Neuroprotective Properties of Linagliptin: Focus on Biochemical Mechanisms in Cerebral Ischemia, Vascular Dysfunction and Certain Neurodegenerative Diseases

 , , ,
, , ,
Abstract
1. Introduction
2. Inflammation and Oxidative Stress
3. Cerebral Blood Flow
4. Neurodegeneration
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Global Health Estimates 2015: Deaths by Cause, Age, Sex, by Country and by Region, 2000–2015; World Health Organization: Geneva, Switzerland, 2016.
- Ahmad, M.; Dar, N.J.; Bhat, Z.S.; Hussain, A.; Shah, A.; Liu, H.; Graham, S.H. Inflammation in ischemic stroke: Mechanisms, consequences and possible drug targets. CNS Neurol. Disord. Drug Targets 2014, 13, 1378–1396. [Google Scholar] [CrossRef] [PubMed]
- Doupis, J. Linagliptin: From bench to bedside. Drug Des. Devel. Ther. 2014, 8, 431–446. [Google Scholar] [CrossRef] [PubMed]
- Vella, A. Mechanism of action of DPP-4 inhibitors--new insights. J. Clin. Endocrinol. Metab. 2012, 97, 2626–2628. [Google Scholar] [CrossRef] [PubMed]
- Wiciński, M.; Wódkiewicz, E.; Słupski, M.; Walczak, M.; Socha, M.; Malinowski, B.; Pawlak-Osińska, K. Neuroprotective Activity of Sitagliptin via Reduction of Neuroinflammation beyond the Incretin Effect: Focus on Alzheimer’s Disease. Biomed. Res. Int. 2018, 2018, 6091014. [Google Scholar] [CrossRef] [PubMed]
- Vella, A.; Bock, G.; Giesler, P.D.; Burton, D.B.; Serra, D.B.; Saylan, M.L.; Deacon, C.F.; Foley, J.E.; Rizza, R.A.; Camilleri, M. The effect of dipeptidyl peptidase-4 inhibition on gastric volume, satiation and enteroendocrine secretion in type 2 diabetes: A double-blind, placebo-controlled crossover study. Clin. Endocrinol. 2008, 69, 737–744. [Google Scholar] [CrossRef]
- Omar, B.; Ahrén, B. Pleiotropic mechanisms for the glucose-lowering action of DPP-4 inhibitors. Diabetes 2014, 63, 2196–2202. [Google Scholar] [CrossRef]
- Retlich, S.; Duval, V.; Graefe-Mody, U.; Friedrich, C.; Patel, S.; Jaehde, U.; Staab, A. Population Pharmacokinetics and Pharmacodynamics of Linagliptin in Patients with Type 2 Diabetes Mellitus. Clin. Pharmacokinet. 2015, 54, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Graefe-Mody, U.; Retlich, S.; Friedrich, C. Clinical pharmacokinetics and pharmacodynamics of linagliptin. Clin. Pharmacokinet. 2012, 51, 411–427. [Google Scholar] [CrossRef]
- Metzmann, K.; Schnell, D.; Jungnik, A.; Ring, A.; Theodor, R.; Hohl, K.; Meinicke, T.; Friedrich, C. Effect of food and tablet-dissolution characteristics on the bioavailability of linagliptin fixed-dose combination with metformin: Evidence from two randomized trials. Int. J. Clin. Pharmacol. Ther. 2014, 52, 549–563. [Google Scholar] [CrossRef]
- Kornelius, E.; Lin, C.L.; Chang, H.H.; Li, H.H.; Huang, W.N.; Yang, Y.S.; Lu, Y.L.; Peng, C.H.; Huang, C.N. DPP-4 Inhibitor Linagliptin Attenuates Aβ-induced Cytotoxicity through Activation of AMPK in Neuronal Cells. CNS Neurosci. Ther. 2015, 21, 549–557. [Google Scholar] [CrossRef]
- Ma, M.; Hasegawa, Y.; Koibuchi, N.; Toyama, K.; Uekawa, K.; Nakagawa, T.; Lin, B.; Kim-Mitsuyama, S. DPP-4 inhibition with linagliptin ameliorates cognitive impairment and brain atrophy induced by transient cerebral ischemia in type 2 diabetic mice. Cardiovasc. Diabetol. 2015, 14, 54. [Google Scholar] [CrossRef] [PubMed]
- Mi, D.H.; Fang, H.J.; Zheng, G.H.; Liang, X.H.; Ding, Y.R.; Liu, X.; Liu, L.P. DPP-4 inhibitors promote proliferation and migration of rat brain microvascular endothelial cells under hypoxic/high-glucose conditions, potentially through the SIRT1/HIF-1/VEGF pathway. CNS Neurosci. Ther. 2018, 25, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Inagaki, M.; Tsuji, M.; Gocho, T.; Handa, K.; Hasegawa, H.; Yura, A.; Kawakami, T.; Ohsawa, I.; Goto, Y.; et al. Linagliptin has Wide-Ranging Anti-Inflammatory Points of Action in Human Umbilical Vein Endothelial Cells. Jpn. Clin. Med. 2016, 7, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Waltereit, R.; Weller, M. Signaling from cAMP/PKA to MAPK and synaptic plasticity. Mol. Neurobiol. 2003, 27, 99–106. [Google Scholar] [CrossRef]
- Tarkowski, E.; Rosengren, L.; Blomstrand, C.; Wikkelsö, C.; Jensen, C.; Ekholm, S.; Tarkowski, A. Intrathecal release of pro- and anti-inflammatory cytokines during stroke. Clin. Exp. Immunol. 1997, 110, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Salheen, S.M.; Panchapakesan, U.; Pollock, C.A.; Woodman, O.L. The Dipeptidyl Peptidase-4 Inhibitor Linagliptin Preserves Endothelial Function in Mesenteric Arteries from Type 1 Diabetic Rats without Decreasing Plasma Glucose. PLoS ONE 2015, 10, e0143941. [Google Scholar] [CrossRef] [PubMed]
- Darsalia, V.; Ortsäter, H.; Olverling, A.; Darlöf, E.; Wolbert, P.; Nyström, T.; Klein, T.; Sjöholm, Å.; Patrone, C. The DPP-4 inhibitor linagliptin counteracts stroke in the normal and diabetic mouse brain: A comparison with glimepiride. Diabetes 2013, 62, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Darsalia, V.; Olverling, A.; Larsson, M.; Mansouri, S.; Nathanson, D.; Nyström, T.; Klein, T.; Sjöholm, Å.; Patrone, C. Linagliptin enhances neural stem cell proliferation after stroke in type 2 diabetic mice. Regul. Pept. 2014, 190–191, 25–31. [Google Scholar] [CrossRef]
- Elbaz, E.M.; Senousy, M.A.; El-Tanbouly, D.M.; Sayed, R.H. Neuroprotective effect of linagliptin against cuprizone-induced demyelination and behavioural dysfunction in mice: A pivotal role of AMPK/SIRT1 and JAK2/STAT3/NF-κB signalling pathway modulation. Toxicol. Appl. Pharmacol. 2018, 352, 153–161. [Google Scholar] [CrossRef]
- Kosaraju, J.; Holsinger, R.M.D.; Guo, L.; Tam, K.Y. Linagliptin, a Dipeptidyl Peptidase-4 Inhibitor, Mitigates Cognitive Deficits and Pathology in the 3xTg-AD Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2017, 54, 6074–6084. [Google Scholar] [CrossRef]
- Salim, H.M.; Fukuda, D.; Higashikuni, Y.; Tanaka, K.; Hirata, Y.; Yagi, S.; Soeki, T.; Shimabukuro, M.; Sata, M. Dipeptidyl peptidase-4 inhibitor, linagliptin, ameliorates endothelial dysfunction and atherogenesis in normoglycemic apolipoprotein-E deficient mice. Vascul. Pharmacol. 2016, 79, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Hardigan, T.; Abdul, Y.; Ergul, A. Linagliptin reduces effects of ET-1 and TLR2-mediated cerebrovascular hyperreactivity in diabetes. Life Sci. 2016, 159, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Chiazza, F.; Tammen, H.; Pintana, H.; Lietzau, G.; Collino, M.; Nyström, T.; Klein, T.; Darsalia, V.; Patrone, C. The effect of DPP-4 inhibition to improve functional outcome after stroke is mediated by the SDF-1α/CXCR4 pathway. Cardiovasc. Diabetol. 2018, 17, 60. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Hasegawa, H.; Tsuji, M.; Oguchi, T.; Mihara, M.; Suzuki, H.; Nishida, K.; Inoue, M.; Shimizu, T.; Ohsawa, I.; et al. Linagliptin inhibits lipopolysaccharide-stimulated interleukin-6 production, intranuclear p65 expression, and p38 mitogen-activated protein kinase phosphorylation in human umbilical vein endothelial cells. Ren. Replace. Ther. 2016, 2, 17. [Google Scholar] [CrossRef][Green Version]
- Shigiyama, F.; Kumashiro, N.; Miyagi, M.; Iga, R.; Kobayashi, Y.; Kanda, E.; Uchino, H.; Hirose, T. Linagliptin improves endothelial function in patients with type 2 diabetes: A randomized study of linagliptin effectiveness on endothelial function. J. Diabetes Investig. 2017, 8, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Hanke, M.L.; Kielian, T. Toll-like receptors in health and disease in the brain: Mechanisms and therapeutic potential. Clin. Sci. 2011, 121, 367–387. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Cho, S. Microglia and Monocyte-Derived Macrophages in Stroke. Neurotherapeutics 2016, 13, 702–718. [Google Scholar] [CrossRef] [PubMed]
- Saqib, U.; Sarkar, S.; Suk, K.; Mohammad, O.; Baig, M.S.; Savai, R. Phytochemicals as modulators of M1-M2 macrophages in inflammation. Oncotarget 2018, 9, 17937–17950. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Leak, R.K.; Shi, Y.; Suenaga, J.; Gao, Y.; Zheng, P.; Chen, J. Microglial and macrophage polarization—new prospects for brain repair. Nat. Rev. Neurol. 2015, 11, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.Y.; Liu, L.; Yang, Q.W. Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Prog. Neurobiol. 2016, 142, 23–44. [Google Scholar] [CrossRef] [PubMed]
- Yamadera, S.; Nakamura, Y.; Inagaki, M.; Kenmotsu, S.; Nohara, T.; Sato, N.; Oguchi, T.; Tsuji, M.; Ohsawa, I.; Gotoh, H.; et al. Linagliptin inhibits lipopolysaccharide-induced inflammation in human U937 monocytes. Inflamm. Regen. 2018, 38, 13. [Google Scholar] [CrossRef] [PubMed]
- Vila, N.; Castillo, J.; Dávalos, A.; Chamorro, A. Proinflammatory cytokines and early neurological worsening in ischemic stroke. Stroke 2000, 31, 2325–2329. [Google Scholar] [CrossRef] [PubMed]
- Zaremba, J.; Losy, J. Early TNF-alpha levels correlate with ischaemic stroke severity. Acta Neurol. Scand. 2001, 104, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T. The nuclear factor NF-κB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rey, E.; Chorny, A.; Delgado, M. Regulation of immune tolerance by anti-inflammatory neuropeptides. Nat. Rev. Immunol. 2007, 7, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Dai, D.; Wang, X.; Ding, Z.; Mehta, J.L. DPP-4 inhibitors repress NLRP3 inflammasome and interleukin-1beta via GLP-1 receptor in macrophages through protein kinase C pathway. Cardiovasc. Drugs Ther. 2014, 28, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Bonora, B.M.; Cappellari, R.; Menegazzo, L.; Vedovato, M.; Iori, E.; Marescotti, M.C.; Albiero, M.; Avogaro, A. Acute Effects of Linagliptin on Progenitor Cells, Monocyte Phenotypes, and Soluble Mediators in Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 748–756. [Google Scholar] [CrossRef]
- Thomas, G.; Tacke, R.; Hedrick, C.C.; Hanna, R.N. Nonclassical patrolling monocyte function in the vasculature. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Bellavance, M.A.; Gosselin, D.; Yong, V.W.; Stys, P.K.; Rivest, S. Patrolling monocytes play a critical role in CX3CR1-mediated neuroprotection during excitotoxicity. Brain Struct. Funct. 2015, 220, 1759–1776. [Google Scholar] [CrossRef]
- Audoy-Rémus, J.; Richard, J.F.; Soulet, D.; Zhou, H.; Kubes, P.; Vallières, L. Rod-Shaped monocytes patrol the brain vasculature and give rise to perivascular macrophages under the influence of proinflammatory cytokines and angiopoietin-2. J. Neurosci. 2008, 28, 10187–10199. [Google Scholar] [CrossRef]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Madden, J.A. Role of the vascular endothelium and plaque in acute ischemic stroke. Neurology 2012, 79 (Suppl. 1), S58–S62. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Harmful and Beneficial Role of ROS. Oxid. Med. Cell. Longev. 2016, 2016, 7909186. [Google Scholar] [CrossRef] [PubMed]
- Lastra, G.; Syed, S.; Kurukulasuriya, L.R.; Manrique, C.; Sowers, J.R. Type 2 diabetes mellitus and hypertension: An update. Endocrinol. Metab. Clin. N. Am. 2014, 43, 103–122. [Google Scholar] [CrossRef]
- Palmer, R.M.; Ashton, D.S.; Moncada, S. Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature 1988, 333, 664. [Google Scholar] [CrossRef] [PubMed]
- Rafieian-Kopaei, M.; Asgary, S.; Adelnia, A.; Setorki, M.; Khazaei, M.; Kazemi, S.; Shamsi, F. The effects of cornelian cherry on atherosclerosis and atherogenic factors in hypercholesterolemic rabbits. J. Med. Plants Res. 2011, 5, 2670–2676. [Google Scholar]
- Kibbe, M.; Billiar, T.; Tzeng, E. Inducible nitric oxide synthase and vascular injury. Cardiovasc. Res. 1999, 43, 650–657. [Google Scholar] [CrossRef]
- Godo, S.; Shimokawa, H. Endothelial Functions. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e108–e114. [Google Scholar] [CrossRef]
- Terpolilli, N.A.; Moskowitz, M.A.; Plesnila, N. Nitric oxide: Considerations for the treatment of ischemic stroke. J. Cereb. Blood Flow Metab. 2012, 32, 1332–1346. [Google Scholar] [CrossRef]
- Vellecco, V.; Mitidieri, E.; Gargiulo, A.; Brancaleone, V.; Matassa, D.; Klein, T.; Esposito, F.; Cirino, G.; Bucci, M. Vascular effects of linagliptin in non-obese diabetic mice are glucose-independent and involve positive modulation of the endothelial nitric oxide synthase (eNOS)/caveolin-1 (CAV-1) pathway. Diabetes Obes. Metab. 2016, 18, 1236–1243. [Google Scholar] [CrossRef]
- Jyoti, U.; Kansal, S.K.; Kumar, P.; Goyal, S. Possible vasculoprotective role of linagliptin against sodium arsenite-induced vascular endothelial dysfunction. Naunyn Schmiedeberg’s Arch Pharmacol. 2016, 389, 167–175. [Google Scholar] [CrossRef]
- Theodorou, K.; Boon, R.A. Endothelial Cell Metabolism in Atherosclerosis. Front. Cell Dev. Biol. 2018, 6, 82. [Google Scholar] [CrossRef]
- Soler, E.P.; Ruiz, V.C. Epidemiology and risk factors of cerebral ischemia and ischemic heart diseases: Similarities and differences. Curr. Cardiol. Rev. 2010, 6, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Hajra, L.; Evans, A.I.; Chen, M.; Hyduk, S.J.; Collins, T.; Cybulsky, M.I. The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc. Natl. Acad. Sci. USA 2000, 97, 9052–9057. [Google Scholar] [CrossRef]
- Tabas, I.; García-Cardeña, G.; Owens, G.K. Recent insights into the cellular biology of atherosclerosis. J. Cell Biol. 2015, 209, 13–22. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef]
- Cahill, P.A.; Redmond, E.M. Vascular endothelium—Gatekeeper of vessel health. Atherosclerosis 2016, 248, 97–109. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, X.M.; Luo, Q.Q.; Huang, S.; Yang, Q.W.; Wang, F.X.; Ke, Y.; Qian, Z.M. CX3CL1/CX3CR1-mediated microglia activation plays a detrimental role in ischemic mice brain via p38MAPK/PKC pathway. J. Cereb. Blood Flow Metab. 2015, 35, 1623–1631. [Google Scholar] [CrossRef]
- Yuan, B.; Shi, H.; Zheng, K.; Su, Z.; Su, H.; Zhong, M.; He, X.; Zhou, C.; Chen, H.; Xiong, Q.; et al. MCP-1-mediated activation of microglia promotes white matter lesions and cognitive deficits by chronic cerebral hypoperfusion in mice. Mol. Cell Neurosci. 2017, 78, 52–58. [Google Scholar] [CrossRef]
- Gao, H.H.; Gao, L.B.; Wen, J.M. Correlations of MCP-1 −2518A>G polymorphism and serum levels with cerebral infarction risk: A meta-analysis. DNA Cell Biol. 2014, 33, 522–530. [Google Scholar] [CrossRef]
- Grozdanov, V.; Bliederhaeuser, C.; Ruf, W.P.; Roth, V.; Fundel-Clemens, K.; Zondler, L.; Brenner, D.; Martin-Villalba, A.; Hengerer, B.; Kassubek, J.; et al. Inflammatory dysregulation of blood monocytes in Parkinson’s disease patients. Acta Neuropathol. 2014, 128, 651–663. [Google Scholar] [CrossRef]
- Whiteley, W.; Jackson, C.; Lewis, S.; Lowe, G.; Rumley, A.; Sandercock, P.; Wardlaw, J.; Dennis, M.; Sudlow, C. Inflammatory markers and poor outcome after stroke: A prospective cohort study and systematic review of interleukin-6. PLoS Med. 2009, 6, e1000145. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, W.; Edvinsson, L.; Xu, C.B. Apolipoprotein B of low-density lipoprotein impairs nitric oxide-mediated endothelium-dependent relaxation in rat mesenteric arteries. Eur. J. Pharmacol. 2014, 725, 10–17. [Google Scholar] [CrossRef]
- Dong, H.; Chen, W.; Wang, X.; Pi, F.; Wu, Y.; Pang, S.; Xie, Y.; Xia, F.; Zhang, Q. Apolipoprotein A1, B levels, and their ratio and the risk of a first stroke: A meta-analysis and case-control study. Metab. Brain Dis. 2015, 30, 1319–1330. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; de Winther, M.P.; Weber, C.; Daemen, M.J.; Lutgens, E.; Soehnlein, O. Atherosclerotic plaque destabilization: Mechanisms, models, and therapeutic strategies. Circ. Res. 2014, 114, 214–226. [Google Scholar] [CrossRef]
- Wang, Y.; Ge, P.; Zhu, Y. TLR2 and TLR4 in the brain injury caused by cerebral ischemia and reperfusion. Mediat. Inflamm. 2013, 2013, 124614. [Google Scholar] [CrossRef]
- Ziegler, G.; Harhausen, D.; Schepers, C.; Hoffmann, O.; Röhr, C.; Prinz, V.; König, J.; Lehrach, H.; Nietfeld, W.; Trendelenburg, G. TLR2 has a detrimental role in mouse transient focal cerebral ischemia. Biochem. Biophys. Res. Commun. 2007, 359, 574–579. [Google Scholar] [CrossRef]
- Lv, M.; Liu, Y.; Zhang, J.; Sun, L.; Liu, Z.; Zhang, S.; Wang, B.; Su, D.; Su, Z. Roles of inflammation response in microglia cell through Toll-like receptors 2/interleukin-23/interleukin-17 pathway in cerebral ischemia/reperfusion injury. Neuroscience 2011, 176, 162–172. [Google Scholar] [CrossRef]
- Heiss, W.D. The ischemic penumbra: How does tissue injury evolve? Ann. N. Y. Acad Sci. 2012, 1268, 26–34. [Google Scholar] [CrossRef]
- Pushie, M.J.; Crawford, A.M.; Sylvain, N.J.; Hou, H.; Hackett, M.J.; George, G.N.; Kelly, M.E. Revealing the Penumbra through Imaging Elemental Markers of Cellular Metabolism in an Ischemic Stroke Model. ACS Chem. Neurosci. 2018, 9, 886–893. [Google Scholar] [CrossRef]
- Hankey, G.J. Stroke. Lancet 2017, 389, 641–654. [Google Scholar] [CrossRef]
- Aggarwal, N.T.; Schneider, J.A.; Wilson, R.S.; Beck, T.L.; Evans, D.A.; Carli, C.D. Characteristics of MR infarcts associated with dementia and cognitive function in the elderly. Neuroepidemiology 2012, 38, 41–47. [Google Scholar] [CrossRef]
- Zhang, L.; Chopp, M.; Zhang, Y.; Xiong, Y.; Li, C.; Sadry, N.; Rhaleb, I.; Lu, M.; Zhang, Z.G. Diabetes Mellitus Impairs Cognitive Function in Middle-Aged Rats and Neurological Recovery in Middle-Aged Rats After Stroke. Stroke 2016, 47, 2112–2118. [Google Scholar] [CrossRef]
- Darsalia, V.; Larsson, M.; Lietzau, G.; Nathanson, D.; Nyström, T.; Klein, T.; Patrone, C. Gliptin-mediated neuroprotection against stroke requires chronic pretreatment and is independent of glucagon-like peptide-1 receptor. Diabetes Obes. Metab. 2016, 18, 537–541. [Google Scholar] [CrossRef]
- Thangarajah, H.; Yao, D.; Chang, E.I.; Shi, Y.; Jazayeri, L.; Vial, I.N.; Galiano, R.D.; Du, X.L.; Grogan, R.; Galvez, M.G.; et al. The molecular basis for impaired hypoxia-induced VEGF expression in diabetic tissues. Proc. Natl. Acad. Sci. USA 2009, 106, 13505–13510. [Google Scholar] [CrossRef]
- Craige, S.M.; Chen, K.; Pei, Y.; Li, C.; Huang, X.; Chen, C.; Shibata, R.; Sato, K.; Walsh, K.; Keaney, J.F., Jr. NADPH oxidase 4 promotes endothelial angiogenesis through endothelial nitric oxide synthase activation. Circulation 2011, 124, 731–740. [Google Scholar] [CrossRef]
- Joo, H.Y.; Yun, M.; Jeong, J.; Park, E.R.; Shin, H.J.; Woo, S.R.; Jung, J.K.; Kim, Y.M.; Park, J.J.; Kim, J.; et al. SIRT1 deacetylates and stabilizes hypoxia-inducible factor-1α (HIF-1α) via direct interactions during hypoxia. Biochem. Biophys. Res. Commun. 2015, 462, 294–300. [Google Scholar] [CrossRef]
- Laemmle, A.; Lechleiter, A.; Roh, V.; Schwarz, C.; Portmann, S.; Furer, C.; Keogh, A.; Tschan, M.P.; Candinas, D.; Vorburger, S.A.; et al. Inhibition of SIRT1 impairs the accumulation and transcriptional activity of HIF-1α protein under hypoxic conditions. PLoS ONE 2012, 7, e33433. [Google Scholar] [CrossRef]
- Wiciński, M.; Socha, M.; Walczak, M.; Wódkiewicz, E.; Malinowski, B.; Rewerski, S.; Górski, K.; Pawlak-Osińska, K. Beneficial Effects of Resveratrol Administration-Focus on Potential Biochemical Mechanisms in Cardiovascular Conditions. Nutrients 2018, 10. [Google Scholar] [CrossRef]
- Rostène, W.; Kitabgi, P.; Parsadaniantz, S.M. Chemokines: A new class of neuromodulator? Nat. Rev. Neurosci. 2007, 8, 895–903. [Google Scholar] [CrossRef]
- Cheng, X.; Wang, H.; Zhang, X.; Zhao, S.; Zhou, Z.; Mu, X.; Zhao, C.; Teng, W. The Role of SDF-1/CXCR4/CXCR7 in Neuronal Regeneration after Cerebral Ischemia. Front. Neurosci. 2017, 11, 590. [Google Scholar] [CrossRef]
- Doitsidou, M.; Reichman-Fried, M.; Stebler, J.; Köprunner, M.; Dörries, J.; Meyer, D.; Esguerra, C.V.; Leung, T.; Raz, E. Guidance of primordial germ cell migration by the chemokine SDF-1. Cell 2002, 111, 647–659. [Google Scholar] [CrossRef]
- Laske, C.; Stellos, K.; Stransky, E.; Seizer, P.; Akcay, O.; Eschweiler, G.W.; Leyhe, T.; Gawaz, M. Decreased plasma and cerebrospinal fluid levels of stem cell factor in patients with early Alzheimer’s disease. J. Alzheimer’s Dis. 2008, 15, 451–460. [Google Scholar] [CrossRef]
- Li, Y.; Huang, J.; He, X.; Tang, G.; Tang, Y.H.; Liu, Y.; Lin, X.; Lu, Y.; Yang, G.Y.; Wang, Y. Postacute stromal cell-derived factor-1α expression promotes neurovascular recovery in ischemic mice. Stroke 2014, 45, 1822–1829. [Google Scholar] [CrossRef]
- Selvaraj, U.M.; Ortega, S.B.; Hu, R.; Gilchrist, R.; Kong, X.; Partin, A.; Plautz, E.J.; Klein, R.S.; Gidday, J.M.; Stowe, A.M. Preconditioning-induced CXCL12 upregulation minimizes leukocyte infiltration after stroke in ischemia-tolerant mice. J. Cereb. Blood Flow Metab. 2017, 37, 801–813. [Google Scholar] [CrossRef]
- Kwon, H.S.; Kim, Y.S.; Park, H.H.; Choi, H.; Lee, K.Y.; Lee, Y.J.; Heo, S.H.; Chang, D.I.; Koh, S.H. Increased VEGF and decreased SDF-1α in patients with silent brain infarction are associated with better prognosis after first-ever acute lacunar stroke. J. Stroke Cerebrovasc. Dis. 2015, 24, 704–710. [Google Scholar] [CrossRef]
- Huang, J.; Li, Y.; Tang, Y.; Tang, G.; Yang, G.Y.; Wang, Y. CXCR4 antagonist AMD3100 protects blood-brain barrier integrity and reduces inflammatory response after focal ischemia in mice. Stroke 2013, 44, 190–197. [Google Scholar] [CrossRef]
- De Vos, A.; Bjerke, M.; Brouns, R.; De Roeck, N.; Jacobs, D.; Van den Abbeele, L.; Guldolf, K.; Zetterberg, H.; Blennow, K.; Engelborghs, S.; et al. Neurogranin and tau in cerebrospinal fluid and plasma of patients with acute ischemic stroke. BMC Neurol. 2017, 17, 170. [Google Scholar] [CrossRef]
- Gallwitz, B.; Rosenstock, J.; Rauch, T.; Bhattacharya, S.; Patel, S.; von Eynatten, M.; et al. 2-year efficacy and safety of linagliptin compared with glimepiride in patients with type 2 diabetes inadequately controlled on metformin: A randomised, double-blind, non-inferiority trial. Lancet 2012, 380, 475–483. [Google Scholar] [CrossRef]
- Li, Y.R.; Tsai, S.S.; Chen, D.Y.; Chen, S.T.; Sun, J.H.; Chang, H.Y.; Chen, T.H. Linagliptin and cardiovascular outcomes in type 2 diabetes after acute coronary syndrome or acute ischemic stroke. Cardiovasc. Diabetol. 2018, 17, 2. [Google Scholar] [CrossRef]
- Marx, N.; Rosenstock, J.; Kahn, S.E.; Zinman, B.; Kastelein, J.J.; Lachin, J.M.; Patel, S. Design and baseline characteristics of the CARdiovascular outcome trial of LINAgliptin versus glimepiride in type 2 diabetes (CAROLINA®). Diabetes Vasc. Dis. Res. 2015, 12, 164–174. [Google Scholar] [CrossRef]
- Rosenstock, J.; Perkovic, V.; Alexander, J.H.; Cooper, M.E.; Marx, N.; Pencina, M.J.; Pfarr, E. Rationale, design, and baseline characteristics of the CArdiovascular safety and Renal Microvascular outcomE study with LINAgliptin (CARMELINA®): A randomized, double-blind, placebo-controlled clinical trial in patients with type 2 diabetes and high cardio-renal risk. Cardiovasc. Diabetol. 2018, 17, 39. [Google Scholar]
- Cunningham, E.L.; McGuinness, B.; Herron, B.; Passmore, A.P. Dementia. Ulster Med. J. 2015, 84, 79–87. [Google Scholar]
- Iqbal, K.; Liu, F.; Gong, C.X.; Grundke-Iqbal, I. Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef]
- Yilmaz, U. [Alzheimer’s disease]. Radiologe 2015, 55, 386–388. [Google Scholar] [CrossRef]
- Wiciński, M.; Socha, M.; Malinowski, B.; Wódkiewicz, E.; Walczak, M.; Górski, K.; Słupski, M.; Pawlak-Osińska, K. Liraglutide and its Neuroprotective Properties-Focus on Possible Biochemical Mechanisms in Alzheimer’s Disease and Cerebral Ischemic Events. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Hong, J.T. NF-kB as a mediator of brain inflammation in AD. CNS Neurol. Disord. Drug Targets 2017. [Google Scholar] [CrossRef]
- van Dam, P.S.; Aleman, A. Insulin-like growth factor-I, cognition and brain aging. Eur. J. Pharmacol. 2004, 490, 87–95. [Google Scholar] [CrossRef]
- Stockhorst, U.; de Fries, D.; Steingrueber, H.J.; Scherbaum, W.A. Insulin and the CNS: Effects on food intake, memory, and endocrine parameters and the role of intranasal insulin administration in humans. Physiol. Behav. 2004, 83, 47–54. [Google Scholar] [CrossRef]
- Craft, S.; Watson, G.S. Insulin and neurodegenerative disease: Shared and specific mechanisms. Lancet Neurol. 2004, 3, 169–178. [Google Scholar] [CrossRef]
- Bedse, G.; Di Domenico, F.; Serviddio, G.; Cassano, T. Aberrant insulin signaling in Alzheimer’s disease: Current knowledge. Front. Neurosci. 2015, 9, 204. [Google Scholar] [CrossRef]
- Pérez, A.; Morelli, L.; Cresto, J.C.; Castaño, E.M. Degradation of soluble amyloid beta-peptides 1-40, 1-42, and the Dutch variant 1-40Q by insulin degrading enzyme from Alzheimer disease and control brains. Neurochem. Res. 2000, 25, 247–255. [Google Scholar] [CrossRef]
- De Felice, F.G.; Vieira, M.N.; Bomfim, T.R.; Decker, H.; Velasco, P.T.; Lambert, M.P.; Viola, K.L.; Zhao, W.Q.; Ferreira, S.T.; Klein, W.L. Protection of synapses against Alzheimer’s-linked toxins: Insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 1971–1976. [Google Scholar] [CrossRef]
- Tokutake, T.; Kasuga, K.; Yajima, R.; Sekine, Y.; Tezuka, T.; Nishizawa, M.; Ikeuchi, T. Hyperphosphorylation of Tau induced by naturally secreted amyloid-β at nanomolar concentrations is modulated by insulin-dependent Akt-GSK3β signaling pathway. J. Biol. Chem. 2012, 287, 35222–35233. [Google Scholar] [CrossRef]
- Metaxas, A.; Kempf, S.J. Neurofibrillary tangles in Alzheimer’s disease: Elucidation of the molecular mechanism by immunohistochemistry and tau protein phospho-proteomics. Neural Regen. Res. 2016, 11, 1579–1581. [Google Scholar] [CrossRef]
- Dziedzic, T. Systemic inflammation as a therapeutic target in acute ischemic stroke. Expert Rev. Neurother. 2015, 15, 523–531. [Google Scholar] [CrossRef]
- Chitnis, T.; Weiner, H.L. CNS inflammation and neurodegeneration. J. Clin. Investig. 2017, 127, 3577–3587. [Google Scholar] [CrossRef]
- Holmes, C. Review: Systemic inflammation and Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2013, 39, 51–68. [Google Scholar] [CrossRef]
- Elwood, E.; Lim, Z.; Naveed, H.; Galea, I. The effect of systemic inflammation on human brain barrier function. Brain Behav. Immun. 2017, 62, 35–40. [Google Scholar] [CrossRef]
- Chen, W.W.; Zhang, X.; Huang, W.J. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef]
- Yan, Z.; Gibson, S.A.; Buckley, J.A.; Qin, H.; Benveniste, E.N. Role of the JAK/STAT signaling pathway in regulation of innate immunity in neuroinflammatory diseases. Clin. Immunol. 2018, 189, 4–13. [Google Scholar] [CrossRef]
- Nicolas, C.S.; Amici, M.; Bortolotto, Z.A.; Doherty, A.; Csaba, Z.; Fafouri, A.; Dournaud, P.; Gressens, P.; Collingridge, G.L.; Peineau, S. The role of JAK-STAT signaling within the CNS. JAK-STAT 2013, 2, e22925. [Google Scholar] [CrossRef]
- Ivanenkov, Y.A.; Balakin, K.V.; Lavrovsky, Y. Small molecule inhibitors of NF-kB and JAK/STAT signal transduction pathways as promising anti-inflammatory therapeutics. Mini Rev. Med. Chem. 2011, 11, 55–78. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef]
- Sun, C.; Zhang, F.; Ge, X.; Yan, T.; Chen, X.; Shi, X.; Zhai, Q. SIRT1 improves insulin sensitivity under insulin-resistant conditions by repressing PTP1B. Cell Metab. 2007, 6, 307–319. [Google Scholar] [CrossRef]
- Greco, S.J.; Hamzelou, A.; Johnston, J.M.; Smith, M.A.; Ashford, J.W.; Tezapsidis, N. Leptin boosts cellular metabolism by activating AMPK and the sirtuins to reduce tau phosphorylation and β-amyloid in neurons. Biochem. Biophys. Res. Commun. 2011, 414, 170–174. [Google Scholar] [CrossRef]
- Vázquez-Manrique, R.P.; Farina, F.; Cambon, K.; Dolores Sequedo, M.; Parker, A.J.; Millán, J.M.; Weiss, A.; Déglon, N.; Neri, C. AMPK activation protects from neuronal dysfunction and vulnerability across nematode, cellular and mouse models of Huntington’s disease. Hum. Mol. Genet. 2016, 25, 1043–1058. [Google Scholar] [CrossRef]
- Wiciński, M.; Malinowski, B.; Węclewicz, M.M.; Grześk, E.; Grześk, G. Anti-atherogenic properties of resveratrol: 4-week resveratrol administration associated with serum concentrations of SIRT1, adiponectin, S100A8/A9 and VSMCs contractility in a rat model. Exp. Ther. Med. 2017, 13, 2071–2078. [Google Scholar] [CrossRef]
- Stefano, G.B.; Challenger, S.; Kream, R.M. Hyperglycemia-associated alterations in cellular signaling and dysregulated mitochondrial bioenergetics in human metabolic disorders. Eur. J. Nutr. 2016, 55, 2339–2345. [Google Scholar] [CrossRef]
- Khan, M.A.; Schultz, S.; Othman, A.; Fleming, T.; Lebrón-Galán, R.; Rades, D.; Clemente, D.; Nawroth, P.P.; Schwaninger, M. Hyperglycemia in Stroke Impairs Polarization of Monocytes/Macrophages to a Protective Noninflammatory Cell Type. J. Neurosci. 2016, 36, 9313–9325. [Google Scholar] [CrossRef]
- Shao, B.; Bayraktutan, U. Hyperglycaemia promotes human brain microvascular endothelial cell apoptosis via induction of protein kinase C-ßI and prooxidant enzyme NADPH oxidase. Redox Biol. 2014, 2, 694–701. [Google Scholar] [CrossRef]
- Wijesekara, N.; Ahrens, R.; Sabale, M.; Wu, L.; Ha, K.; Verdile, G.; Fraser, P.E. Amyloid-β and islet amyloid pathologies link Alzheimer’s disease and type 2 diabetes in a transgenic model. FASEB J. 2017, 31, 5409–5418. [Google Scholar] [CrossRef]
- Bruno, A.; Williams, L.S.; Kent, T.A. How important is hyperglycemia during acute brain infarction? Neurologist 2004, 10, 195–200. [Google Scholar] [CrossRef]
- Mi, D.; Wang, P.; Yang, B.; Pu, Y.; Yang, Z.; Liu, L. Correlation of hyperglycemia with mortality after acute ischemic stroke. Ther. Adv. Neurol. Disord. 2017, 11, 1756285617731686. [Google Scholar] [CrossRef]
- Carvalho, C.; Katz, P.S.; Dutta, S.; Katakam, P.V.; Moreira, P.I.; Busija, D.W. Increased susceptibility to amyloid-β toxicity in rat brain microvascular endothelial cells under hyperglycemic conditions. J. Alzheimer’s Dis. 2014, 38, 75–83. [Google Scholar] [CrossRef]
- Huang, J.; Liu, B.; Yang, C.; Chen, H.; Eunice, D.; Yuan, Z. Acute hyperglycemia worsens ischemic stroke-induced brain damage via high mobility group box-1 in rats. Brain Res. 2013, 1535, 148–155. [Google Scholar] [CrossRef]
- Sato, T.; Iwaki, M.; Shimogaito, N.; Wu, X.; Yamagishi, S.; Takeuchi, M. TAGE (toxic AGEs) theory in diabetic complications. Curr. Mol. Med. 2006, 6, 351–358. [Google Scholar] [CrossRef]


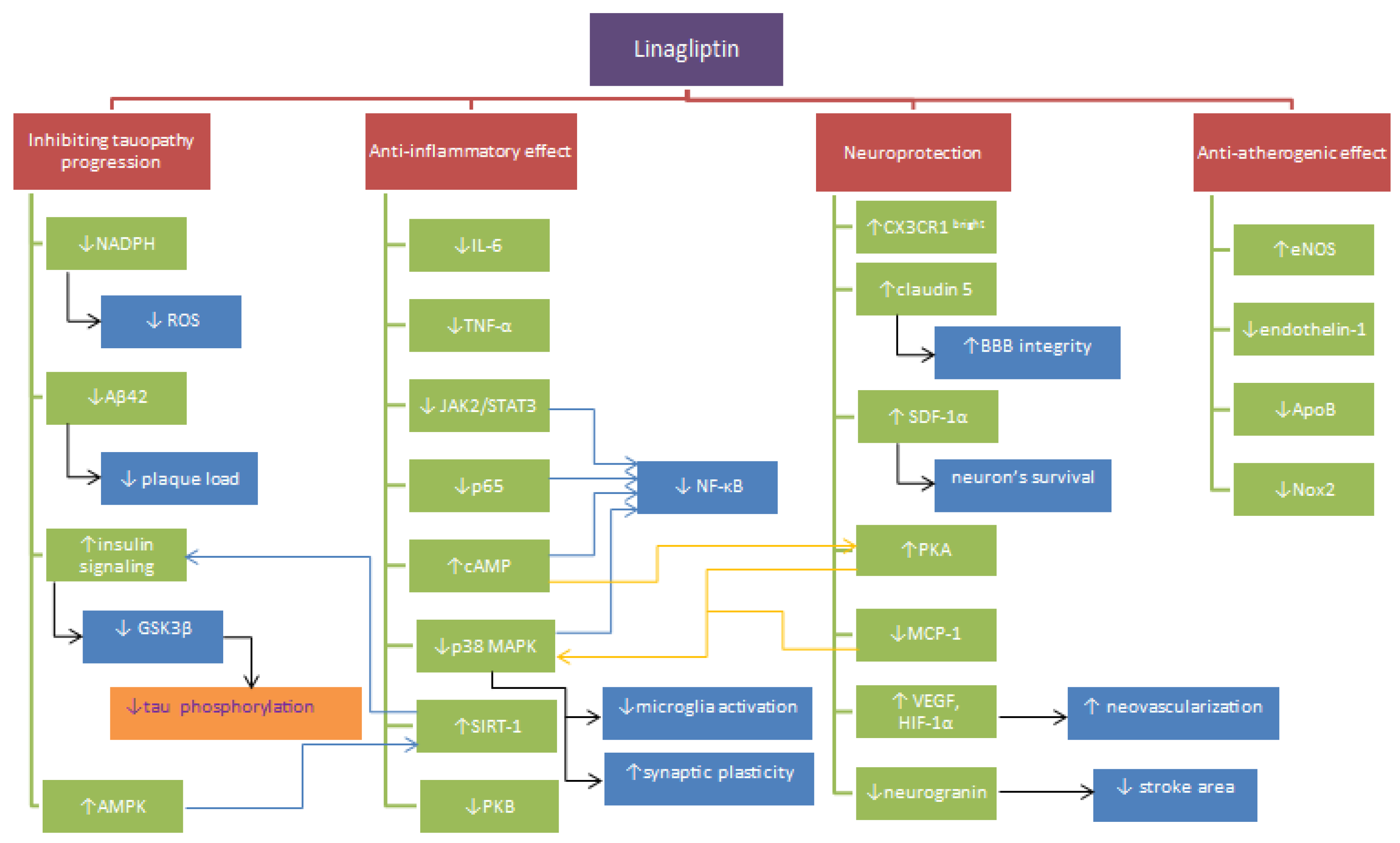{kind=link}
| Authors | Subject of Study | Dose of Linagliptin | Results |
|---|---|---|---|
| Kornelius et al. (2015) [11] | SK-N-MC human neuronal cells | 10–100 μM of linagliptin for 24 h | ↓ Aβ-induced cytotoxicity, ↓ GSK3β, ↓ ROS, ↓ hyper p-tau |
| Ma et al. (2015) [12] | rBMVECs | 0.083 g/kg diet for 8 weeks ater BCCAO | ↓ cognitive impairment, ↓ stroke volume, ↓ COS |
| Mi et al. (2018) [13] | rBMVECs | 40 nM | ↑ VEGF, ↑ eNOS, ↑ HIF-1α, ↑ SIRT1, |
| Nakamura et al. (2016) [14] | HUVECs | 1, 5, 10, 50, and 100 nM 1 h prior to incubation with LPS | ↓ IL-6, ↓ p-p38 MAPK ↓ p65 |
| Nakamura et al. (2016) [15] | HUVECs | 1 h 50 nM after 1 h 1 μg/mL LPS together with 50 nM linagliptin, | ↑ PKA, ↑ PKC, ↑ cAMP, ↓ PKB ↓ ROS |
| Yamadera et al. (2018) [16] | U937 cells | 1, 5, 10, 50, or 100 nM | ↓ IL-6, ↓ TNF-α |
| Salheen et al. (2015) [17] | STZ-induced diabetic rats | 2 mg/kg/ day for 4 weeks | ↑ NO, ↑ EDR, ↓ NADPH, ↑ Nox2 |
| Darsalia et al. (2013) [18] | C57BL/6 mice | 10 mg/kg/day for 4 weeks before and 3 weeks after MCAO | ↑ survival of neurons |
| Darsalia et al. (2014) [19] | C57BL/6 mice | 10 mg/kg/day for 4 weeks before and 3 weeks after MCAO | ↑ NSCs proliferation |
| Elbaz et al. (2018) [20] | C57BL/6 mice | 10 mg/kg/day for 3 weeks after 2 (from 3) weeks cuprizone administration | ↓ p-JAK2, ↑ p-AMPK, ↓ p-STAT3, ↓ NF-κB p65, ↑ SIRT1. |
| Kosaraju et al. (2017) [21] | 3xTg-AD mouse | 5, 10, and 20 mg/kg/day for 8 weeks. | ↑ Cognitive Performance, ↓ Aβ42, ↓ hyper p-tau |
| Salim et al. (2016) [22] | ApoE(−/−) mice | 10 mg/kg/day for 20 weeks | ↓ VCAM-1 ↓ MCP-1 ↓ NADPH |
| Hardigan et al. (2016) [23] | Male type-2 diabetic GK rats | 83 mg/kg for one week, next 166mg/kg for three weeks | ↓ ET-1, ↓ TLR2 |
| Chiazza al. (2018) [24] | C57BL/6 mice | varied at every stage of the experiment | ↑ post stroke rehabilitation ↑ SDF-1α ↓ stroke volume |
| Fadini et al. (2016) [25] | Diabetes type 2 patients | 5 mg per day for 4 days | ↑ SDF-1α, ↑ CX3CR1bright, ↓ MCP-1, ↓ CCL22, ↓ IL-12 |
| Shigiyama et al. (2015) [26] | Diabetes type 2 patients | 750 mg/day metformin + 5 mg/day linagliptin for 16 weeks | ↓ Apo B |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiciński, M.; Górski, K.; Walczak, M.; Wódkiewicz, E.; Słupski, M.; Pawlak-Osińska, K.; Malinowski, B. Neuroprotective Properties of Linagliptin: Focus on Biochemical Mechanisms in Cerebral Ischemia, Vascular Dysfunction and Certain Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 4052. https://doi.org/10.3390/ijms20164052
Wiciński M, Górski K, Walczak M, Wódkiewicz E, Słupski M, Pawlak-Osińska K, Malinowski B. Neuroprotective Properties of Linagliptin: Focus on Biochemical Mechanisms in Cerebral Ischemia, Vascular Dysfunction and Certain Neurodegenerative Diseases. International Journal of Molecular Sciences. 2019; 20(16):4052. https://doi.org/10.3390/ijms20164052
Chicago/Turabian StyleWiciński, Michał, Karol Górski, Maciej Walczak, Eryk Wódkiewicz, Maciej Słupski, Katarzyna Pawlak-Osińska, and Bartosz Malinowski. 2019. "Neuroprotective Properties of Linagliptin: Focus on Biochemical Mechanisms in Cerebral Ischemia, Vascular Dysfunction and Certain Neurodegenerative Diseases" International Journal of Molecular Sciences 20, no. 16: 4052. https://doi.org/10.3390/ijms20164052
APA StyleWiciński, M., Górski, K., Walczak, M., Wódkiewicz, E., Słupski, M., Pawlak-Osińska, K., & Malinowski, B. (2019). Neuroprotective Properties of Linagliptin: Focus on Biochemical Mechanisms in Cerebral Ischemia, Vascular Dysfunction and Certain Neurodegenerative Diseases. International Journal of Molecular Sciences, 20(16), 4052. https://doi.org/10.3390/ijms20164052