 2, 616–624. [Google Scholar] [CrossRef]
- Vij, M.; Shanmugam, N.P.; Reddy, M.S.; Sankaranarayanan, S.; Rela, M. Paediatric hepatocellular carcinoma in tight junction protein 2 (TJP2) deficiency. Virchows. Arch. 2017, 471, 679–683. [Google Scholar] [CrossRef]
- Dixon, P.H.; Sambrotta, M.; Chambers, J.; Taylor-Harris, P.; Syngelaki, A.; Nicolaides, K.; Knisely, A.S.; Thompson, R.J.; Williamson, C. An expanded role for heterozygous mutations of ABCB4, ABCB11, ATP8B1, ABCC2 and TJP2 in intrahepatic cholestasis of pregnancy. Sci. Rep. 2017, 7, 11823. [Google Scholar] [CrossRef]
- Stanton, P.G. Regulation of the blood-testis barrier. Semin. Cell Dev. Biol. 2016, 59, 166–173. [Google Scholar] [CrossRef]
- Ortega-Olvera, J.M.; Winkler, R.; Quitanilla-Vega, B.; Shibayama, M.; Chavez-Munguia, B.; Martin-Tapia, D.; Alarcon, L.; Gonzalez-Mariscal, L. The organophosphate pesticide methamidophos opens the blood-testis barrier and covalently binds to ZO-2 in mice. Toxicol. Appl. Pharmacol. 2018, 360, 257–272. [Google Scholar] [CrossRef]
- Sambrotta, M.; Thompson, R.J. Mutations in TJP2, encoding zona occludens 2, and liver disease. Tissue Barriers 2015, 3, e1026537. [Google Scholar] [CrossRef]
- Ben-Yosef, T.; Belyantseva, I.A.; Saunders, T.L.; Hughes, E.D.; Kawamoto, K.; Van Itallie, C.M.; Beyer, L.A.; Halsey, K.; Gardner, D.J.; Wilcox, E.R.; et al. Claudin 14 knockout mice, a model for autosomal recessive deafness DFNB29, are deaf due to cochlear hair cell degeneration. Hum. Mol. Genet. 2003, 12, 2049–2061. [Google Scholar] [CrossRef]
- Kitajiri, S.; Katsuno, T.; Sasaki, H.; Ito, J.; Furuse, M.; Tsukita, S. Deafness in occludin-deficient mice with dislocation of tricellulin and progressive apoptosis of the hair cells. Biol. Open 2014, 3, 759–766. [Google Scholar] [CrossRef]
- Kamitani, T.; Sakaguchi, H.; Tamura, A.; Miyashita, T.; Yamazaki, Y.; Tokumasu, R.; Inamoto, R.; Matsubara, A.; Mori, N.; Hisa, Y.; et al. Deletion of Tricellulin Causes Progressive Hearing Loss Associated with Degeneration of Cochlear Hair Cells. Sci. Rep. 2015, 5, 18402. [Google Scholar] [CrossRef]
- Morozko, E.L.; Nishio, A.; Ingham, N.J.; Chandra, R.; Fitzgerald, T.; Martelletti, E.; Borck, G.; Wilson, E.; Riordan, G.P.; Wangemann, P.; et al. ILDR1 null mice, a model of human deafness DFNB42, show structural aberrations of tricellular tight junctions and degeneration of auditory hair cells. Hum. Mol. Genet. 2015, 24, 609–624. [Google Scholar] [CrossRef]
- Nakano, Y.; Kim, S.H.; Kim, H.M.; Sanneman, J.D.; Zhang, Y.; Smith, R.J.; Marcus, D.C.; Wangemann, P.; Nessler, R.A.; Banfi, B. A claudin-9-based ion permeability barrier is essential for hearing. Plos Genet. 2009, 5, e1000610. [Google Scholar] [CrossRef]
- Gow, A.; Davies, C.; Southwood, C.M.; Frolenkov, G.; Chrustowski, M.; Ng, L.; Yamauchi, D.; Marcus, D.C.; Kachar, B. Deafness in Claudin 11-null mice reveals the critical contribution of basal cell tight junctions to stria vascularis function. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 7051–7062. [Google Scholar] [CrossRef]
- Kitajiri, S.; Miyamoto, T.; Mineharu, A.; Sonoda, N.; Furuse, K.; Hata, M.; Sasaki, H.; Mori, Y.; Kubota, T.; Ito, J.; et al. Compartmentalization established by claudin-11-based tight junctions in stria vascularis is required for hearing through generation of endocochlear potential. J. Cell Sci. 2004, 117, 5087–5096. [Google Scholar] [CrossRef]
- Islas, S.; Vega, J.; Ponce, L.; Gonzalez-Mariscal, L. Nuclear localization of the tight junction protein ZO-2 in epithelial cells. Exp. Cell Res. 2002, 274, 138–148. [Google Scholar] [CrossRef]
- Traweger, A.; Fuchs, R.; Krizbai, I.A.; Weiger, T.M.; Bauer, H.C.; Bauer, H. The tight junction protein ZO-2 localizes to the nucleus and interacts with the heterogeneous nuclear ribonucleoprotein scaffold attachment factor-B. J. Biol Chem. 2003, 278, 2692–2700. [Google Scholar] [CrossRef]
- Amaya, E.; Alarcon, L.; Martin-Tapia, D.; Cuellar-Perez, F.; Cano-Cortina, M.; Ortega-Olvera, J.M.; Cisneros, B.; Rodriguez, A.; Gamba, G.; Gonzalez-Mariscal, L. Activation of the Ca2+ sensing receptor and the PKC/WNK4 downstream signaling cascade induces arrival of ZO-2 to tight junctions and its separation from 14-3-3. Mol. Biol. Cell 2019. In press. [Google Scholar] [CrossRef]
- Tapia, R.; Huerta, M.; Islas, S.; Avila-Flores, A.; Lopez-Bayghen, E.; Weiske, J.; Huber, O.; Gonzalez-Mariscal, L. Zona occludens-2 inhibits cyclin D1 expression and cell proliferation and exhibits changes in localization along the cell cycle. Mol. Biol. Cell 2009, 20, 1102–1117. [Google Scholar] [CrossRef]
- Lopez-Bayghen, E.; Jaramillo, B.E.; Huerta, M.; Betanzos, A.; Gonzalez-Mariscal, L. TJ Proteins That Make Round Trips to the Nucleus. In Tight Junctions; Gonzalez-Mariscal, L., Ed.; Springer US: Boston, MA, USA, 2006; pp. 76–100. [Google Scholar] [CrossRef]
- Quiros, M.; Alarcon, L.; Ponce, A.; Giannakouros, T.; Gonzalez-Mariscal, L. The intracellular fate of zonula occludens 2 is regulated by the phosphorylation of SR repeats and the phosphorylation/O-GlcNAcylation of S257. Mol. Biol. Cell 2013, 24, 2528–2543. [Google Scholar] [CrossRef]
- Meerschaert, K.; Tun, M.P.; Remue, E.; De Ganck, A.; Boucherie, C.; Vanloo, B.; Degeest, G.; Vandekerckhove, J.; Zimmermann, P.; Bhardwaj, N.; et al. The PDZ2 domain of zonula occludens-1 and -2 is a phosphoinositide binding domain. Cell Mol. Life Sci. 2009, 66, 3951–3966. [Google Scholar] [CrossRef]
- Gonzalez-Mariscal, L.; Ponce, A.; Alarcon, L.; Jaramillo, B.E. The tight junction protein ZO-2 has several functional nuclear export signals. Exp. Cell Res. 2006, 312, 3323–3335. [Google Scholar] [CrossRef]
- Chamorro, D.; Alarcon, L.; Ponce, A.; Tapia, R.; Gonzalez-Aguilar, H.; Robles-Flores, M.; Mejia-Castillo, T.; Segovia, J.; Bandala, Y.; Juaristi, E.; et al. Phosphorylation of zona occludens-2 by protein kinase C epsilon regulates its nuclear exportation. Mol. Biol. Cell 2009, 20, 4120–4129. [Google Scholar] [CrossRef]
- Wetzel, F.; Mittag, S.; Cano-Cortina, M.; Wagner, T.; Kramer, O.H.; Niedenthal, R.; Gonzalez-Mariscal, L.; Huber, O. SUMOylation regulates the intracellular fate of ZO-2. Cell. Mol. Life Sci. CMLS 2016, 74, 373–392. [Google Scholar] [CrossRef]
- Gonzalez-Mariscal, L.; Contreras, R.G.; Bolivar, J.J.; Ponce, A.; Chavez De Ramirez, B.; Cereijido, M. Role of calcium in tight junction formation between epithelial cells. Am. J. Physiol. 1990, 259, C978–C986. [Google Scholar] [CrossRef]
- Jouret, F.; Wu, J.; Hull, M.; Rajendran, V.; Mayr, B.; Schofl, C.; Geibel, J.; Caplan, M.J. Activation of the Ca(2)+-sensing receptor induces deposition of tight junction components to the epithelial cell plasma membrane. J. Cell Sci. 2013, 126, 5132–5142. [Google Scholar] [CrossRef]
- Jaramillo, B.E.; Ponce, A.; Moreno, J.; Betanzos, A.; Huerta, M.; Lopez-Bayghen, E.; Gonzalez-Mariscal, L. Characterization of the tight junction protein ZO-2 localized at the nucleus of epithelial cells. Exp. Cell Res. 2004, 297, 247–258. [Google Scholar] [CrossRef]
- Lechuga, S.; Alarcon, L.; Solano, J.; Huerta, M.; Lopez-Bayghen, E.; Gonzalez-Mariscal, L. Identification of ZASP, a novel protein associated to Zona occludens-2. Exp. Cell Res. 2010, 316, 3124–3139. [Google Scholar] [CrossRef]
- Hong, E.A.; Gautrey, H.L.; Elliott, D.J.; Tyson-Capper, A.J. SAFB1- and SAFB2-mediated transcriptional repression: Relevance to cancer. Biochem. Soc. Trans. 2012, 40, 826–830. [Google Scholar] [CrossRef]
- Lin, J.; Xu, P.; LaVallee, P.; Hoidal, J.R. Identification of proteins binding to E-Box/Ku86 sites and function of the tumor suppressor SAFB1 in transcriptional regulation of the human xanthine oxidoreductase gene. J. Biol. Chem. 2008, 283, 29681–29689. [Google Scholar] [CrossRef]
- Huerta, M.; Munoz, R.; Tapia, R.; Soto-Reyes, E.; Ramirez, L.; Recillas-Targa, F.; Gonzalez-Mariscal, L.; Lopez-Bayghen, E. Cyclin D1 is transcriptionally down-regulated by ZO-2 via an E box and the transcription factor c-Myc. Mol. Biol. Cell 2007, 18, 4826–4836. [Google Scholar] [CrossRef]
- Townson, S.M.; Kang, K.; Lee, A.V.; Oesterreich, S. Structure-function analysis of the estrogen receptor alpha corepressor scaffold attachment factor-B1: Identification of a potent transcriptional repression domain. J. Biol. Chem. 2004, 279, 26074–26081. [Google Scholar] [CrossRef]
- Nikolakaki, E.; Kohen, R.; Hartmann, A.M.; Stamm, S.; Georgatsou, E.; Giannakouros, T. Cloning and characterization of an alternatively spliced form of SR protein kinase 1 that interacts specifically with scaffold attachment factor-B. J. Biol. Chem. 2001, 276, 40175–40182. [Google Scholar] [CrossRef]
- McCracken, S.; Longman, D.; Marcon, E.; Moens, P.; Downey, M.; Nickerson, J.A.; Jessberger, R.; Wilde, A.; Caceres, J.F.; Emili, A.; et al. Proteomic analysis of SRm160-containing complexes reveals a conserved association with cohesin. J. Biol. Chem. 2005, 280, 42227–42236. [Google Scholar] [CrossRef]
- Tsai, Y.C.; Greco, T.M.; Boonmee, A.; Miteva, Y.; Cristea, I.M. Functional proteomics establishes the interaction of SIRT7 with chromatin remodeling complexes and expands its role in regulation of RNA polymerase I transcription. Mol. Cell. Proteom. MCP 2012, 11, 60–76. [Google Scholar] [CrossRef]
- Havugimana, P.C.; Hart, G.T.; Nepusz, T.; Yang, H.; Turinsky, A.L.; Li, Z.; Wang, P.I.; Boutz, D.R.; Fong, V.; Phanse, S.; et al. A census of human soluble protein complexes. Cell 2012, 150, 1068–1081. [Google Scholar] [CrossRef]
- Tang, Y.; Puri, A.; Ricketts, M.D.; Rai, T.S.; Hoffmann, J.; Hoi, E.; Adams, P.D.; Schultz, D.C.; Marmorstein, R. Identification of an ubinuclein 1 region required for stability and function of the human HIRA/UBN1/CABIN1/ASF1a histone H3.3 chaperone complex. Biochemistry 2012, 51, 2366–2377. [Google Scholar] [CrossRef]
- Lupo, J.; Conti, A.; Sueur, C.; Coly, P.A.; Coute, Y.; Hunziker, W.; Burmeister, W.P.; Germi, R.; Manet, E.; Gruffat, H.; et al. Identification of new interacting partners of the shuttling protein ubinuclein (Ubn-1). Exp. Cell Res. 2012, 318, 509–520. [Google Scholar] [CrossRef]
- Bernstein, K.A.; Gallagher, J.E.; Mitchell, B.M.; Granneman, S.; Baserga, S.J. The small-subunit processome is a ribosome assembly intermediate. Eukaryot Cell 2004, 3, 1619–1626. [Google Scholar] [CrossRef]
- Leung, J.W.; Makharashvili, N.; Agarwal, P.; Chiu, L.Y.; Pourpre, R.; Cammarata, M.B.; Cannon, J.R.; Sherker, A.; Durocher, D.; Brodbelt, J.S.; et al. ZMYM3 regulates BRCA1 localization at damaged chromatin to promote DNA repair. Genes Dev. 2017, 31, 260–274. [Google Scholar] [CrossRef]
- Bonnelye, E.; Vanacker, J.M.; Dittmar, T.; Begue, A.; Desbiens, X.; Denhardt, D.T.; Aubin, J.E.; Laudet, V.; Fournier, B. The ERR-1 orphan receptor is a transcriptional activator expressed during bone development. Mol. Endocrinol. 1997, 11, 905–916. [Google Scholar] [CrossRef][Green Version]
- Wan, C.; Borgeson, B.; Phanse, S.; Tu, F.; Drew, K.; Clark, G.; Xiong, X.; Kagan, O.; Kwan, J.; Bezginov, A.; et al. Panorama of ancient metazoan macromolecular complexes. Nature 2015, 525, 339–344. [Google Scholar] [CrossRef]
- Shaheen, R.; Tasak, M.; Maddirevula, S.; Abdel-Salam, G.M.H.; Sayed, I.S.M.; Alazami, A.M.; Al-Sheddi, T.; Alobeid, E.; Phizicky, E.M.; Alkuraya, F.S. PUS7 mutations impair pseudouridylation in humans and cause intellectual disability and microcephaly. Hum. Genet. 2019, 138, 231–239. [Google Scholar] [CrossRef]
- Betanzos, A.; Huerta, M.; Lopez-Bayghen, E.; Azuara, E.; Amerena, J.; Gonzalez-Mariscal, L. The tight junction protein ZO-2 associates with Jun, Fos and C/EBP transcription factors in epithelial cells. Exp. Cell Res. 2004, 292, 51–66. [Google Scholar] [CrossRef]
- Kao, S.H.; Wang, W.L.; Chen, C.Y.; Chang, Y.L.; Wu, Y.Y.; Wang, Y.T.; Wang, S.P.; Nesvizhskii, A.I.; Chen, Y.J.; Hong, T.M.; et al. GSK3beta controls epithelial-mesenchymal transition and tumor metastasis by CHIP-mediated degradation of Slug. Oncogene 2014, 33, 3172–3182. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Ting, L.; Bruckner, R.J.; Gebreab, F.; Gygi, M.P.; Szpyt, J.; Tam, S.; Zarraga, G.; Colby, G.; Baltier, K.; et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell 2015, 162, 425–440. [Google Scholar] [CrossRef]
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Dominguez-Calderon, A.; Avila-Flores, A.; Ponce, A.; Lopez-Bayghen, E.; Calderon-Salinas, J.V.; Luis Reyes, J.; Chavez-Munguia, B.; Segovia, J.; Angulo, C.; Ramirez, L.; et al. ZO-2 silencing induces renal hypertrophy through a cell cycle mechanism and the activation of YAP and the mTOR pathway. Mol. Biol. Cell 2016, 27, 1581–1595. [Google Scholar] [CrossRef]
- Palmer, H.G.; Gonzalez-Sancho, J.M.; Espada, J.; Berciano, M.T.; Puig, I.; Baulida, J.; Quintanilla, M.; Cano, A.; de Herreros, A.G.; Lafarga, M.; et al. Vitamin D(3) promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition of beta-catenin signaling. J. Cell Biol. 2001, 154, 369–387. [Google Scholar] [CrossRef]
- Bautista-Garcia, P.; Reyes, J.L.; Martin, D.; Namorado, M.C.; Chavez-Munguia, B.; Soria-Castro, E.; Huber, O.; Gonzalez-Mariscal, L. Zona occludens-2 protects against podocyte dysfunction induced by ADR in mice. Am. J. Physiol. Ren. Physiol. 2013, 304, F77–F87. [Google Scholar] [CrossRef]
- Meng, Z.; Moroishi, T.; Guan, K.L. Mechanisms of Hippo pathway regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. YAP/TAZ incorporation in the beta-catenin destruction complex orchestrates the Wnt response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef]
- Zhao, B.; Li, L.; Tumaneng, K.; Wang, C.Y.; Guan, K.L. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev. 2010, 24, 72–85. [Google Scholar] [CrossRef]
- Kusch, A.; Tkachuk, S.; Tkachuk, N.; Patecki, M.; Park, J.K.; Dietz, R.; Haller, H.; Dumler, I. The tight junction protein ZO-2 mediates proliferation of vascular smooth muscle cells via regulation of Stat1. Cardiovasc. Res. 2009, 83, 115–122. [Google Scholar] [CrossRef]
- Oka, T.; Remue, E.; Meerschaert, K.; Vanloo, B.; Boucherie, C.; Gfeller, D.; Bader, G.D.; Sidhu, S.S.; Vandekerckhove, J.; Gettemans, J.; et al. Functional complexes between YAP2 and ZO-2 are PDZ domain-dependent, and regulate YAP2 nuclear localization and signalling. Biochem J. 2010, 432, 461–472. [Google Scholar] [CrossRef]
- Diehl, J.A.; Cheng, M.; Roussel, M.F.; Sherr, C.J. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998, 12, 3499–3511. [Google Scholar] [CrossRef]
- Klein, P.S.; Melton, D.A. A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. USA 1996, 93, 8455–8459. [Google Scholar] [CrossRef]
- Hedgepeth, C.M.; Conrad, L.J.; Zhang, J.; Huang, H.C.; Lee, V.M.; Klein, P.S. Activation of the Wnt signaling pathway: A molecular mechanism for lithium action. Dev. Biol. 1997, 185, 82–91. [Google Scholar] [CrossRef]
- Qiao, X.; Roth, I.; Feraille, E.; Hasler, U. Different effects of ZO-1, ZO-2 and ZO-3 silencing on kidney collecting duct principal cell proliferation and adhesion. Cell Cycle 2014, 13, 3059–3075. [Google Scholar] [CrossRef]
- Traweger, A.; Lehner, C.; Farkas, A.; Krizbai, I.A.; Tempfer, H.; Klement, E.; Guenther, B.; Bauer, H.C.; Bauer, H. Nuclear Zonula occludens-2 alters gene expression and junctional stability in epithelial and endothelial cells. Differ. Res. Biol. Divers. 2008, 76, 99–106. [Google Scholar] [CrossRef]
- Iqbal, M.A.; Gupta, V.; Gopinath, P.; Mazurek, S.; Bamezai, R.N. Pyruvate kinase M2 and cancer: An updated assessment. Febs Lett. 2014, 588, 2685–2692. [Google Scholar] [CrossRef]
- Bojarski, C.; Weiske, J.; Schoneberg, T.; Schroder, W.; Mankertz, J.; Schulzke, J.D.; Florian, P.; Fromm, M.; Tauber, R.; Huber, O. The specific fates of tight junction proteins in apoptotic epithelial cells. J. Cell Sci. 2004, 117, 2097–2107. [Google Scholar] [CrossRef]
- Chaitanya, G.V.; Steven, A.J.; Babu, P.P. PARP-1 cleavage fragments: Signatures of cell-death proteases in neurodegeneration. Cell Commun Signal. 2010, 8, 31. [Google Scholar] [CrossRef]
- Liu, J.; Yuan, Q.; Ling, X.; Tan, Q.; Liang, H.; Chen, J.; Lin, L.; Xiao, Y.; Chen, W.; Liu, L.; et al. PARP1 may be involved in hydroquinoneinduced apoptosis by poly ADPribosylation of ZO2. Mol. Med. Rep. 2017, 16, 8076–8084. [Google Scholar] [CrossRef]
- Kazmierczak, M.; Harris, S.L.; Kazmierczak, P.; Shah, P.; Starovoytov, V.; Ohlemiller, K.K.; Schwander, M. Progressive Hearing Loss in Mice Carrying a Mutation in Usp53. J. Neurosci. : Off. J. Soc. Neurosci. 2015, 35, 15582–15598. [Google Scholar] [CrossRef]
- Maddirevula, S.; Alhebbi, H.; Alqahtani, A.; Algoufi, T.; Alsaif, H.S.; Ibrahim, N.; Abdulwahab, F.; Barr, M.; Alzaidan, H.; Almehaideb, A.; et al. Identification of novel loci for pediatric cholestatic liver disease defined by KIF12, PPM1F, USP53, LSR, and WDR83OS pathogenic variants. Genet. Med. : Off. J. Am. Coll. Med. Genet. 2019, 21, 1164–1172. [Google Scholar] [CrossRef]
- Oka, T.; Schmitt, A.P.; Sudol, M. Opposing roles of angiomotin-like-1 and zona occludens-2 on pro-apoptotic function of YAP. Oncogene 2012, 31, 128–134. [Google Scholar] [CrossRef]
- Fanning, A.S.; Van Itallie, C.M.; Anderson, J.M. Zonula occludens-1 and -2 regulate apical cell structure and the zonula adherens cytoskeleton in polarized epithelia. Mol. Biol. Cell 2012, 23, 577–590. [Google Scholar] [CrossRef]
- Mattagajasingh, S.N.; Huang, S.C.; Hartenstein, J.S.; Benz, E.J., Jr. Characterization of the interaction between protein 4.1R and ZO-2. A possible link between the tight junction and the actin cytoskeleton. J. Biol Chem 2000, 275, 30573–30585. [Google Scholar] [CrossRef]
- Baines, A.J.; Lu, H.C.; Bennett, P.M. The Protein 4.1 family: Hub proteins in animals for organizing membrane proteins. Biochim. Et Biophys. Acta 2014, 1838, 605–619. [Google Scholar] [CrossRef]
- Monteiro, A.C.; Sumagin, R.; Rankin, C.R.; Leoni, G.; Mina, M.J.; Reiter, D.M.; Stehle, T.; Dermody, T.S.; Schaefer, S.A.; Hall, R.A.; et al. JAM-A associates with ZO-2, afadin, and PDZ-GEF1 to activate Rap2c and regulate epithelial barrier function. Mol. Biol. Cell 2013, 24, 2849–2860. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Umeda, K.; Wada, M.; Nada, S.; Okada, M.; Tsukita, S.; Tsukita, S. ZO-1- and ZO-2-dependent integration of myosin-2 to epithelial zonula adherens. Mol. Biol. Cell 2008, 19, 3801–3811. [Google Scholar] [CrossRef]
- Umeda, K.; Matsui, T.; Nakayama, M.; Furuse, K.; Sasaki, H.; Furuse, M.; Tsukita, S. Establishment and characterization of cultured epithelial cells lacking expression of ZO-1. J. Biol Chem 2004, 279, 44785–44794. [Google Scholar] [CrossRef]
- Cartagena-Rivera, A.X.; Van Itallie, C.M.; Anderson, J.M.; Chadwick, R.S. Apical surface supracellular mechanical properties in polarized epithelium using noninvasive acoustic force spectroscopy. Nat. Commun. 2017, 8, 1030. [Google Scholar] [CrossRef]
- Van Itallie, C.M.; Fanning, A.S.; Bridges, A.; Anderson, J.M. ZO-1 stabilizes the tight junction solute barrier through coupling to the perijunctional cytoskeleton. Mol. Biol. Cell 2009, 20, 3930–3940. [Google Scholar] [CrossRef]
- Tokuda, S.; Higashi, T.; Furuse, M. ZO-1 knockout by TALEN-mediated gene targeting in MDCK cells: Involvement of ZO-1 in the regulation of cytoskeleton and cell shape. PLoS ONE 2014, 9, e104994. [Google Scholar] [CrossRef]
- Spadaro, D.; Le, S.; Laroche, T.; Mean, I.; Jond, L.; Yan, J.; Citi, S. Tension-Dependent Stretching Activates ZO-1 to Control the Junctional Localization of Its Interactors. Curr. Biol. : Cb 2017, 27, 3783–3795.e8. [Google Scholar] [CrossRef]
- Handorf, A.M.; Zhou, Y.; Halanski, M.A.; Li, W.J. Tissue stiffness dictates development, homeostasis, and disease progression. Organogenesis 2015, 11, 1–15. [Google Scholar] [CrossRef]
- Shinto, O.; Yashiro, M.; Kawajiri, H.; Shimizu, K.; Shimizu, T.; Miwa, A.; Hirakawa, K. Inhibitory effect of a TGFbeta receptor type-I inhibitor, Ki26894, on invasiveness of scirrhous gastric cancer cells. Br. J. Cancer 2010, 102, 844–851. [Google Scholar] [CrossRef]
- Jaffe, A.B.; Kaji, N.; Durgan, J.; Hall, A. Cdc42 controls spindle orientation to position the apical surface during epithelial morphogenesis. J. Cell Biol. 2008, 183, 625–633. [Google Scholar] [CrossRef]
- Etienne-Manneville, S.; Hall, A. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCzeta. Cell 2001, 106, 489–498. [Google Scholar] [CrossRef]
- Gupta, G.D.; Coyaud, E.; Goncalves, J.; Mojarad, B.A.; Liu, Y.; Wu, Q.; Gheiratmand, L.; Comartin, D.; Tkach, J.M.; Cheung, S.W.; et al. A Dynamic Protein Interaction Landscape of the Human Centrosome-Cilium Interface. Cell 2015, 163, 1484–1499. [Google Scholar] [CrossRef]
- Monnich, M.; Borgeskov, L.; Breslin, L.; Jakobsen, L.; Rogowski, M.; Doganli, C.; Schroder, J.M.; Mogensen, J.B.; Blinkenkjaer, L.; Harder, L.M.; et al. CEP128 Localizes to the Subdistal Appendages of the Mother Centriole and Regulates TGF-beta/BMP Signaling at the Primary Cilium. Cell Rep. 2018, 22, 2584–2592. [Google Scholar] [CrossRef]
- Kashihara, H.; Chiba, S.; Kanno, S.I.; Suzuki, K.; Yano, T.; Tsukita, S. Cep128 associates with Odf2 to form the subdistal appendage of the centriole. Genes Cells : Devoted Mol. Cell. Mech. 2019, 24, 231–243. [Google Scholar] [CrossRef]
- Hung, H.F.; Hehnly, H.; Doxsey, S. The Mother Centriole Appendage Protein Cenexin Modulates Lumen Formation through Spindle Orientation. Curr. Biol. : Cb 2016, 26, 793–801. [Google Scholar] [CrossRef]
- Ganapathi Sankaran, D.; Stemm-Wolf, A.J.; Pearson, C.G. CEP135 isoform dysregulation promotes centrosome amplification in breast cancer cells. Mol. Biol. Cell 2019, 30, 1230–1244. [Google Scholar] [CrossRef]
- Gromley, A.; Jurczyk, A.; Sillibourne, J.; Halilovic, E.; Mogensen, M.; Groisman, I.; Blomberg, M.; Doxsey, S. A novel human protein of the maternal centriole is required for the final stages of cytokinesis and entry into S phase. J. Cell Biol. 2003, 161, 535–545. [Google Scholar] [CrossRef]
- Liu, B.; Preisig, P. TGF-beta1-mediated hypertrophy involves inhibiting pRB phosphorylation by blocking activation of cyclin E kinase. Am. J. Physiol. 1999, 277, F186–F194. [Google Scholar]
- Liu, B.; Preisig, P.A. Compensatory renal hypertrophy is mediated by a cell cycle-dependent mechanism. Kidney Int. 2002, 62, 1650–1658. [Google Scholar] [CrossRef]
- Jurkovitz, C.T.; England, B.K.; Ebb, R.G.; Mitch, W.E. Influence of ammonia and pH on protein and amino acid metabolism in LLC-PK1 cells. Kidney Int. 1992, 42, 595–601. [Google Scholar] [CrossRef][Green Version]
- Ling, H.; Vamvakas, S.; Gekle, M.; Schaefer, L.; Teschner, M.; Schaefer, R.M.; Heidland, A. Role of lysosomal cathepsin activities in cell hypertrophy induced by NH4Cl in cultured renal proximal tubule cells. J. Am. Soc. Nephrol. : Jasn 1996, 7, 73–80. [Google Scholar]
- Glaunsinger, B.A.; Weiss, R.S.; Lee, S.S.; Javier, R. Link of the unique oncogenic properties of adenovirus type 9 E4-ORF1 to a select interaction with the candidate tumor suppressor protein ZO-2. Embo J. 2001, 20, 5578–5586. [Google Scholar] [CrossRef]
- Thomas, M.; Myers, M.P.; Massimi, P.; Guarnaccia, C.; Banks, L. Analysis of Multiple HPV E6 PDZ Interactions Defines Type-Specific PDZ Fingerprints That Predict Oncogenic Potential. Plos Pathog. 2016, 12, e1005766. [Google Scholar] [CrossRef]
- Hernandez-Monge, J.; Garay, E.; Raya-Sandino, A.; Vargas-Sierra, O.; Diaz-Chavez, J.; Popoca-Cuaya, M.; Lambert, P.F.; Gonzalez-Mariscal, L.; Gariglio, P. Papillomavirus E6 oncoprotein up-regulates occludin and ZO-2 expression in ovariectomized mice epidermis. Exp. Cell Res. 2013, 319, 2588–2603. [Google Scholar] [CrossRef]
- Grm, H.S.; Banks, L. Degradation of hDlg and MAGIs by human papillomavirus E6 is E6-AP-independent. J. Gen. Virol. 2004, 85, 2815–2819. [Google Scholar] [CrossRef]
- Lee, S.S.; Glaunsinger, B.; Mantovani, F.; Banks, L.; Javier, R.T. Multi-PDZ domain protein MUPP1 is a cellular target for both adenovirus E4-ORF1 and high-risk papillomavirus type 18 E6 oncoproteins. J. Virol. 2000, 74, 9680–9693. [Google Scholar] [CrossRef]
- Nakagawa, S.; Huibregtse, J.M. Human scribble (Vartul) is targeted for ubiquitin-mediated degradation by the high-risk papillomavirus E6 proteins and the E6AP ubiquitin-protein ligase. Mol. Cell. Biol. 2000, 20, 8244–8253. [Google Scholar] [CrossRef]
- Fink, C.; Weigel, R.; Hembes, T.; Lauke-Wettwer, H.; Kliesch, S.; Bergmann, M.; Brehm, R.H. Altered expression of ZO-1 and ZO-2 in Sertoli cells and loss of blood-testis barrier integrity in testicular carcinoma in situ. Neoplasia 2006, 8, 1019–1027. [Google Scholar] [CrossRef]
- Luczka, E.; Syne, L.; Nawrocki-Raby, B.; Kileztky, C.; Hunziker, W.; Birembaut, P.; Gilles, C.; Polette, M. Regulation of membrane-type 1 matrix metalloproteinase expression by zonula occludens-2 in human lung cancer cells. Clin. Exp. Metastasis 2013. [Google Scholar] [CrossRef]
- Saito, K.; Enya, K.; Oneyama, C.; Hikita, T.; Okada, M. Proteomic identification of ZO-1/2 as a novel scaffold for Src/Csk regulatory circuit. Biochem. Biophys. Res. Commun. 2008, 366, 969–975. [Google Scholar] [CrossRef]
- Chlenski, A.; Ketels, K.V.; Engeriser, J.L.; Talamonti, M.S.; Tsao, M.S.; Koutnikova, H.; Oyasu, R.; Scarpelli, D.G. zo-2 gene alternative promoters in normal and neoplastic human pancreatic duct cells. Int J. Cancer 1999, 83, 349–358. [Google Scholar] [CrossRef]
- Chlenski, A.; Ketels, K.V.; Tsao, M.S.; Talamonti, M.S.; Anderson, M.R.; Oyasu, R.; Scarpelli, D.G. Tight junction protein ZO-2 is differentially expressed in normal pancreatic ducts compared to human pancreatic adenocarcinoma. Int. J. Cancer 1999, 82, 137–144. [Google Scholar] [CrossRef]
- Chlenski, A.; Ketels, K.V.; Korovaitseva, G.I.; Talamonti, M.S.; Oyasu, R.; Scarpelli, D.G. Organization and expression of the human zo-2 gene (tjp-2) in normal and neoplastic tissues. Biochim. Et Biophys. Acta 2000, 1493, 319–324. [Google Scholar] [CrossRef]
- Kim, Y.J.; Jung, Y.D.; Kim, T.O.; Kim, H.S. Alu-related transcript of TJP2 gene as a marker for colorectal cancer. Gene 2013, 524, 268–274. [Google Scholar] [CrossRef]
- Tokes, A.M.; Szasz, A.M.; Juhasz, E.; Schaff, Z.; Harsanyi, L.; Molnar, I.A.; Baranyai, Z.; Besznyak, I., Jr.; Zarand, A.; Salamon, F.; et al. Expression of tight junction molecules in breast carcinomas analysed by array PCR and immunohistochemistry. Pathol. Oncol. Res. 2012, 18, 593–606. [Google Scholar] [CrossRef]
- Kato, Y.; Yashiro, M.; Noda, S.; Tendo, M.; Kashiwagi, S.; Doi, Y.; Nishii, T.; Matsuoka, J.; Fuyuhiro, Y.; Shinto, O.; et al. Establishment and characterization of a new hypoxia-resistant cancer cell line, OCUM-12/Hypo, derived from a scirrhous gastric carcinoma. Br. J. Cancer 2010, 102, 898–907. [Google Scholar] [CrossRef]
- Pope, W.B.; Chen, J.H.; Dong, J.; Carlson, M.R.; Perlina, A.; Cloughesy, T.F.; Liau, L.M.; Mischel, P.S.; Nghiemphu, P.; Lai, A.; et al. Relationship between gene expression and enhancement in glioblastoma multiforme: Exploratory DNA microarray analysis. Radiology 2008, 249, 268–277. [Google Scholar] [CrossRef]
- Deroo, B.J.; Korach, K.S. Estrogen receptors and human disease. J. Clin. Investig. 2006, 116, 561–570. [Google Scholar] [CrossRef]
- Acconcia, F.; Marino, M. The Effects of 17beta-estradiol in Cancer are Mediated by Estrogen Receptor Signaling at the Plasma Membrane. Front. Physiol. 2011, 2, 30. [Google Scholar] [CrossRef]
- Doi, Y.; Yashiro, M.; Yamada, N.; Amano, R.; Noda, S.; Hirakawa, K. VEGF-A/VEGFR-2 signaling plays an important role for the motility of pancreas cancer cells. Ann. Surg. Oncol. 2012, 19, 2733–2743. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Disease | Alteration | Location within ZO-2 | Features of Disease | Reference |
|---|---|---|---|---|---|
| Human | ADNSHL | A112T | PDZ1 | [19] | |
| T1188A | TEL | ||||
| G694E | GuK | [20] | |||
| Genomic duplication at chromosome 9p13.3-q21.13 | [4] | ||||
| Familial hypercholanemia | V48A | PDZ1 | Patients with itching, elevated bile acid concentration and fat malabsorption. TJP2 mutation combined with bile acid Coenzyme A (BAAT) mutation | [21] | |
| Intrahepatic cholestasis | I875T | GuK | 71-year-old patient | [22] | |
| R322W | PDZ2 | Disease since early infancy | |||
| A256T | ZO-2 deficiency | [23] | |||
| S296A | |||||
| Y261S | |||||
| A454G | |||||
| A664S | |||||
| Q318G | |||||
| A632fs | |||||
| S1136A | |||||
| T880S | [24] | ||||
| N814Q | Disease since early infancy that leads to hepatocellular carcinoma | [25] | |||
| A273fs | [26] | ||||
| Exon 18 deletion | [27] | ||||
| T62M | PDZ1 | Intrahepatic cholestasis of pregnancy (ICP) | [28] | ||
| T626S | SH3 | ||||
| Mouse | ZO-2 KO | Reduced male fertility | [16] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Mariscal, L.; Gallego-Gutiérrez, H.; González-González, L.; Hernández-Guzmán, C. ZO-2 Is a Master Regulator of Gene Expression, Cell Proliferation, Cytoarchitecture, and Cell Size. Int. J. Mol. Sci. 2019, 20, 4128. https://doi.org/10.3390/ijms20174128
González-Mariscal L, Gallego-Gutiérrez H, González-González L, Hernández-Guzmán C. ZO-2 Is a Master Regulator of Gene Expression, Cell Proliferation, Cytoarchitecture, and Cell Size. International Journal of Molecular Sciences. 2019; 20(17):4128. https://doi.org/10.3390/ijms20174128
Chicago/Turabian StyleGonzález-Mariscal, Lorenza, Helios Gallego-Gutiérrez, Laura González-González, and Christian Hernández-Guzmán. 2019. "ZO-2 Is a Master Regulator of Gene Expression, Cell Proliferation, Cytoarchitecture, and Cell Size" International Journal of Molecular Sciences 20, no. 17: 4128. https://doi.org/10.3390/ijms20174128
APA StyleGonzález-Mariscal, L., Gallego-Gutiérrez, H., González-González, L., & Hernández-Guzmán, C. (2019). ZO-2 Is a Master Regulator of Gene Expression, Cell Proliferation, Cytoarchitecture, and Cell Size. International Journal of Molecular Sciences, 20(17), 4128. https://doi.org/10.3390/ijms20174128