Comparative Analysis of miRNA Abundance Revealed the Function of Vvi-miR828 in Fruit Coloring in Root Restriction Cultivation Grapevine (Vitis vinifera L.)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Result
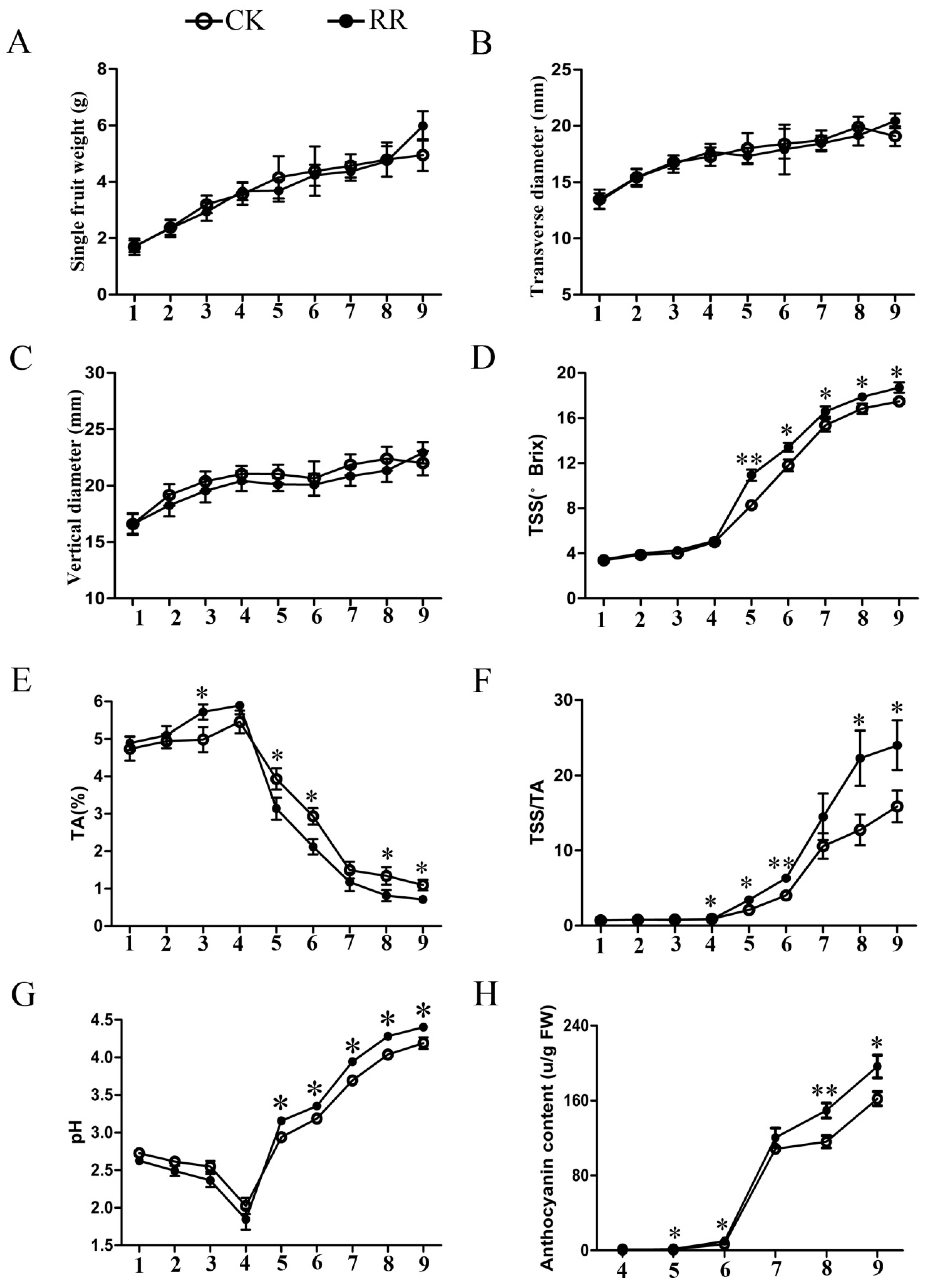
2.1. Measurement of Soluble Solids, Titratable Acid, pH, and Anthocyanin Content in Grape Berries
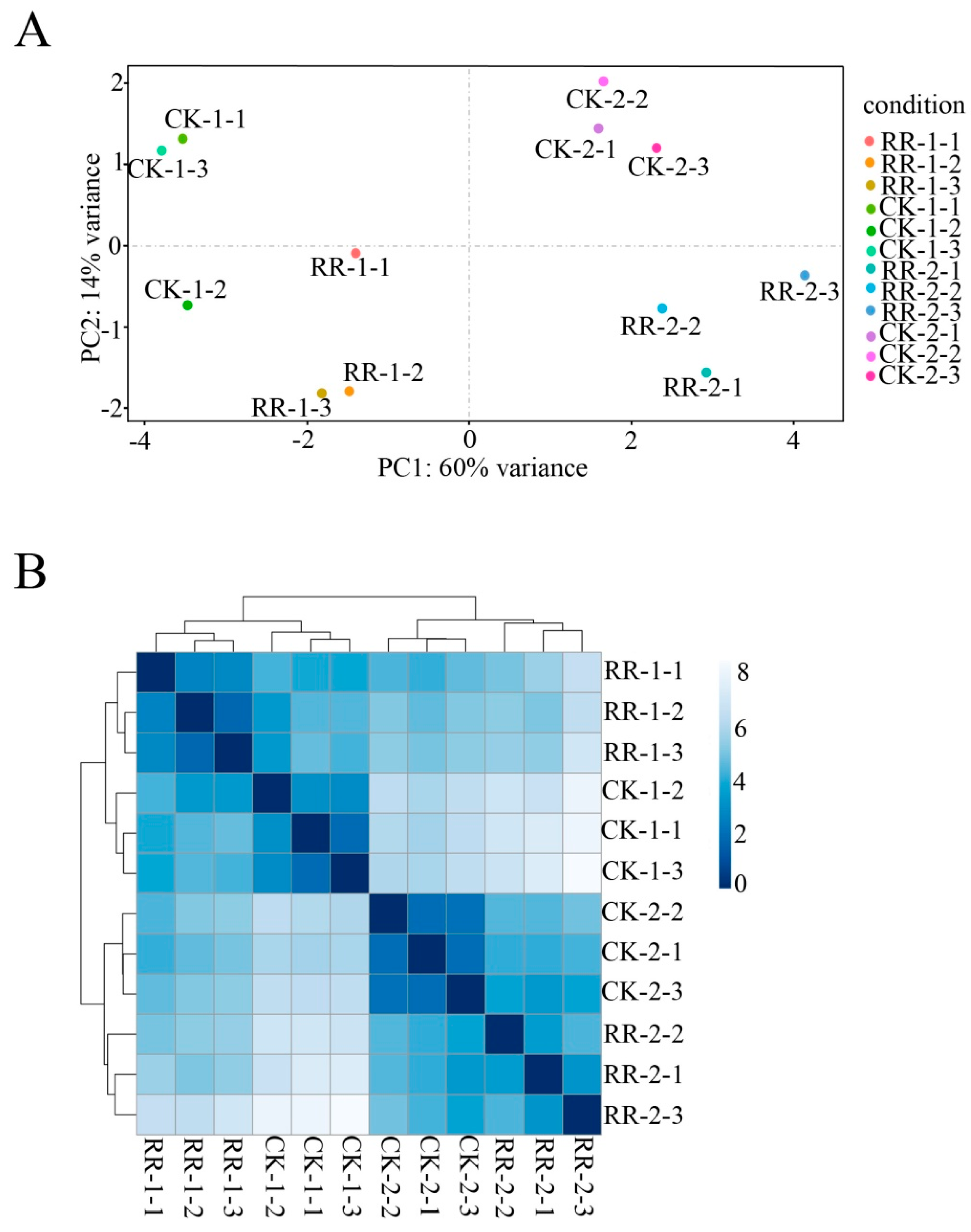
2.2. Sequencing and Annotation of Grape Fruit Small RNAs
2.3. Known and Novel miRNAs
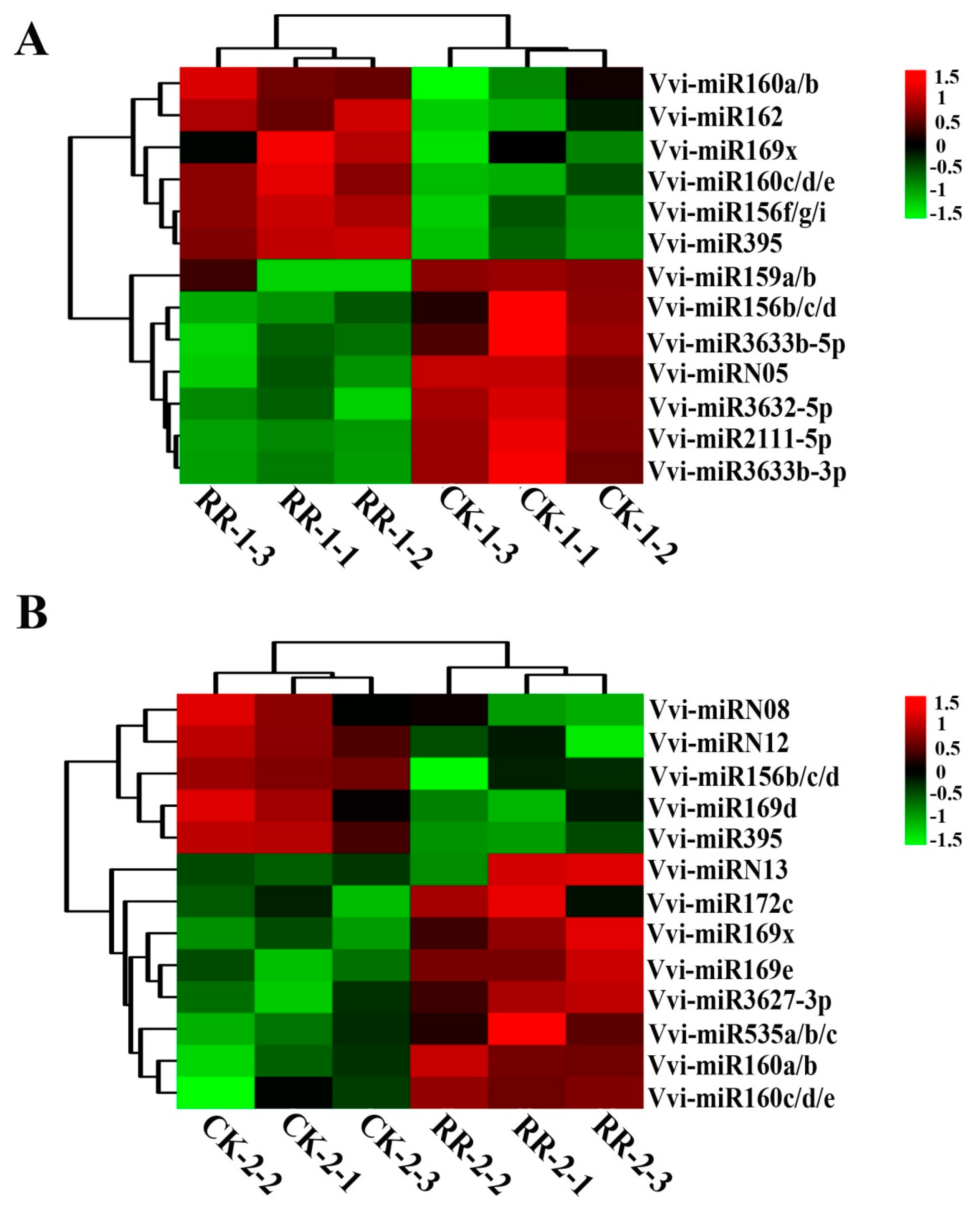
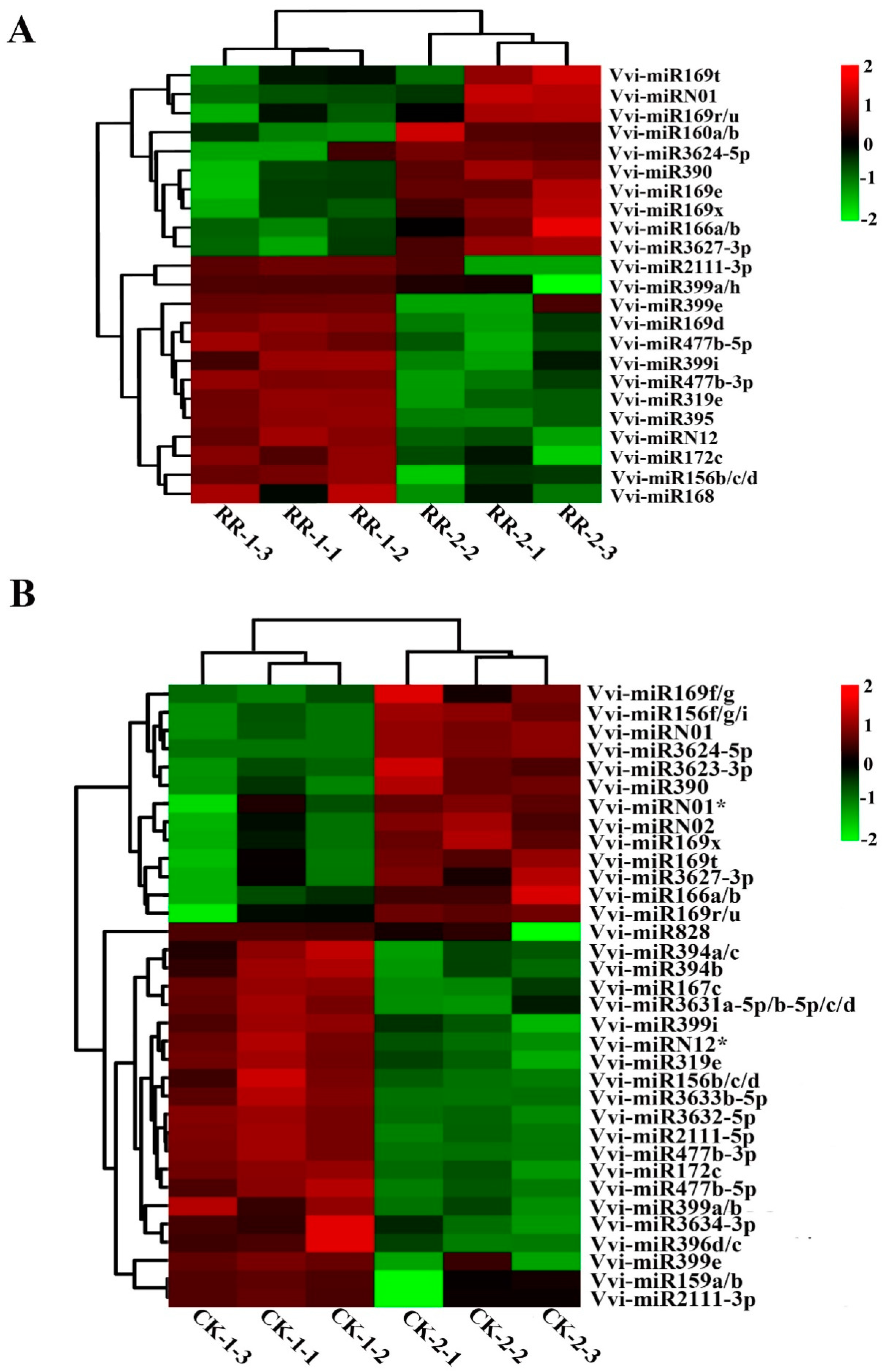
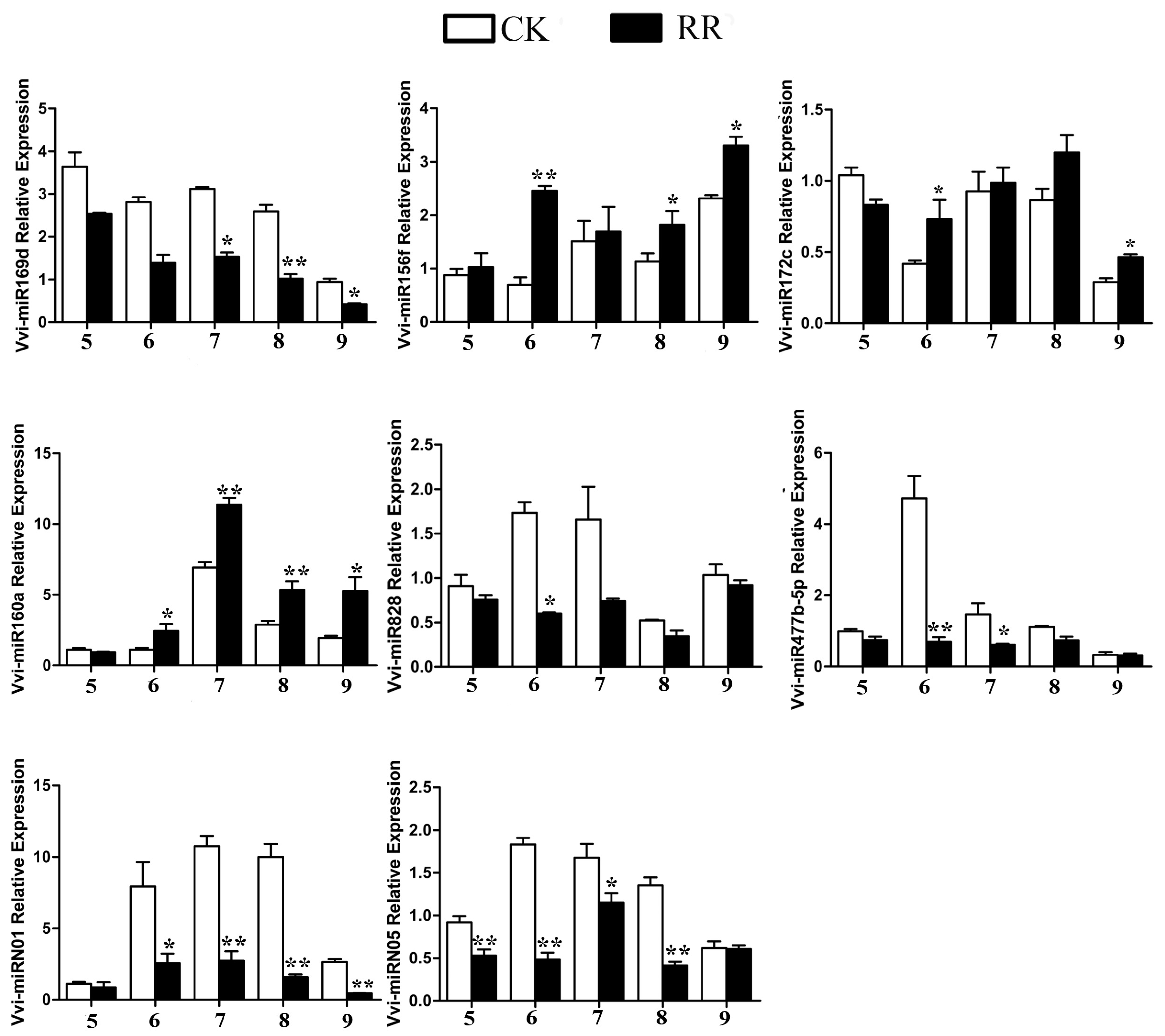
2.4. Differentially Expressed miRNAs Between RR and CK
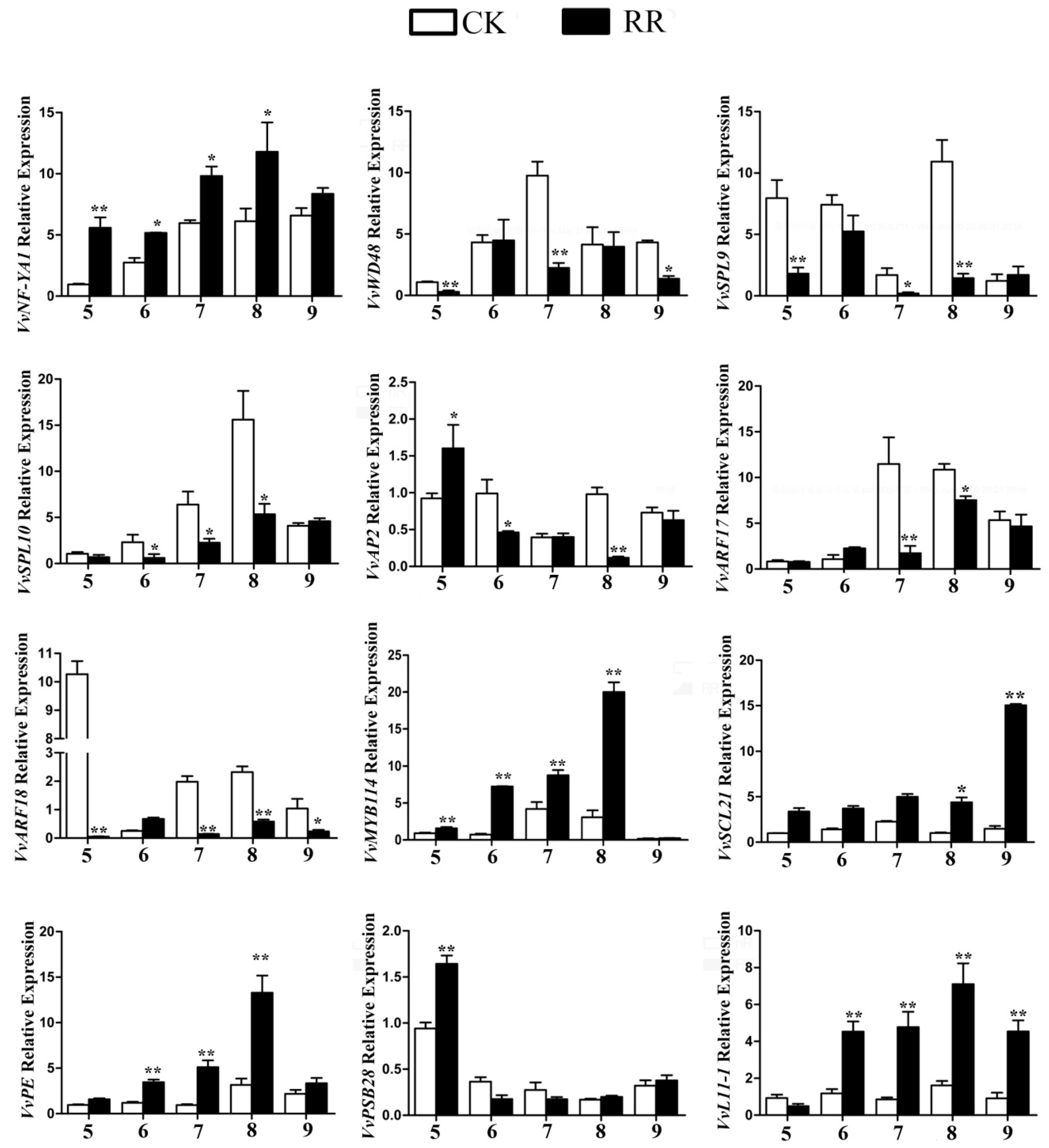
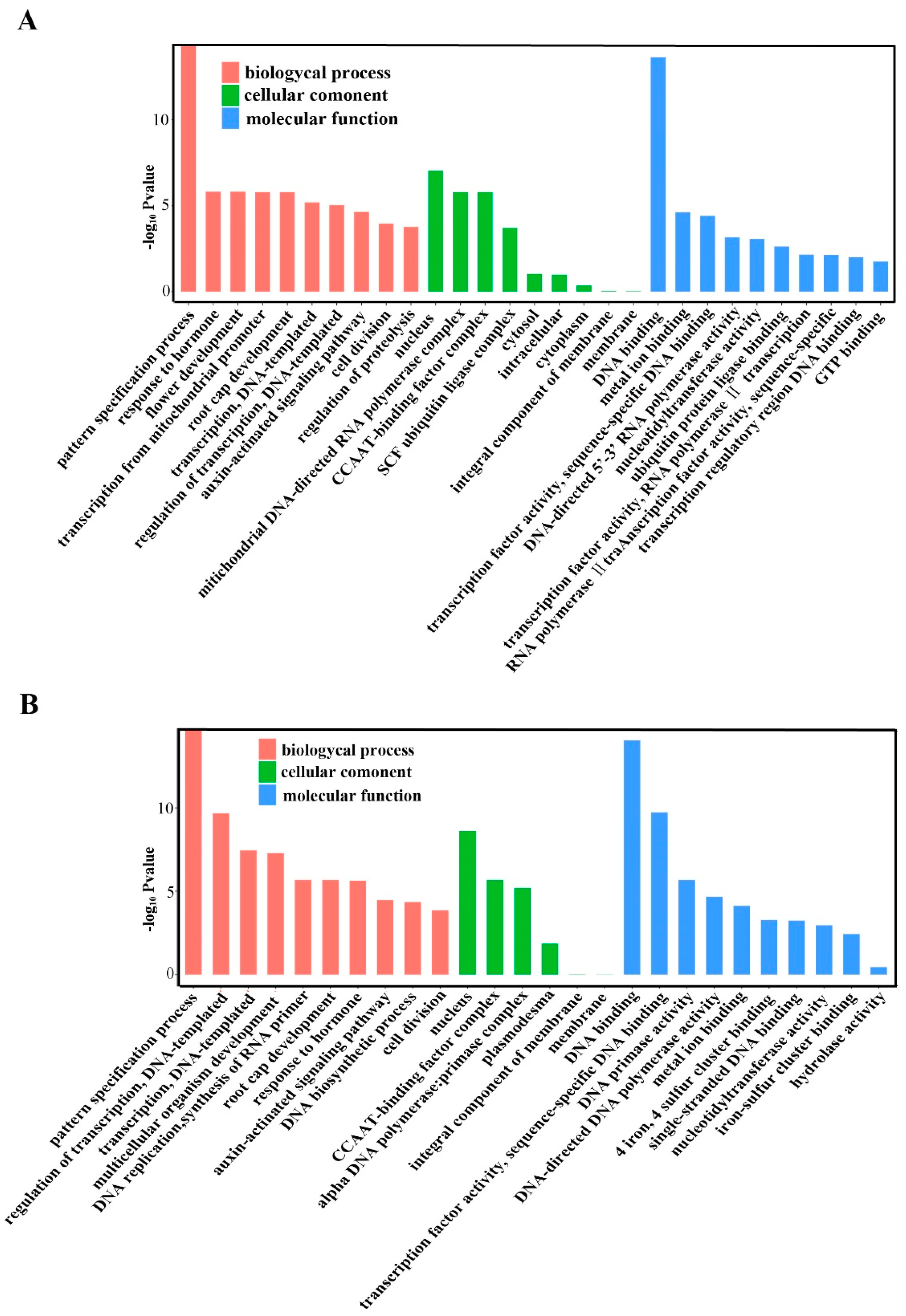
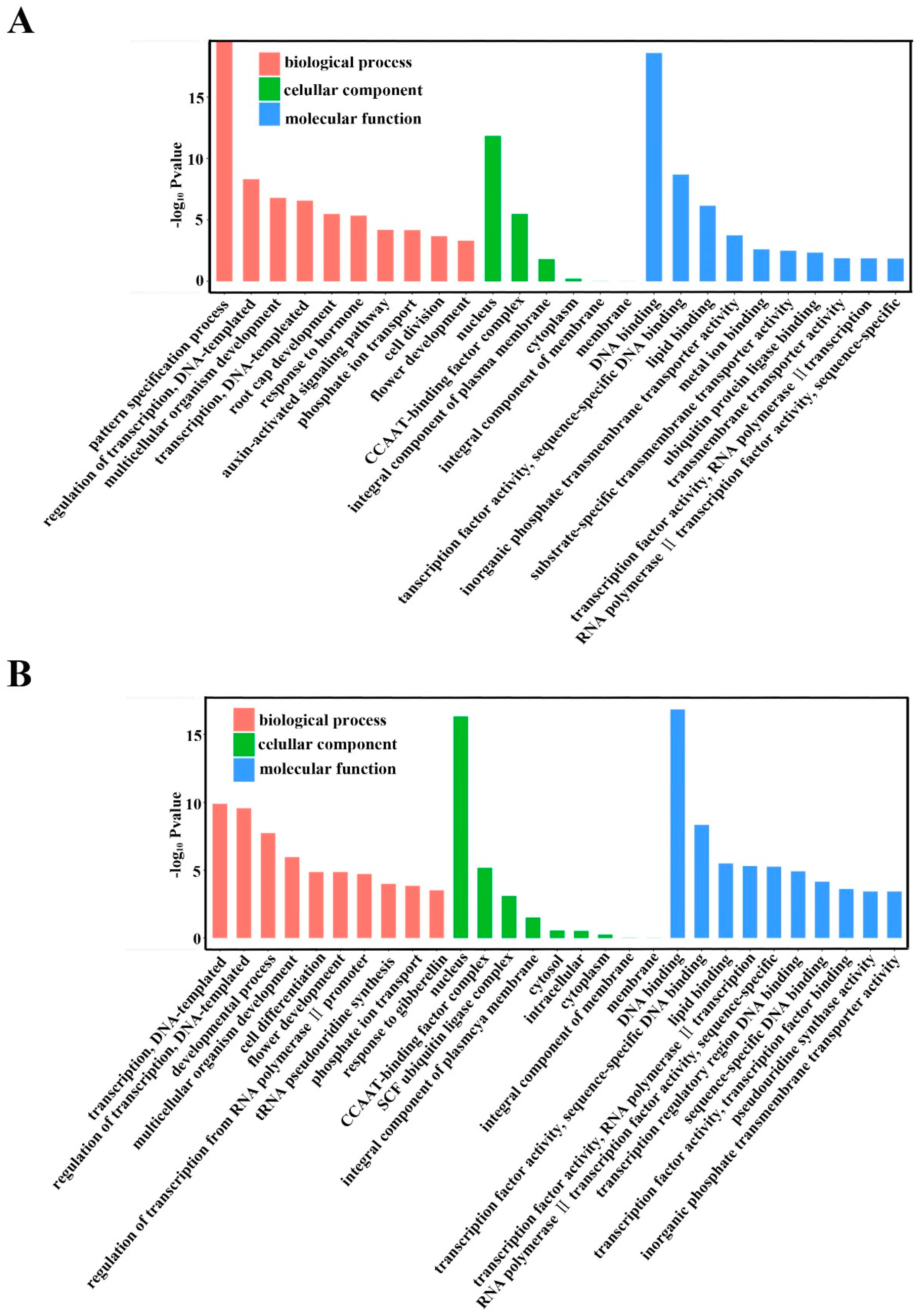
2.5. Annotation of Potential Targets of Differentially Expressed miRNAs
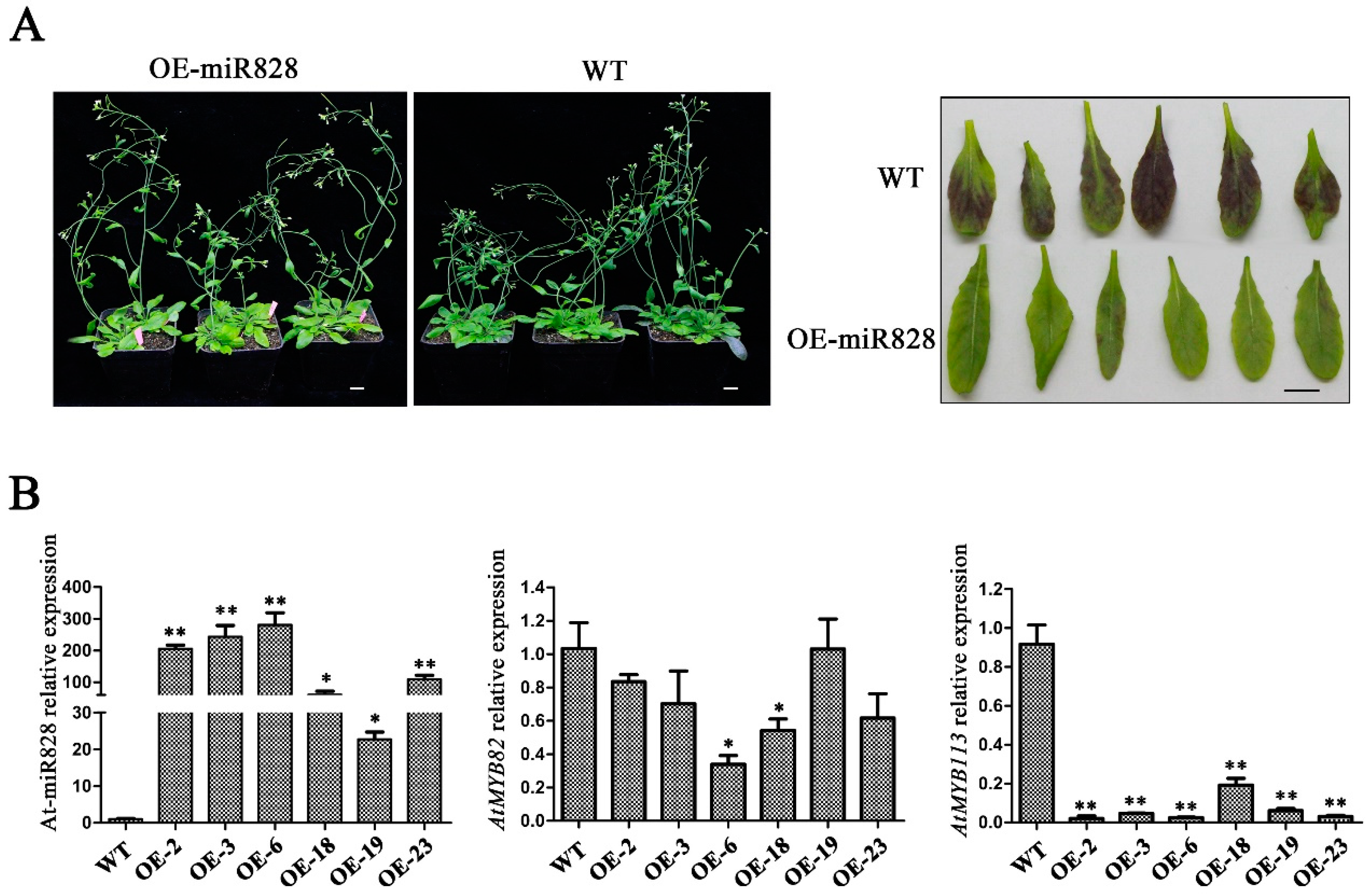
2.6. Phenotypic Characterization of Vvi-miR828-OE Lines
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Measurement of the Fruit Size
4.3. Determination of Fruit Quality Index
4.4. RNA Extraction and cDNA Synthesis
4.5. Small RNA Library Construction and Sequencing
4.6. Bioinformatic Analysis of miRNAs
4.7. Analysis of Differentially Expressed miRNAs
4.8. Prediction of Target Genes for Known and Novel mRNAs and GO Analyses
4.9. Quantitative RT-PCR (qRT-PCR) Analysis
4.10. Plant Material and Growth Conditions
4.11. Construction of Plant Expression Cassettes, Plant Transformation and Characterization of Transgenic Plants
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ismail, M.R.; Noor, K.M. Growth, water retations and physiological processes of starfruit (Averrhoa carambola L.) plants under root growth restriction. Sci. Hortic. 1996, 66, 51–58. [Google Scholar] [CrossRef]
- Wang, S.; Okamoto, G.; Hirano, K.; Lu, J.; Zhang, C. Effects of restricted rooting volume on vine growth and berry development of ‘Kyoho’ grapevines. Am. J. Enol. Vitic. 2001, 52, 248–253. [Google Scholar]
- Imai, M.; Watanabe, M.; Suyama, K.; Osada, T.; Sakai, D.; Kawada, H.; Matsumae, M.; Mochida, L. Delayed accumulation of activated macrophages and inhibition of remyelination after spinal cord injury in an adult rodent model. J. Neurosurg. Spine 2008, 8, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; He, J.J.; Bai, Y. Root restriction affected anthocyanin composition and up-regulated the transcription of their biosynthetic genes during berry development in ‘Summer Black’ grape. Acta Physiol. Plant. 2013, 35, 2205–2217. [Google Scholar] [CrossRef]
- Mataa, M.; Tominaga, S. Effects of root restriction on tree development in Ponkan mandarin (Citrus reticulata Blanco). J. Am. Soc. Hortic. Sci. 1998, 123, 651–655. [Google Scholar] [CrossRef]
- White, M.D.; Tustin, D.S.; Foote, K.F.; Campbell, J.M. Growth of young sweet cherry trees in response to root restriction using root control bags. Acta Hortic. 2001, 557, 391–398. [Google Scholar] [CrossRef]
- Hooijdonk, B.; Woolley, D.; Warrington, I.; Tustin, S. Architectural development of ‘Royal Gala’ apple scions in response to rootstock, root restriction, and benzylaminopurine. Acta Hortic. 2006, 727, 561–568. [Google Scholar] [CrossRef]
- Bartel, B.; Bartel, D.P. MicroRNAs: At the root of plant development? Plant Physiol. 2003, 132, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Chen, X. A microRNA as translational repressor of APETALA2 in Arabidopsis flower development. Science 2004, 303, 2022–2025. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Baloch, I.A.; Barozia, M.Y.; Din, M. MiRNAs: The Mega regulators in eukaryotic genomes. Pure Appl. Biol. 2013, 2, 334–335. [Google Scholar] [CrossRef]
- Ma, C.; Lu, Y.; Bai, S.L.; Zhang, W.N.; Duan, X.W.; Meng, D.; Wang, Z.G.; Wang, A.D.; Zhou, Z.S.; Li, T.Z. Cloning and characterization of miRNAs and their targets, including a novel miRNA-targeted NBS-LRR proteinclass gene in apple (Golden Delicious). Mol. Plant 2014, 7, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Yang, J.; Cheng, Q.; Mao, A.; Zhang, J.; Wang, S.; Weng, Y.; Wen, C. Comparative analysis of miRNA and mRNA abundance in determinate cucumber by high-throughput sequencing. PLoS ONE 2018, 13, e0190691. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, L.; Chen, D.; Wu, X.; Huang, D.; Chen, L.; Li, L.; Deng, X.; Xu, Q. Genome-wide comparison of microRNAs and their targeted transcripts among leaf, flower and fruit of sweet orange. BMC Genom. 2014, 15, 695. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.L.; Saito, T.; Ito, A.; Tuan, P.A.; Xu, Y.; Teng, Y.; Moriquchi, T. Small RNA and PARE sequencing in flower bud reveal the involvement of sRNAs in endodormancy release of Japanese pear (Pyrus pyrifolia ‘Kosui’). BMC Genom. 2016, 17, 230. [Google Scholar] [CrossRef]
- Pantaleo, V.; Szittya, G.; Moxon, S.; Miozzi, L.; Moulton, V.; Dalmay, T.; Burqyan, J. Identification of grapevine microRNAs and their targets using high-throughput sequencing and degradome analysis. Plant J. 2010, 62, 960–976. [Google Scholar]
- Saminathan, T.; Bodunrin, A.; Singh, N.V.; Devarajan, R.; Nimmakayalla, P.; Jeff, M.; Aradhya, M.; Reddy, U.K. Genome-wide identification of microRNAs in pomegranate (Punica granatum, L.) by high-throughput sequencing. BMC Plant Biol. 2016, 16, 122. [Google Scholar] [CrossRef]
- Llave, C.; Kasschau, K.D.; Rector, M.A.; Carrington, J.C. Endogenous and silencing-associated small RNAs in plants. Plant Cell 2002, 14, 1605–1619. [Google Scholar] [CrossRef]
- Lindow, M.; Krogh, A. Computational evidence for hundreds of non-conserved plant microRNAs. BMC Genom. 2005, 6, 119–128. [Google Scholar] [CrossRef]
- Rajagopalan, R.; Vaucheret, H.; Trejo, J.; Bartel, D.P. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 2006, 20, 3407–3425. [Google Scholar] [CrossRef]
- Jones-Rhoades, M.W.; Bartel, D.P.; Bartel, B. MicroRNAs and their regulatory roles in plants. Annu. Rev. Plant Biol. 2006, 57, 19–53. [Google Scholar] [CrossRef] [PubMed]
- Mallory, A.C.; Vaucheret, H. Functions of microRNAs and related small RNAs in plants. Nat. Genet. 2006, 38, S31–S36. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, Q.; Pan, X. MicroRNAs and their regulatory roles in animals and plants. J. Cell Physiol. 2007, 210, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.W.; Wang, L.J.; Mao, Y.B.; Cai, W.J.; Xue, H.W.; Chen, X.Y. Control of root cap formation by MicroRNA-targeted auxin response factors in Arabidopsis. Plant Cell 2005, 17, 2204–2216. [Google Scholar] [CrossRef] [PubMed]
- Boualem, A.; Laporte, P.; Jovanovic, M.; Laffont, C.; Plet, J.; Combier, J.P.; Niebel, A.; Crespi, M.; Fruqier, F. MicroRNA166 controls root and nodule development in Medicago truncatula. Plant J. 2008, 54, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Bao, M.; Bian, H.; Zha, Y.; Li, F.; Sun, Y.; Bai, B.; Chen, Z.; Wang, J.; Zhu, M.; Han, N. miR396a-Mediated basic helix-loop-helix transcription factor bHLH74 repression acts as a regulator for root growth in Arabidopsis seedlings. Plant Cell Physiol. 2014, 55, 1343–1353. [Google Scholar] [CrossRef]
- Sorin, C.; Declerck, M.; Christ, A.; Blein, T.; Ma, L.; Lelandais-Briere, C.; Njo, M.F.; Beeckman, T.; Crespi, M.; Hartmann, C. A miR169 isoform regulates specific NF-YA targets and root architecture in Arabidopsis. New Phytol. 2014, 202, 1197–1211. [Google Scholar] [CrossRef]
- Knauer, S.; Holt, A.L.; Rubio-Somoza, I.; Tucker, E.J.; Hinze, A.; Pisch, M.; Javelle, M.; Timmerman, M.C.; Tucker, M.R.; Laux, T. A protodermal miR394 signal defines a region of stem cell competence in the Arabidopsis shoot meristem. Dev. Cell 2013, 24, 125–132. [Google Scholar] [CrossRef]
- Zhu, H.; Hu, F.; Wang, R.; Zhou, X.; Sze, S.H.; Liou, L.W.; Barefoot, A.; Dickman, M.; Zhang, X. Arabidopsis Argonaute10 specifically sequesters miR166/165 to regulate shoot apical meristem development. Cell 2011, 145, 242–256. [Google Scholar] [CrossRef]
- Kidner, C.A.; Martienssen, R.A. Spatially restricted microRNA directs leaf polarity through ARGONAUTE1. Nature 2004, 428, 81–84. [Google Scholar] [CrossRef]
- Xu, L.; Yang, L.; Huang, H. Transcriptional, post-transcriptional and post-translational regulations of gene expression during leaf polarity formation. Cell Res. 2007, 17, 512–519. [Google Scholar] [CrossRef]
- Chitwood, D.H.; Guo, M.; Nogueira, F.T.; Timmermans, M.C. Establishing leaf polarity: The role of small RNAs and positional signals in the shoot apex. Development 2007, 134, 813–823. [Google Scholar] [CrossRef]
- Mallory, A.C.; Dugas, D.V.; Bartel, D.P.; Bartel, B. MicroRNA regulation of NAC-domain targets is required for proper formation and separation of adjacent embryonic, vegetative, and floral organs. Curr. Biol. 2004, 14, 1035–1046. [Google Scholar] [CrossRef]
- Teotia, S.; Tang, G. To bloom or not to bloom: Role of microRNAs in plant flowering. Mol. Plant 2015, 8, 359–377. [Google Scholar] [CrossRef]
- Juarez, M.T.; Kui, J.S.; Thomas, J.; Heller, B.A.; Timmermans, M.C. microRNA-mediated repression of rolled leaf1 specifies maize leaf polarity. Nature 2004, 428, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Schwab, R.; Palatnik, J.F.; Riester, M.; Schommer, C.; Schmid, M.; Weiqel, D. Specific effects of microRNAs on the plant transcriptome. Dev. Cell 2005, 8, 517–527. [Google Scholar] [CrossRef]
- Berger, Y.; Harpaz-Saad, S.; Brand, A.; Melnik, H.; Sirding, N.; Alvarez, J.P.; Zinder, M.; Samach, A.; Eshed, Y.; Ori, N. The NAC-domain transcription factor GOBLET specifies leaflet boundaries in compound tomato leaves. Development 2009, 136, 823–832. [Google Scholar] [CrossRef]
- Bertolini, E.; Verelst, W.; Horner, D.S.; Gianfranceschi, L.; Piccolo, V.; Inzé, D.; Pè, M.E.; Mica, E. Addressing the role of microRNAs in reprogramming leaf growth during drought stress in Brachypodium distachyon. Mol. Plant 2013, 6, 423–443. [Google Scholar] [CrossRef]
- Millar, A.A.; Gubler, F. The Arabidopsis GAMYB-like genes, MYB33 and MYB65, are microRNA-regulated genes that redundantly facilitate anther development. Plant Cell 2005, 17, 705–721. [Google Scholar] [CrossRef]
- Gandikota, M.; Birkenbihl, R.P.; Hohmann, S.; Cardon, G.H.; Saedler, H.; Huijser, P. The miRNA156/157 recognition element in the 3’ UTR of the Arabidopsis SBP box gene SPL3 prevents early flowering by translational inhibition in seedlings. Plant J. 2007, 49, 683–693. [Google Scholar] [CrossRef]
- Palatnik, J.F.; Wollmann, H.; Schommer, C.; Schwab, R.; Boisbouvier, J.; Rodriquez, R.; Warthmann, N.; Allen, E.; Dezulian, T.; Huson, D.; et al. Sequence and expression differences underlie functional specialization of Arabidopsis microRNAs miR159 and miR319. Dev. Cell 2007, 13, 115–125. [Google Scholar] [CrossRef]
- Mathieu, J.; Yant, L.J.; Murdter, F.; Küttner, F.; Schmid, M. Repression of flowering by the miR172 target SMZ. PLoS Biol. 2009, 7, e1000148. [Google Scholar] [CrossRef]
- Wu, G.; Park, M.Y.; Conway, S.R.; Wang, J.W.; Weiqel, D.; Poethiq, R.S. The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell 2009, 138, 750–759. [Google Scholar] [CrossRef]
- Gou, J.Y.; Felippes, F.F.; Liu, C.J.; Weiqel, D.; Wang, J.W. Negative regulation of anthocyanin biosynthesis in Arabidopsis by a miR156-targeted SPL transcription factor. Plant Cell 2011, 23, 1512–1522. [Google Scholar] [CrossRef]
- Xia, R.; Zhu, H.; An, Y.; Beers, E.P.; Liu, Z. Apple miRNAs and tasiRNAs with novel regulatory networks. Genome Biol. 2012, 13, R47. [Google Scholar] [CrossRef]
- Liu, Z.B.; Zhang, Y.P.; Ou, L.J.; Kang, L.; Liu, Y.; Lv, J.; Wei, G.; Yang, B.; Yang, S.; Chen, W.; et al. Identification and characterization of novel microRNAs for fruit development and quality in hot pepper (Capsicum annuum L.). Gene 2016, 608, 66–72. [Google Scholar] [CrossRef]
- Li, D.; Liu, Z.; Gao, L.; Wang, L.F.; Gao, M.J.; Jiao, Z.J.; Qiao, H.L.; Yang, J.W.; Chen, M.; Yao, L.G.; et al. Genome-wide identification and characterization of microRNAs in developing grains of Zea mays, L. PLoS ONE 2016, 11, e0153168. [Google Scholar] [CrossRef]
- Leng, F.; Tang, D.D.; Lin, Q.; Cao, J.P.; Wu, D.; Wang, S.P.; Sun, C.D. Transcriptomic analyses of ascorbic acid and carotenoid metabolites influenced by root restriction during grape berry development and ripening. J. Aqric. Food Chem. 2017, 65, 2008–2016. [Google Scholar] [CrossRef]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Sung, G.H.; Spatafora, J.W.; Carrington, J.C. Evolution of microRNA genes by inverted duplication of target gene sequences in Arabidopsis thaliana. Nat. Genet. 2004, 36, 1282–1290. [Google Scholar] [CrossRef]
- Mi, S.J.; Cai, T.; Hu, Y.G.; Chen, Y.M.; Hodges, E.; Ni, F.R.; Wu, L.; Li, S.; Zhou, H.Y.; Long, C.Z.; et al. Sorting od small RNAs into Arabidopsis argonaute complexes is directed by the 5′ terminal nucleotide. Cell 2008, 133, 116–127. [Google Scholar] [CrossRef]
- Sunkar, R.; Jagadeeswaran, G. In silico identification of conserved microRNAs in large number of diverse plant species. BMC Plant Biol. 2008, 8, 37. [Google Scholar] [CrossRef]
- Adai, A.; Johnson, C.; Mlotshwa, S.; Archer-Evans, S.; Manocha, V.; Vance, V.; Sundaresan, V. Computational prediction of miRNAs in Arabidopsis thaliana. Genome Res. 2005, 15, 78–91. [Google Scholar] [CrossRef]
- Jin, W.; Li, N.; Zhang, B.; Wu, F.L.; Li, W.J.; Guo, A.G.; Deng, Z.Y. Identification and verification of microRNA in wheat (Triticum aestivum). J. Plant Res. 2008, 121, 351–355. [Google Scholar] [CrossRef]
- Lu, S.; Sun, Y.H.; Chiang, V.L. Stress-responsive microRNAs in Populus. Plant J. 2008, 55, 131–151. [Google Scholar] [CrossRef]
- Morin, R.D.; O’Connor, M.D.; Griffith, M.; Kuchenbauer, F.; Delaney, A.; Prabhu, A.L.; Zhao, Y.; McDonald, H.; Zeng, T.; Hirst, M.; et al. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res. 2008, 18, 610–621. [Google Scholar] [CrossRef]
- Yang, F.X.; Cai, J.; Yang, Y.; Liu, Z.B. Overexpression of microRNA828 reduces anthocyanin accumulation in Arabidopsis. Plant Cell Tissue Organ. Cult. 2013, 115, 159–167. [Google Scholar] [CrossRef]
- Liu, Q.; Chen, Y.Q. Insights into the mechanism of plant development: Interactions of miRNAs pathway with phytohormone response. Biochem. Biophys. Res. Commun. 2009, 384, 1–5. [Google Scholar] [CrossRef]
- Henderson, I.R.; Zhang, X.; Lu, C.; Johnson, L.; Meyers, B.C.; Green, P.J.; Jacobsen, S.E. Dissecting Arabidopsis thaliana DICER function in small RNA processing, gene silencing and DNA methylation patterning. Nat. Gene. 2006, 38, 721–725. [Google Scholar] [CrossRef]
- Morin, R.D.; Aksay, G.; Dolgosheina, E.; Ebhardt, H.A.; Maqrini, V.; Mardis, E.R.; Sahinalp, S.C.; Unrau, P.J. Comparative analysis of the small RNA transcriptomes of Pinus contorta and Oryza sativa. Genome Res. 2008, 18, 571–584. [Google Scholar] [CrossRef]
- Hake, S. MicroRNAs: A role in plant development. Curr. Biol. 2003, 13, R851–R852. [Google Scholar] [CrossRef][Green Version]
- Zhang, Y. miRU: An automated plant miRNA target prediction server. Nucleic Acids Res. 2005, 33, W701–W704. [Google Scholar] [CrossRef]
- Hobert, O. Gene regulation by transcription factors and microRNAs. Science 2008, 319, 1785–1786. [Google Scholar] [CrossRef]
- Wang, H.C.; Huang, X.M.; Hu, G.B.; Huang, H.B. Studies on the relationship between anthocyanin biosythesis and related enymes in litchi pericarp. Sci. Agric. Sin. 2004, 37, 2028–2032. [Google Scholar]
- Li, M.F.; Li, X.F.; Han, Z.H.; Shu, H.R.; Li, T.Z. Molecular analysis of two Chinese pear (Pyrus bretschneideri Rehd.) spontaneous self-compatible mutants. Plant Biol. 2009, 11, 774–783. [Google Scholar] [CrossRef]
- Wang, T.; Chen, L.; Zhao, M.; Tian, Q.; Zhang, W.H. Identification of drought responsive microRNAs in Medicago truncatula by genome-wide high-throughput sequencing. BMC Genom. 2011, 12, 367. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Bonnet, E.; Wuyts, J.; Rouze, P.; Yves, V.D.P. Evidence that microRNA precursors, unlike other non-coding RNAs, have lower folding free energies than random sequences. Bioinformatics 2004, 20, 2911–2917. [Google Scholar] [CrossRef]
- Zhang, B.H.; Pan, X.P.; Cox, S.B.; Cobb, G.P.; Anderson, T.A. Evidence that miRNAs are different from other RNAs. Cell Mol. Life Sci. 2006, 63, 246–254. [Google Scholar] [CrossRef]
- Axtell, M.J. Classification and comparison of small RNAs from Plants. Annu. Rev. Plant Biol. 2013, 64, 137–159. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential Expression of RNA-Seq Data at the Gene Level-the DESeq Package; European Molecular Biology Laboratory (EMBL): Heidelberg, Germany, 2012. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Luo, M.; Gao, Z.; Li, H.; Li, Q.; Zhang, C.X.; Xu, W.P.; Song, S.R.; Ma, C.; Wang, S.P. Selection of reference genes for miRNA qRT-PCR under abiotic stress in grapevine. Sci. Rep. 2018, 8, 4444. [Google Scholar] [CrossRef]
- Bechtold, N.; Ellis, J.; Pelletier, G. In planta Agrobacterium mediated gene transfer by infiltration of adult Arabidopsis thaliana plants. CR Acad. Sci. Paris Life Sci. 1993, 316, 1194–1199. [Google Scholar]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Q.; Deng, B.; Gao, J.; Zhao, Z.; Chen, Z.; Song, S.; Wang, L.; Zhao, L.; Xu, W.; Zhang, C.; et al. Comparative Analysis of miRNA Abundance Revealed the Function of Vvi-miR828 in Fruit Coloring in Root Restriction Cultivation Grapevine (Vitis vinifera L.). Int. J. Mol. Sci. 2019, 20, 4058. https://doi.org/10.3390/ijms20164058
Chen Q, Deng B, Gao J, Zhao Z, Chen Z, Song S, Wang L, Zhao L, Xu W, Zhang C, et al. Comparative Analysis of miRNA Abundance Revealed the Function of Vvi-miR828 in Fruit Coloring in Root Restriction Cultivation Grapevine (Vitis vinifera L.). International Journal of Molecular Sciences. 2019; 20(16):4058. https://doi.org/10.3390/ijms20164058
Chicago/Turabian StyleChen, Qiuju, Bohan Deng, Jie Gao, Zhongyang Zhao, Zili Chen, Shiren Song, Lei Wang, Liping Zhao, Wenping Xu, Caixi Zhang, and et al. 2019. "Comparative Analysis of miRNA Abundance Revealed the Function of Vvi-miR828 in Fruit Coloring in Root Restriction Cultivation Grapevine (Vitis vinifera L.)" International Journal of Molecular Sciences 20, no. 16: 4058. https://doi.org/10.3390/ijms20164058
APA StyleChen, Q., Deng, B., Gao, J., Zhao, Z., Chen, Z., Song, S., Wang, L., Zhao, L., Xu, W., Zhang, C., Wang, S., & Ma, C. (2019). Comparative Analysis of miRNA Abundance Revealed the Function of Vvi-miR828 in Fruit Coloring in Root Restriction Cultivation Grapevine (Vitis vinifera L.). International Journal of Molecular Sciences, 20(16), 4058. https://doi.org/10.3390/ijms20164058