Integrated Analysis of miRNA and mRNA Endorses a Twenty miRNAs Signature for Colorectal Carcinoma

 ,
,  ,
,  , ,
, ,  ,
,  ,
,  ,
, 
Abstract
1. Introduction
2. Results
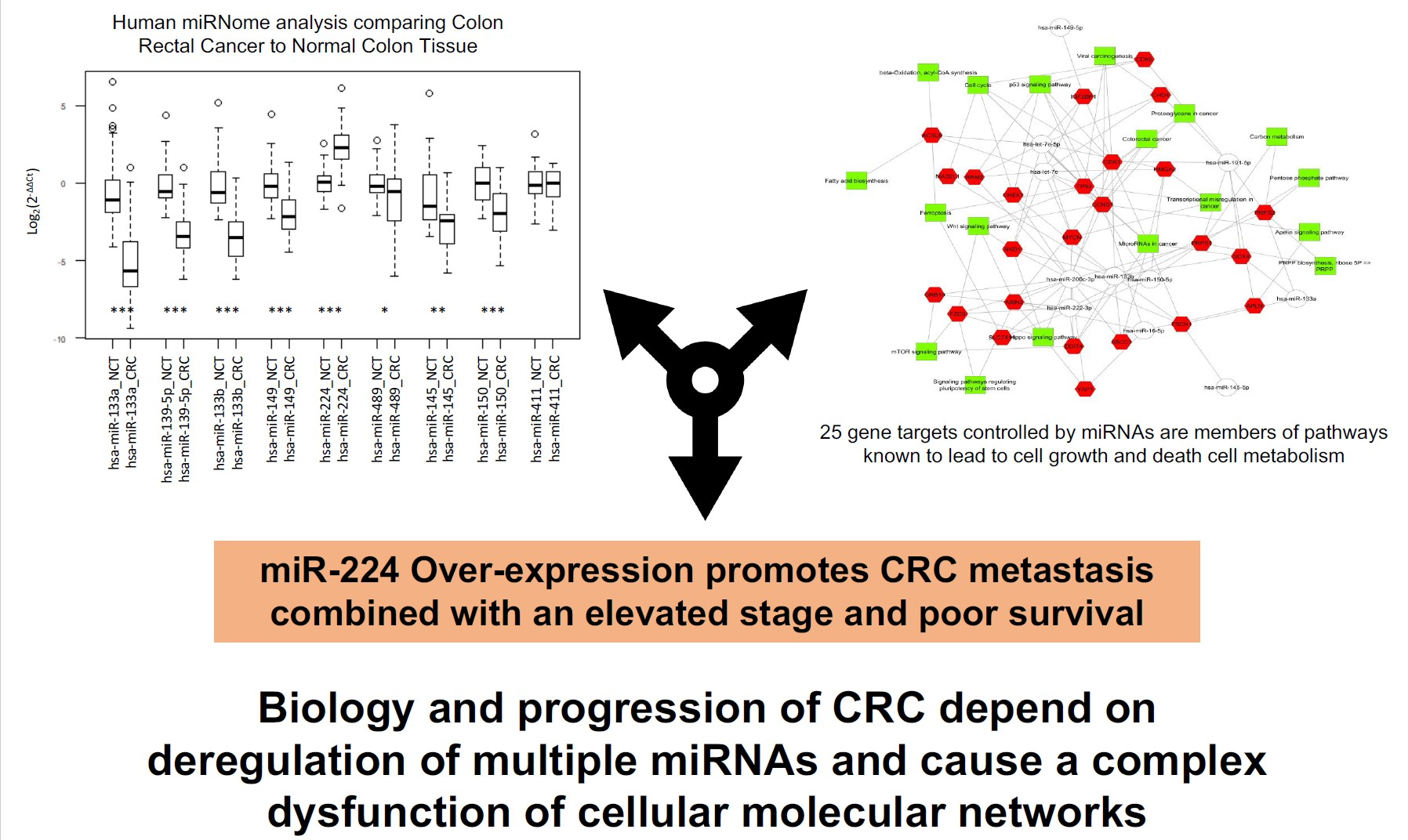
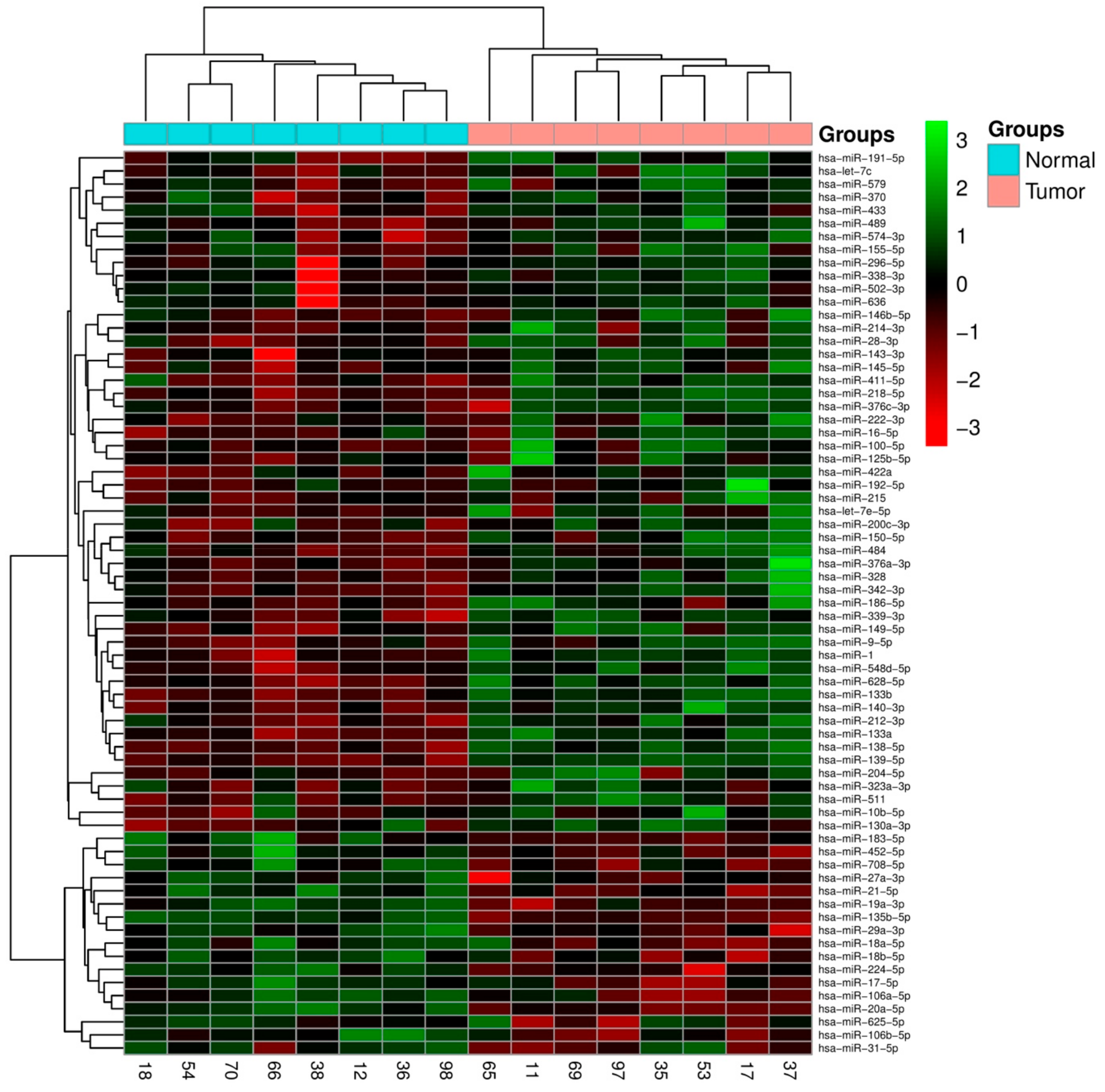
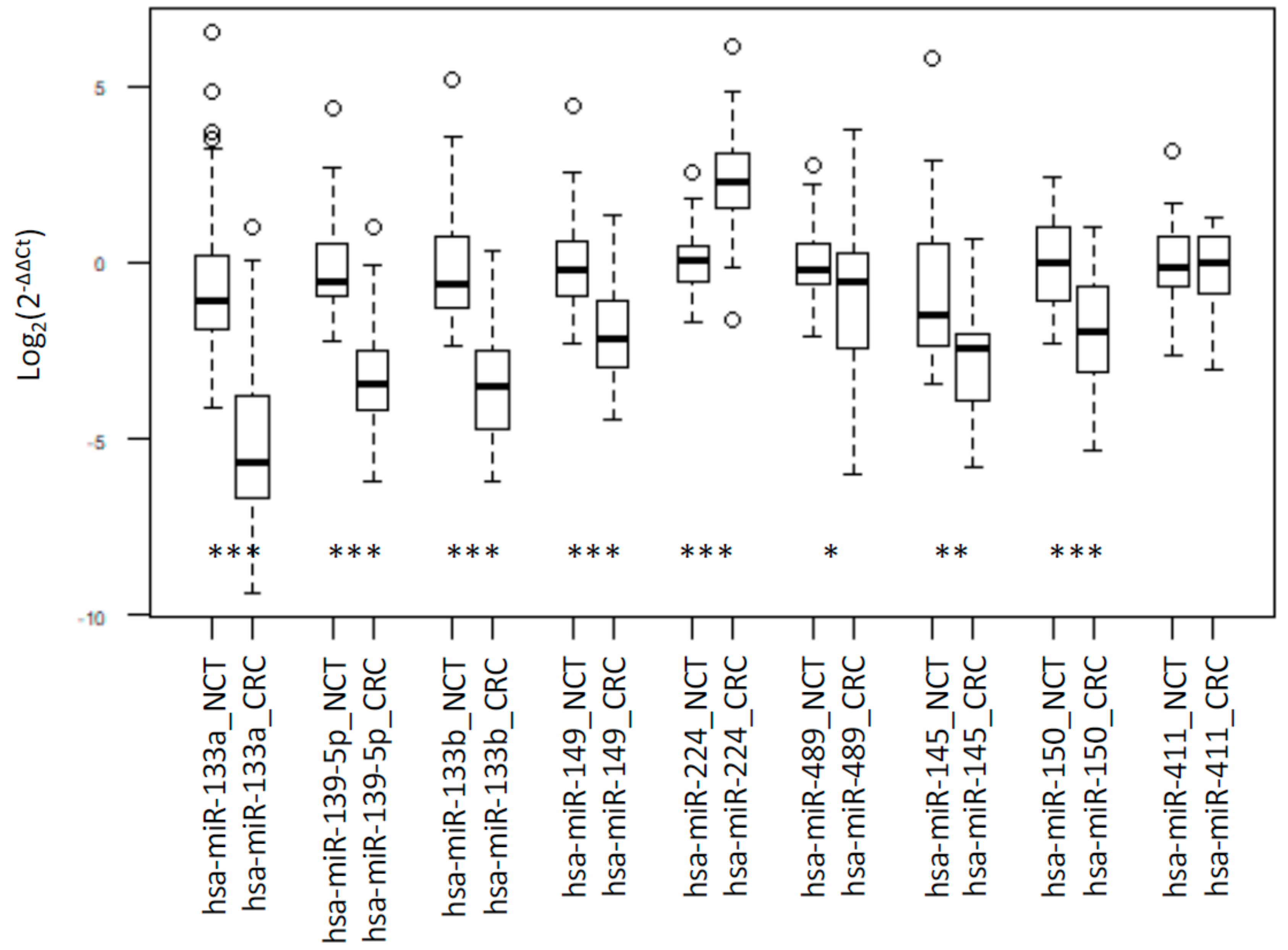
2.1. Integrated Signature of miRNAs in CRC
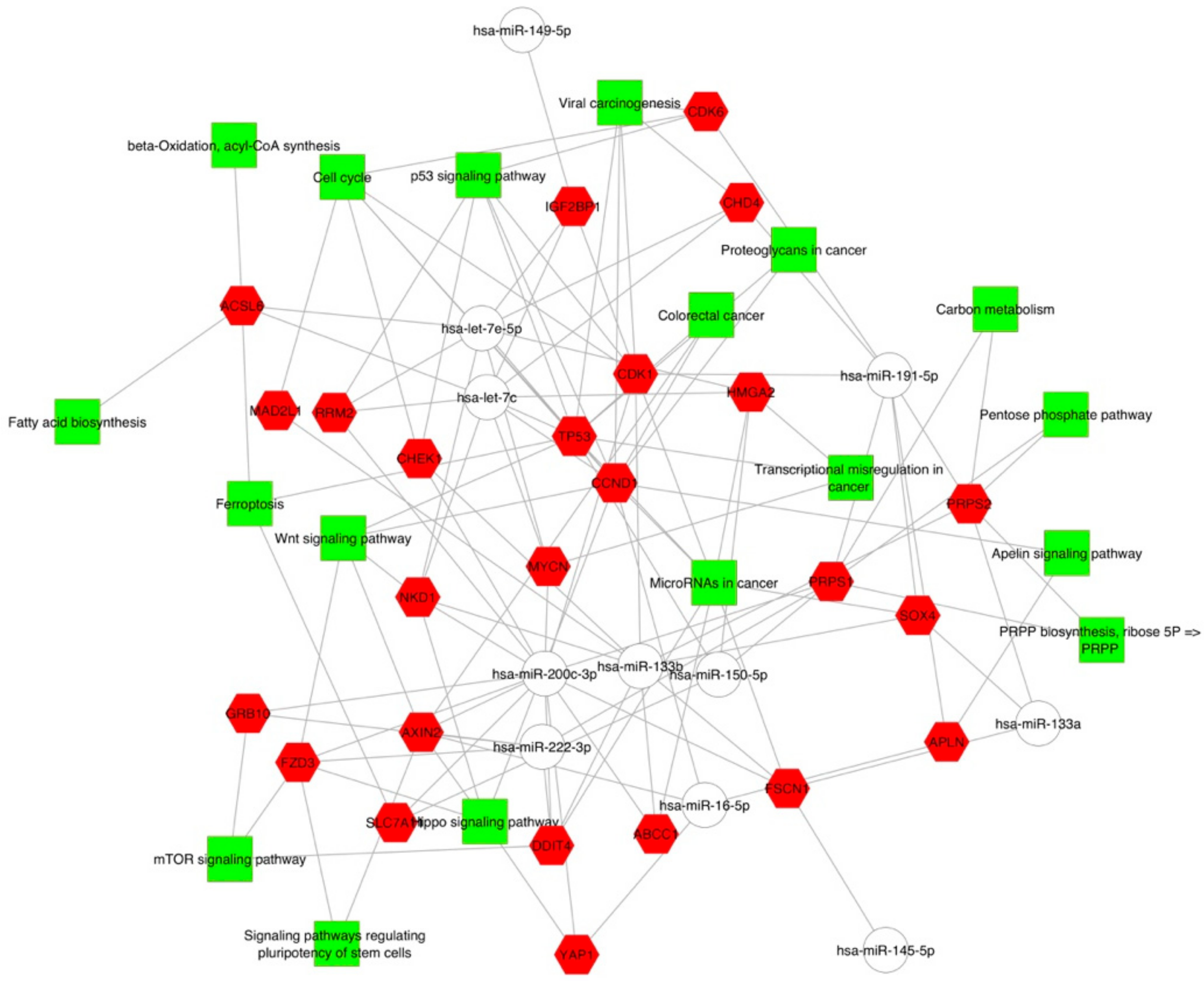
2.2. Gene Targets of miRNAs and Functional Analysis
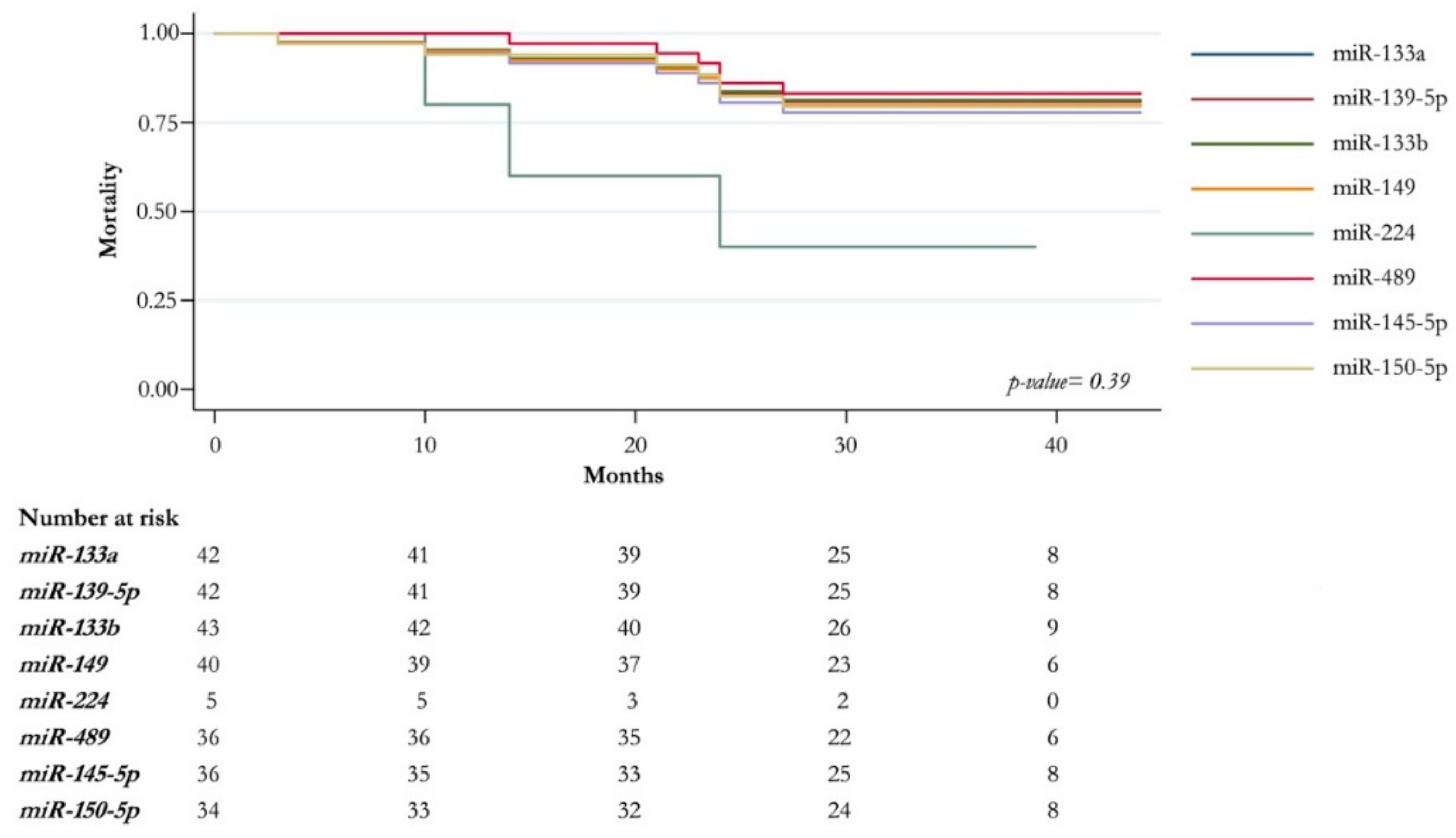
2.3. Association Analysis of Clinic-Pathological Features and miRNAs Expression Level
3. Discussion
4. Materials and Methods
4.1. Patients and Samples
4.2. Human miRNA Card Array and Quantitative Real-Time PCR
4.3. Experimental Identification of miRNA Gene Targets, Gene Ontology and Pathways Mapping
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CRC | Colorectal cancer |
| NCT | Normal colon tissue |
| MiRNAs | MicroRNAs |
| TLDA | TaqMan® Array Human MicroRNA technology |
| SAM | Statistical Analysis of Microarray |
| DE | Differentially expressed |
| GO | Gene Ontology |
| IQR | Interquartile range |
| FC | Fold Change |
| OR | Odds Ratio |
| CI | Confidence Interval |
| PCP | Planar cell polarity |
| PPP | Pentose phosphate pathway |
| FA | Fatty acids |
| CIN FDR | Chromosomal Instability False Discovery Rate |
References
- Siegel, R.L.; Miller, K.D.; Fedewa, S.A.; Ahnen, D.J.; Meester, R.G.S.; Barzi, A.; Jemal, A. Colorectal cancer statistics, 2017. CA. Cancer J. Clin. 2017, 67, 177–193. [Google Scholar] [CrossRef]
- Brenner, H.; Kloor, M.; Pox, C.P. Colorectal Cancer. Lancet. 2014, 383, 1490–1502. [Google Scholar] [CrossRef]
- Riihimaki, M.; Hemminki, A.; Sundquist, J.; Hemminki, K. Patterns of metastasis in colon and rectal cancer. Sci. Rep. 2016, 6, 29765. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Lu, Z. The fate of miRNA* strand through evolutionary analysis: Implication for degradation as merely carrier strand or potential regulatory molecule? PLoS ONE 2010, 5, e11387. [Google Scholar] [CrossRef]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Croce, C.M. The role of microRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Chen, Z.; Guo, Y.; Feng, Y.; Li, Z.; Han, W.; Wang, J.; Zhao, W.; Jiao, Y.; Li, K.; et al. Tumor suppressor MicroRNA-27a in colorectal carcinogenesis and progression by targeting SGPP1 and Smad2. PLoS ONE 2014, 9, e105991. [Google Scholar] [CrossRef]
- Liu, L.; Chen, L.; Xu, Y.; Li, R.; Du, X. MicroRNA-195 promotes apoptosis and suppresses tumorigenicity of human colorectal cancer cells. Biochem. Biophys. Res. Commun. 2010, 400, 236–240. [Google Scholar] [CrossRef]
- Hollis, M.; Nair, K.; Vyas, A.; Chaturvedi, L.S.; Gambhir, S.; Vyas, D. MicroRNAs potential utility in colon cancer: Early detection, prognosis, and chemosensitivity. World J. Gastroenterol. 2015, 21, 8284–8292. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ma, D.; Fesler, A.; Zhai, H.; Leamniramit, A.; Li, W.; Wu, S.; Ju, J. Expression analysis of microRNA as prognostic biomarkers in colorectal cancer. Oncotarget 2017, 8, 52403–52412. [Google Scholar] [CrossRef]
- Pira, G.; Uva, P.; Scanu, A.; Cossu Rocca, P.; Uleri, E.; Piu, C.; Porcu, A.; Carru, C.; Manca, A.; Persico, I.; et al. Landscape of transcriptome variations uncovering known and novel driver events in colorectal carcinoma. Sci. Rep. 2019. under review. [Google Scholar]
- Arends, M.J. Pathways of colorectal carcinogenesis. Appl. Immunohistochem. Mol. Morphol. 2013, 21, 97–102. [Google Scholar] [PubMed]
- Makondi, P.T.; Wei, P.L.; Huang, C.Y.; Chang, Y.J. Development of novel predictive miRNA/ target gene pathways for colorectal cancer distance metastasis to the liver using a bioinformatic approach. PLoS ONE 2019, 14, e0211968. [Google Scholar] [CrossRef] [PubMed]
- Slattery, M.L.; Herrick, J.S.; Mullany, L.E.; Samowitz, W.S.; Sevens, J.R.; Sakoda, L.; Wolff, R.K. The co-regulatory networks of tumor suppressor genes, oncogenes, and miRNAs in colorectal cancer. Genes Chromosom. Cancer 2017, 56, 769–787. [Google Scholar] [CrossRef]
- Baldin, V.; Lukas, J.; Marcote, M.J.; Pagano, M.; Draetta, G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 1993, 7, 812–821. [Google Scholar] [CrossRef]
- Al-Kuraya, K.; Novotny, H.; Bavi, P.; Siraj, A.K.; Uddin, S.; Ezzat, A.; Al Sanea, N.; Al-Dayel, F.; Al-Mana, H.; Sheikh, S.S.; et al. HER2, TOP2A, CCND1, EGFR and C-MYC oncogene amplification in colorectal cancer. J. Clin. Pathol. 2007, 60, 768–772. [Google Scholar] [CrossRef]
- Balcerczak, E.; Pasz-Walczak, G.; Kumor, P.; Panczyk, M.; Kordek, R.; Wierzbicki, R.; Mirowski, M. Cyclin D1 protein and CCND1 gene expression in colorectal cancer. Eur. J. Surg. Oncol. 2005, 31, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Tong, F.; Ying, Y.; Pan, H.; Zhao, W.; Li, H.; Zhan, X. MicroRNA-466 (miR-466) functions as a tumor suppressor and prognostic factor in colorectal cancer (CRC). Bosn. J. Basic Med. Sci. 2018, 18, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Wiesmann, M.; Rohan, M.; Chan, V.; Jefferson, A.B.; Guo, L.; Sakamoto, D.; Caothien, R.H.; Fuller, J.H.; Reinhard, C.; et al. Elevated expression of axin2 and hnkd mRNA provides evidence that Wnt/ beta -catenin signaling is activated in human colon tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 14973–14978. [Google Scholar] [CrossRef] [PubMed]
- Reeves, R. Molecular biology of HMGA proteins: Hubs of nuclear function. Gene 2001, 277, 63–81. [Google Scholar] [CrossRef]
- Madison, B.B.; Jeganathan, A.N.; Mizuno, R.; Winslow, M.M.; Castells, A.; Cuatrecasas, M.; Rustgi, A.K. Let-7 Represses Carcinogenesis and a Stem Cell Phenotype in the Intestine via Regulation of Hmga2. PLoS Genet. 2015, 11, e1005408. [Google Scholar] [CrossRef]
- Wang, X.; Liu, X.; Li, A.Y.J.; Chen, L.; Lai, L.; Lin, H.H.; Hu, S.; Yao, L.; Peng, J.; Loera, S.; et al. Overexpression of HMGA2 promotes metastasis and impacts survival of colorectal cancers. Clin. Cancer Res. 2011, 17, 2570–2580. [Google Scholar] [CrossRef]
- Lederer, M.; Bley, N.; Schleifer, C.; Hüttelmaier, S. The role of the oncofetal IGF2 mRNA-binding protein 3 (IGF2BP3) in cancer. Semin. Cancer Biol. 2014, 29, 3–12. [Google Scholar] [CrossRef]
- Busch, B.; Bley, N.; Müller, S.; Glaß, M.; Misiak, D.; Lederer, M.; Vetter, M.; Strauß, H.G.; Thomssen, C.; Hüttelmaier, S. The oncogenic triangle of HMGA2, LIN28B and IGF2BP1 antagonizes tumor-suppressive actions of the let-7 family. Nucleic Acids Res. 2016, 44, 3845–3864. [Google Scholar] [CrossRef]
- Camargo, F.D.; Gokhale, S.; Johnnidis, J.B.; Fu, D.; Bell, G.W.; Jaenisch, R.; Brummelkamp, T.R. YAP1 Increases Organ Size and Expands Undifferentiated Progenitor Cells. Curr. Biol. 2007, 17, 2054–2060. [Google Scholar] [CrossRef]
- Schlegelmilch, K.; Mohseni, M.; Kirak, O.; Pruszak, J.; Rodriguez, J.R.; Zhou, D.; Kreger, B.T.; Vasioukhin, V.; Avruch, J.; Brummelkamp, T.R.; et al. Yap1 acts downstream of α-catenin to control epidermal proliferation. Cell 2011, 144, 782–795. [Google Scholar] [CrossRef] [PubMed]
- Heallen, T.; Zhang, M.; Wang, J.; Bonilla-Claudio, M.; Klysik, E.; Johnson, R.L.; Martin, J.F. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 2011, 332, 458–461. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Wang, L.; Wang, C.Y.; Yu, J.; Guan, K.L. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 2012, 26, 54–68. [Google Scholar] [CrossRef] [PubMed]
- Tapon, N.; Harvey, K.F.; Bell, D.W.; Wahrer, D.C.R.; Schiripo, T.A.; Haber, D.A.; Hariharan, I.K. Salvador promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell 2002, 110, 467–478. [Google Scholar] [CrossRef]
- Zhang, X.; George, J.; Deb, S.; Degoutin, J.L.; Takano, E.A.; Fox, S.B.; Bowtell, D.D.L.; Harvey, K.F. The Hippo pathway transcriptional co-activator, YAP, is an ovarian cancer oncogene. Oncogene 2011, 30, 2810–2822. [Google Scholar] [CrossRef]
- Varelas, X.; Miller, B.W.; Sopko, R.; Song, S.; Gregorieff, A.; Fellouse, F.A.; Sakuma, R.; Pawson, T.; Hunziker, W.; McNeill, H.; et al. The Hippo Pathway Regulates Wnt/β-Catenin Signaling. Dev. Cell 2010, 18, 479–591. [Google Scholar] [CrossRef] [PubMed]
- Sorli, S.C.; Le Gonidec, S.; Knibiehler, B.; Audigier, Y. Apelin is a potent activator of tumour neoangiogenesis. Oncogene 2007, 26, 7692–7699. [Google Scholar] [CrossRef]
- Picault, F.X.; Chaves-Almagro, C.; Projetti, F.; Prats, H.; Masri, B.; Audigier, Y. Tumour co-expression of apelin and its receptor is the basis of an autocrine loop involved in the growth of colon adenocarcinomas. Eur. J. Cancer 2014, 50, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Cahill, D.P.; Lengauer, C.; Yu, J.; Riggins, G.J.; Willson, J.K.; Markowitz, S.D.; Kinzler, K.W.; Vogelstein, B. Mutations of mitotic checkpoint genes in human cancers. Nature 1998, 392, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Zhu, J.; Ou, C.; Deng, Z.; Chen, M.; Huang, W.; Li, L. MicroRNA-145 inhibits tumour growth and metastasis in colorectal cancer by targeting fascin-1. Br. J. Cancer 2014, 110, 2300–2309. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhu, S.; Song, X.; Sun, X.; Fan, Y.; Liu, J.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T.R.; et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017, 20, 1692–1704. [Google Scholar] [CrossRef] [PubMed]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular Fatty Acid Metabolism and Cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Hu, S.; Wu, J.; Chen, L.; Lu, J.; Wang, X.; Liu, X.; Zhou, B.; Yen, Y. Overexpression of RRM2 decreases thrombspondin-1 and increases VEGF production in human cancer cells in vitro and in vivo: Implication of RRM2 in angiogenesis. Mol. Cancer 2009, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef]
- Chen, W.C.; Wang, C.Y.; Hung, Y.H.; Weng, T.Y.; Yen, M.C.; Lai, M.D. Systematic analysis of gene expression alterations and clinical outcomes for long-chain acyl-coenzyme A synthetase family in cancer. PLoS ONE 2016, 11, e0155660. [Google Scholar]
- Van Horn, C.G.; Caviglia, J.M.; Li, L.O.; Wang, S.; Granger, D.A.; Coleman, R.A. Characterization of recombinant long-chain rat Acyl-CoA synthetase isoforms 3 and 6: Identification of a novel variant of isoform 6. Biochemistry 2005, 44, 1635–1642. [Google Scholar] [CrossRef] [PubMed]
- Mashima, T.; Oh-hara, T.; Sato, S.; Mochizuki, M.; Sugimoto, Y.; Yamazaki, K.; Hamada, J.I.; Tada, M.; Moriuchi, T.; Ishikawa, Y.; et al. p53-defective tumors with a functional apoptosome-mediated pathway: A new therapeutic target. J. Natl. Cancer Inst. 2005, 97, 765–777. [Google Scholar] [CrossRef]
- Hamilton, S.R.; Bosman, F.T.; Boffetta, P. Carcinoma of the colon and rectum. In WHO Classification of Tumours of the Digestive System, 4th ed.; Boman, F.T., Carneiro, F., Hruban, R.H., Theise, N.D., Eds.; IARC: Lyon, France, 2010; Volume 3, pp. 134–181. [Google Scholar]
- Uva, P.; Cossu-Rocca, P.; Loi, F.; Pira, G.; Murgia, L.; Orrù, S.; Floris, M.; Muroni, M.R.; Sanges, F.; Carru, C.; et al. miRNA-135b contributes to triple negative breast cancer molecular heterogeneity: Different expression profile in Basal-like versus non-Basal-like phenotypes. Int. J. Med. Sci. 2018, 15, 536–548. [Google Scholar] [CrossRef]
- Paraskevopoulou, M.D.; Georgakilas, G.; Kostoulas, N.; Vlachos, I.S.; Vergoulis, T.; Reczko, M.; Filippidis, C.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-microT web server v5.0: Service integration into miRNA functional analysis workflows. Nucleic Acids Res. 2013, 41, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The microRNA.org resource: Targets and expression. Nucleic Acids Res. 2008, 36, 149–153. [Google Scholar] [CrossRef]
- Wong, N.; Wang, X. miRDB: An online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015, 43, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Krek, A.; Grün, D.; Poy, M.N.; Wolf, R.; Rosenberg, L.; Epstein, E.J.; MacMenamin, P.; Da Piedade, I.; Gunsalus, K.C.; Stoffel, M.; et al. Combinatorial microRNA target predictions. Nat. Genet. 2005, 37, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Kertesz, M.; Iovino, N.; Unnerstall, U.; Gaul, U.; Segal, E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007, 39, 1278–1284. [Google Scholar] [CrossRef]
- Li, J.H.; Liu, S.; Zhou, H.; Qu, L.H.; Yang, J.H. StarBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, 92–97. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Zuo, Z.; Cai, G.; Kang, S.; Gao, X.; Li, T. miRecords: An integrated resource for microRNA-target interactions. Nucleic Acids Res. 2009, 37, 105–110. [Google Scholar] [CrossRef]
- Hsu, S.D.; Lin, F.M.; Wu, W.Y.; Liang, C.; Huang, W.C.; Chan, W.L.; Tsai, W.T.; Chen, G.Z.; Lee, C.J.; Chiu, C.M.; et al. MiRTarBase: A database curates experimentally validated microRNA-target interactions. Nucleic Acids Res. 2011, 39, 163–169. [Google Scholar] [CrossRef]
- Ihaka, R.; Gentleman, R. R: A Language for Data Analysis and Graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. [Google Scholar]
- Kaimal, V.; Bardes, E.E.; Tabar, S.C.; Jegga, A.G.; Aronow, B.J. ToppCluster: A multiple gene list feature analyzer for comparative enrichment clustering and networkbased dissection of biological systems. Nucleic Acids Res. 2010, 38, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miR-139-5p | miR-133b | miR-149 | miR-224 | miR-489 | miR-145 | miR-150 | |
|---|---|---|---|---|---|---|---|
| miR-133a | 0.87 ** | 0.90 ** | 0.77 ** | −0.52 ** | 0.44 * | 0.74 ** | 0.67 ** |
| miR-139-5p | 0.81 ** | 0.85 ** | −0.44 * | 0.44 * | 0.64 ** | 0.69 ** | |
| miR-133b | 0.77 ** | −0.34 * | 0.37 * | 0.82 ** | 0.71 ** | ||
| miR-149 | −0.22 | 0.38 * | 0.57 ** | 0.49 * | |||
| miR-224 | −0.19 | −0.27 | −0.31 | ||||
| miR-489 | 0.14 | 0.31 | |||||
| miR-145 | 0.64 ** |
| Alive at 28/02/2018, n (%) | 35 (79.6) | |
| Median (IQR) time of survival, months | 31.5 (27.5–38.5) | |
| Localization, n (%) | Right | 24 (54.6) |
| Left | 14 (31.8) | |
| Rectum | 6 (13.6) | |
| Tumor stage, n (%) | I | 11 (25.6) |
| II | 6 (14.0) | |
| III | 20 (46.5) | |
| IV | 6 (14.0) | |
| Histologic grade, n (%) | G1 | 2 (4.6) |
| G2 | 30 (68.2) | |
| G3 | 12 (27.3) | |
| Tumor infiltrating lymphocytes, n (%) | 13 (31.0) | |
| KRAS mutational status, n (%) | 17 (38.6) | |
| MiR-133a, n (%) | Down | 43 (97.7) |
| Up | 1 (2.3) | |
| MiR-139-5p, n (%) | Down | 43 (97.7) |
| Up | 1 (2.3) | |
| MiR-133b, n (%) | Down | 44 (100.0) |
| Up | 0 (0.0) | |
| MiR-149-5p, n (%) | Down | 41 (97.6) |
| Up | 1 (2.4) | |
| MiR-224-5p, n (%) | Down | 6 (14.0) |
| Up | 37 (86.1) | |
| MiR-489, n (%) | Down | 37 (86.1) |
| Up | 6 (14.0) | |
| MiR-145-5p, n (%) | Down | 37 (100.0) |
| Up | 0 (0.0) | |
| MiR-150-5p, n (%) | Down | 35 (94.6) |
| Up | 2 (5.4) | |
| Variables | miR-224-5p | miR-489 | |||
|---|---|---|---|---|---|
| OR (95% CI) | p Value | OR (95% CI) | p Value | ||
| Localization | Right | 1.2 (0.2–6.6) | 0.85 | - | - |
| Left | 0.4 (0.1–2.4) | 0.34 | 0.4 (0.1–2.3) | 0.30 | |
| Rectum | - | - | 5.4 (0.9–34.2) | 0.07 | |
| Tumor stage | I | - | - | 0.5 (0.1–5.0) | 0.57 |
| II | 0.8 (0.1–8.4) | 0.86 | - | - | |
| III | 1.8 (0.3–11.0) | 0.53 | 1.3 (0.2–7.1) | 0.80 | |
| IV | 0.1 (0.0–0.7) | 0.02 | 4 (0.6–29.3) | 0.17 | |
| Histologic grade | G1 | - | - | 7.2 (0.4–134.2) | 0.19 |
| G2 | 1.2 (0.2–7.4) | 0.86 | 0.9 (0.1–5.3) | 0.86 | |
| G3 | 0.6 (0.1–4.1) | 0.64 | 0.5 (0.1–5.2) | 0.59 | |
| Tumor infiltrating lymphocytes | 0.7 (0.1–4.5) | 0.67 | - | - | |
| KRAS mutational status | 3.4 (0.4–32.2) | 0.28 | 4.2 (0.7–26.0) | 0.13 | |
| Variables | miR-224-5p | miR-489 | ||||
|---|---|---|---|---|---|---|
| Down | Up | p Value | Down | Up | p Value | |
| Gender | ||||||
| Male, n (%) | 4 (66.7) | 23 (62.2) | 0.83 | 24 (64.9) | 3 (50.0) | 0.48 |
| Female, n (%) | 2 (33.3) | 14 (37.8) | 0.83 | 13 (35.1) | 3 (50.0) | 0.48 |
| Age at diagnosis | ||||||
| Under 65 years, n (%) | 2 (33.3) | 11 (29.7) | 0.86 | 8 (21.6) | 5 (83.3) | 0.002 |
| Over 66 years, n (%) | 4 (66.7) | 26 (70.3) | 0.86 | 29 (78.4) | 1 (16.7) | 0.002 |
| Localization | ||||||
| Right, n (%) | 3 (50.0) | 20 (54.1) | 1.0 | 21 (56.7) | 2 (33.3) | 0.39 |
| Left, n (%) | 3 (50.0) | 11 (29.7) | 0.37 | 10 (27.0) | 4 (66.7) | 0.08 |
| Rectum, n (%) | 0 (0.0) | 6 (16.2) | 0.57 | 6 (16.2) | 0 (0.0) | 0.57 |
| Histologic grade | ||||||
| G1–G2, n (%) | 4 (66.7) | 28 (75.7) | 0.64 | 27 (73.0) | 5 (83.3) | 0.59 |
| G3, n (%) | 2 (33.3) | 9 (24.3) | 0.64 | 10 (27.0) | 1 (16.7) | 0.59 |
| Depth of invasion | ||||||
| T1–T2, n (%) | 0 (0.0) | 14(37.8) | 0.07 | 12 (32.4) | 2 (33.3) | 0.97 |
| T3–T4, n (%) | 6 (100.0) | 23 (62.2) | 0.07 | 25 (67.6) | 4 (66.7) | 0.97 |
| Nodal status | ||||||
| N0–N1, n (%) | 2 (33.3) | 30 (85.7) | 0.004 | 29 (80.6) | 3 (60.0) | 0.30 |
| N2–N3, n (%) | 4 (66.7) | 5 (14.3) | 0.004 | 7 (19.4) | 2 (40.0) | 0.30 |
| Distant metastasis | ||||||
| Present, n (%) | 3 (50.0) | 34 (91.9) | 0.006 | 33 (89.2) | 4 (66.7) | 0.14 |
| Absent, n (%) | 3 (50.0) | 3 (8.1) | 0.006 | 4 (10.8) | 2 (33.3) | 0.14 |
| Tumor stage | ||||||
| I–II, n (%) | 1 (16.7) | 16 (44.4) | 0.20 | 16 (44.4) | 1 (16.7) | 0.20 |
| III–IV, n (%) | 5 (83.3) | 20 (55.6) | 0.20 | 20 (55.6) | 5 (83.3) | 0.20 |
| Tumor infiltrating lymphocytes | ||||||
| Present, n (%) | 2 (40.0) | 11 (30.6) | 0.67 | 13 (36.1) | 0 (0.0) | 0.10 |
| Absent, n (%) | 3 (60.0) | 25 (69.4) | 0.67 | 23 (63.9) | 5 (100.0) | 0.10 |
| Neoplastic embolization | ||||||
| Present, n (%) | 3 (60.0) | 9 (25.0) | 0.11 | 10 (27.8) | 2 (40.0) | 0.57 |
| Absent, n (%) | 2 (40.0) | 27 (75.0) | 0.11 | 26 (72.2) | 3 (60.0) | 0.57 |
| Perineural invasion | ||||||
| Present, n (%) | 1 (20.0) | 2 (5.6) | 0.25 | 3 (8.3) | 0 (0.0) | 0.50 |
| Absent, n (%) | 4 (80.0) | 34 (94.4) | 0.25 | 33 (91.7) | 5 (100.0) | 0.50 |
| KRAS mutational status | ||||||
| Wild type, n (%) | 5 (83.3) | 22 (59.5) | 0.26 | 25 (67.6) | 2 (33.3) | 0.11 |
| Mutation, n (%) | 1 (16.7) | 15 (40.5) | 0.26 | 12 (32.4) | 4 (66.7) | 0.11 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angius, A.; Uva, P.; Pira, G.; Muroni, M.R.; Sotgiu, G.; Saderi, L.; Uleri, E.; Caocci, M.; Ibba, G.; Cesaraccio, M.R.; et al. Integrated Analysis of miRNA and mRNA Endorses a Twenty miRNAs Signature for Colorectal Carcinoma. Int. J. Mol. Sci. 2019, 20, 4067. https://doi.org/10.3390/ijms20164067
Angius A, Uva P, Pira G, Muroni MR, Sotgiu G, Saderi L, Uleri E, Caocci M, Ibba G, Cesaraccio MR, et al. Integrated Analysis of miRNA and mRNA Endorses a Twenty miRNAs Signature for Colorectal Carcinoma. International Journal of Molecular Sciences. 2019; 20(16):4067. https://doi.org/10.3390/ijms20164067
Chicago/Turabian StyleAngius, Andrea, Paolo Uva, Giovanna Pira, Maria Rosaria Muroni, Giovanni Sotgiu, Laura Saderi, Elena Uleri, Maurizio Caocci, Gabriele Ibba, Maria Rosaria Cesaraccio, and et al. 2019. "Integrated Analysis of miRNA and mRNA Endorses a Twenty miRNAs Signature for Colorectal Carcinoma" International Journal of Molecular Sciences 20, no. 16: 4067. https://doi.org/10.3390/ijms20164067
APA StyleAngius, A., Uva, P., Pira, G., Muroni, M. R., Sotgiu, G., Saderi, L., Uleri, E., Caocci, M., Ibba, G., Cesaraccio, M. R., Serra, C., Carru, C., Manca, A., Sanges, F., Porcu, A., Dolei, A., Scanu, A. M., Cossu Rocca, P., & De Miglio, M. R. (2019). Integrated Analysis of miRNA and mRNA Endorses a Twenty miRNAs Signature for Colorectal Carcinoma. International Journal of Molecular Sciences, 20(16), 4067. https://doi.org/10.3390/ijms20164067