1. Introduction
Two-pore domain potassium (K
2P) channels have a major role in the regulation of cell excitability and membrane potential in excitable and non-excitable cells [
1]. K
2P channels contain two pore-forming loops and four transmembrane domains per subunit, creating dimeric channels [
2]. The TASK (tandem of pore domains in a weak inwardly rectifying K
+ channel [TWIK]-related acid-sensitive K
+ channel) subfamily is integrated by TASK-1 [
3], TASK-3 [
4] (sharing 58.9% of aminoacidic (aa) sequence identity [
5]), and TASK-5 (sharing 51.4 % and 55.1% of aa sequence identity with TASK-1 and TASK-3, respectively) [
6]. Some studies have demonstrated that TASK channels participate in the chemical control of breathing due to their intrinsic pH and O
2 sensitivity [
7,
8,
9]. These channels are expressed in the nervous, cardiovascular, genitourinary, and gastrointestinal systems [
10]. They are involved in chemosensation [
11] and also have a role in the regulation of the immune system [
12]. TASK channels are acid-sensitive and anesthetic-activated members of the K
2P family. They contribute to the effects of general anesthetics due to the activation of background K
+ currents causing a decrease of excitability by neuronal hyperpolarization [
13], which makes these channels prominent molecular targets for these drugs.
K
2P channels exhibit a different topology and 3D-structure in relation with other K
+ channels. The recently released crystallographic structures of some K
2P channels, such as TRAAK (PDBs:3UM7 [
5] and 4I9W [
14]), TREK-1 (PDB:4TWK, 6CQ6 and 6CQ8 [
15]), TREK-2 (PDBs:4BW5, 4XDJ, 4XDK and 4DKL [
16]), and TWIK-1 (PDB:3UKM [
17]), show that these proteins exhibit some features that characterize their unique gating and ion permeation properties. For example, close to the membrane center, the TM2 helix bends approximately 20°. This structural change generates two side-cavities named fenestrations, connecting the pore to the hydrophobic core of the membrane [
5,
17]. These fenestrations have a key role in the modulation of K
2P channels [
18], providing binding pockets for drugs like norfluoxetine in TREK-2 [
16] and bupivacaine in TASK-1 [
19]. It has been proven by molecular dynamic simulations that drugs like A1899 and PK-THPP bind preferentially to TASK-1 and TASK-3 channels with open fenestrations, respectively [
18,
20]. Thus, these hydrophobic cavities are potential drug-binding sites, and also might provide new pathways that could guide blockers into their binding site.
TASK-3 is highly expressed in the hippocampus, cerebellum, and cortex [
21], and some previous studies have described that TASK-3 regulates neurotransmitter function [
22]. The development of new selective TASK-3 modulators could influence the pharmacological treatment of several neurological conditions, such as sleep disorders, neurodegeneration, cognitive impairment, Huntington’s disease, Parkinson’s disease, or major depressive disorder [
23].
Not many promising inhibitory TASK-3 modulators have been reported in the scientific literature [
24]. One of the few reports in this context was made by Coburn et al. [
25]; they informed the use of aminopyrimidine derivatives as potent TASK-3 blockers. Noriega-Navarro et al. [
26] reported the application of dihydropyrrolo[2,1-a]isoquinoline derivatives (DPIs) as novel TASK inhibitors. In this sense, the use of fused heterocyclic-compounds has attracted attention as new TASK modulators. Therefore, the development of simple theoretical/experimental methodologies is necessary to find new compounds with a different chemical nature with potential usefulness as TASK-3 modulators.
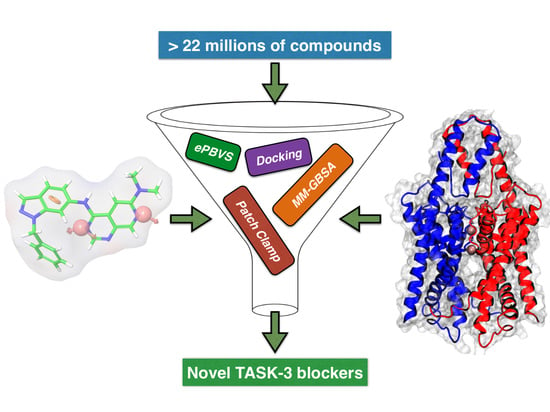
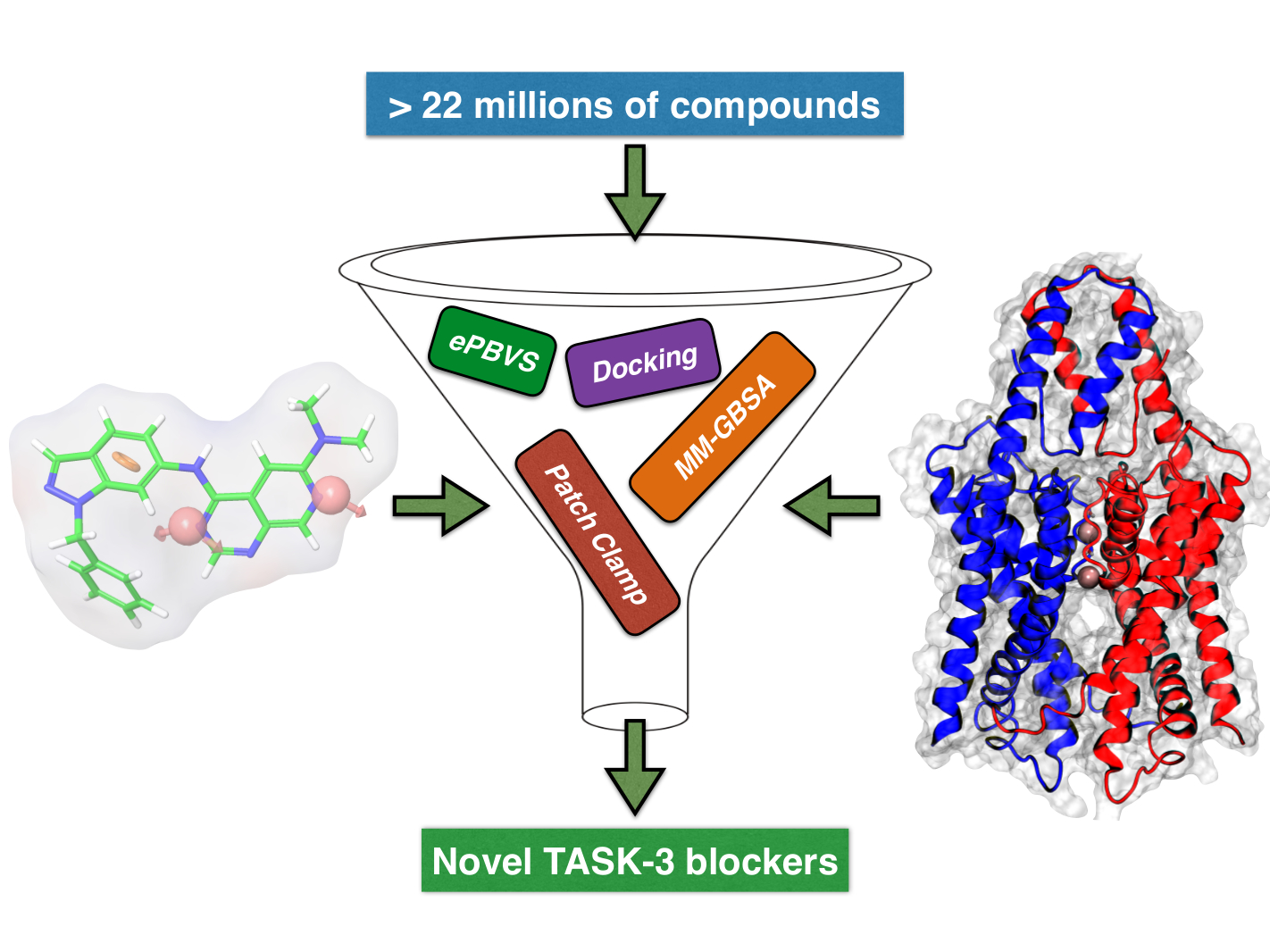
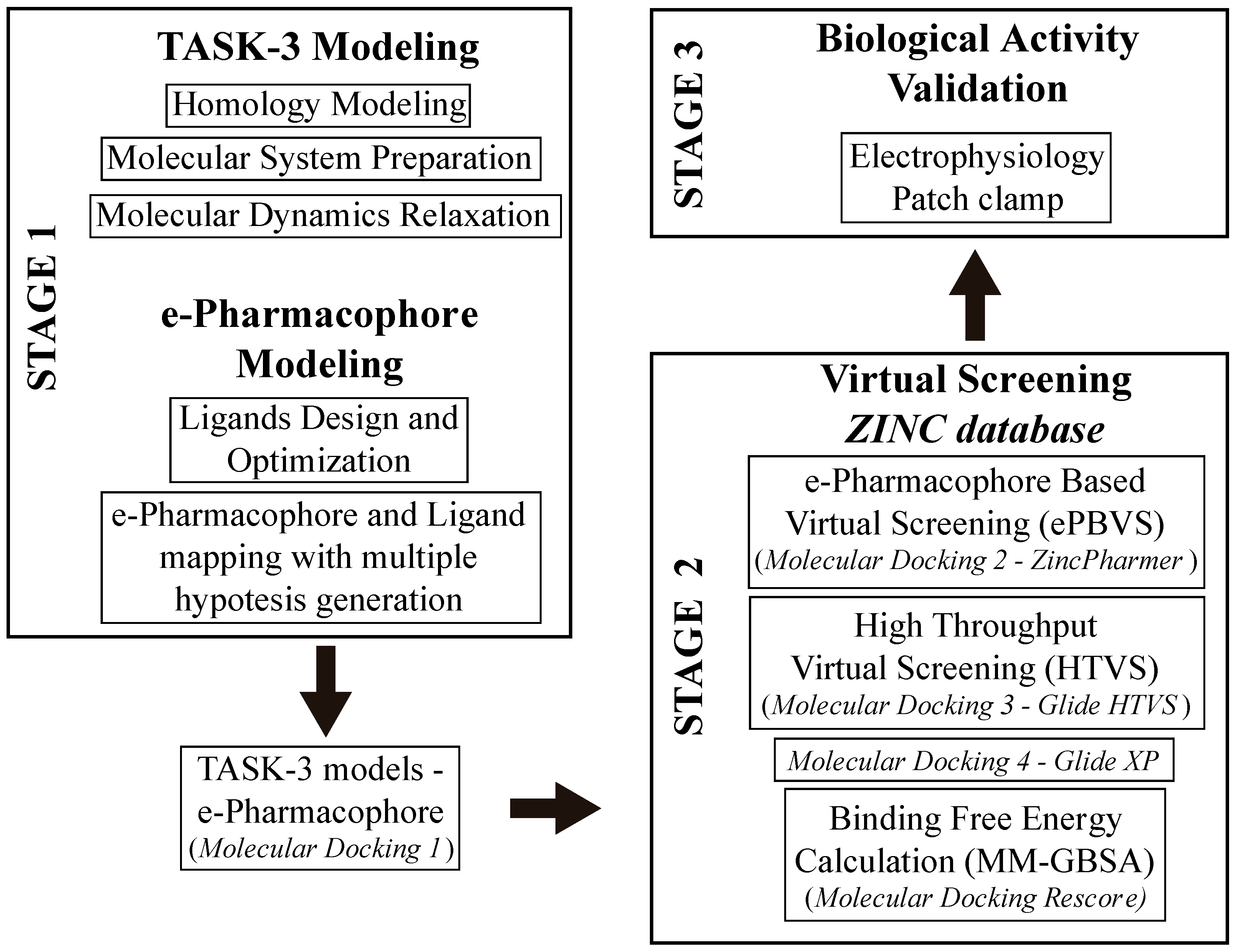
In this study, we developed a systematic pipeline (
Scheme 1) to search novel TASK-3 blockers that includes a pharmacophore-based virtual screening (
e-PBVS). The 19 putative blockers found were screened against human TASK-3 by a patch clamp, obtaining one active ligand that exhibited inhibitory activity against TASK-3 in the μM range. The active ligand was used as a scaffold and a new compound was designed, synthetized, and tested against TASK-3, exhibiting a four-fold higher activity.
3. Discussion
There has been no information of computational screening targeting the central cavity and/or fenestrations of K
2P channels. Recently, Luo et. al. developed a virtual screening but in the extracellular cap of K
2P channels to identify inhibitors targeting this site [
38]. Nevertheless, due to their pharmacological potential as protein targets in diverse diseases, much evidence has been recently accumulated regarding the molecular characteristics underlying the interactions between different compounds and K
2P channels [
39].
In the current work, we proposed a protocol that includes pharmacophore-based virtual screening, docking-based high throughput virtual screening, re-docking to refine poses, and binding free energy calculations to find new inhibitors for TASK-3 channels. The success in the finding of hit compounds (DR1 to DR19) indicates that our assumptions in the ‘e-Pharmacophore hypothesis in conjunction with the binding in our four TASK-3 models’ (derived by using information of the known blockers) were correct and useful for the identification of novel active compounds (e.g., DR16), whose activity can be further optimized (e.g., DR16.1).
We also identified the putative residues involved in the binding site of the studied compounds and our results contain some residues of the previously identified A1899 [
18,
30,
35] and PK-THPP (compound 23 in this study) [
20,
40] binding sites.
DR16 and
DR16.1 share 11 residues in their putative binding site in TASK-3 channels and 63.6% of these residues belong to the binding site of A1899. Within this 63.6%, three residues are also part of the PK-THPP binding site (
Table S2). However, a mutagenesis study is mandatory to confirm the binding site of these new drugs.
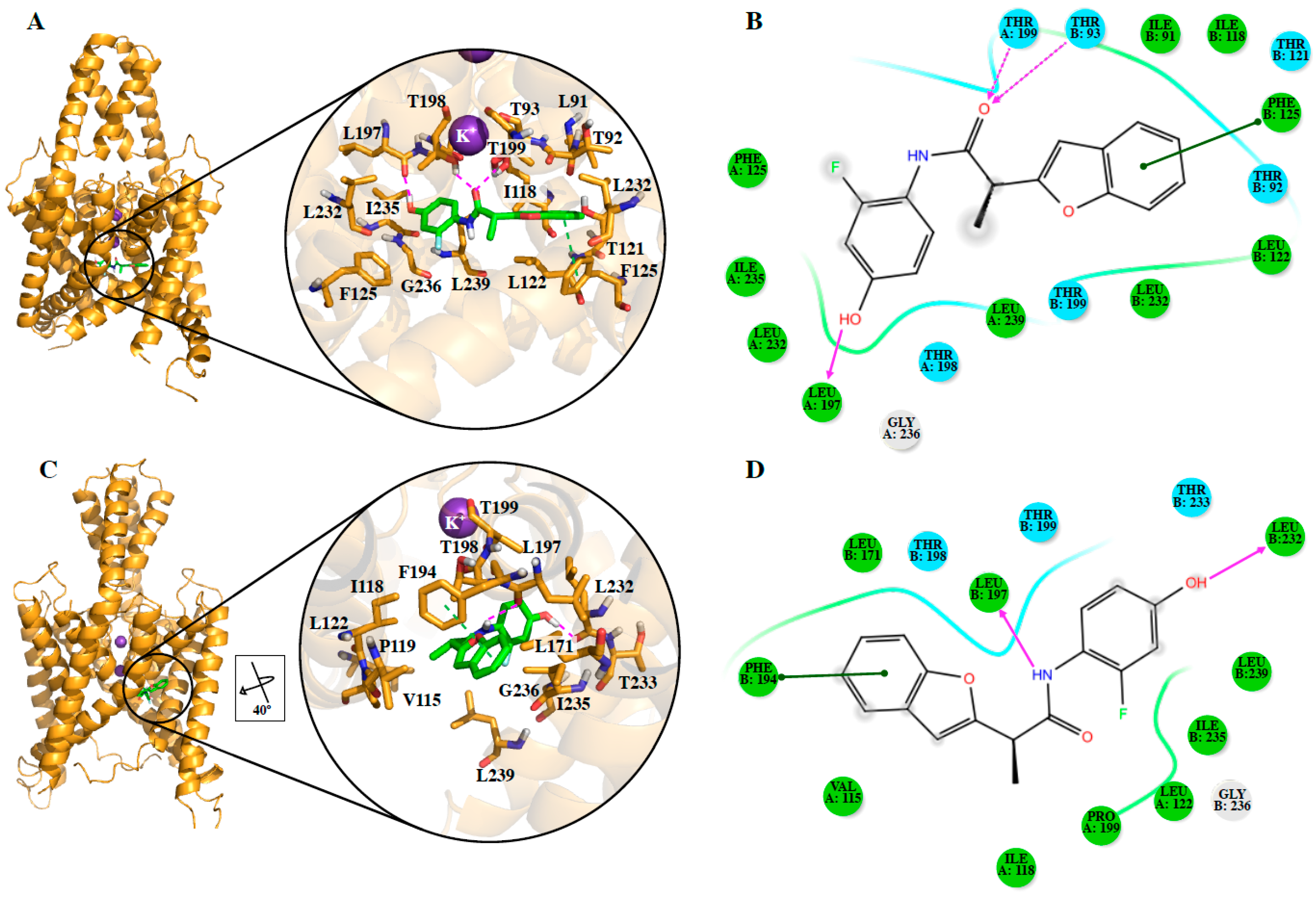
Since it is not possible to know a priori the preferred conformation for including TASK-3 modulators, both closed and opened states of the channel, in the different TASK-3 homology models generated (
Table 1,
Figure S1A), were considered. Analysis of the different fenestration states in T3-trCO (
Figure S1B) shows how the hydrophobic interactions between Leu239 (TM4 segment) with Leu197 (TM2 segment) and Val115 (inner helix 1), as well as between Ile235 (TM4 segment) with Leu197 and Val115, modulate the fenestration opening–closing mechanism. These interactions are in concordance with the results presented by Brohawn et al. [
14], where the residues Leu151, Leu236, Ile279, and Leu283 of TRAAK are implicated in the opening–closing mechanism of the TRAAK fenestration [
41] (TASK-3 residues Val115, Leu197, Ile235, and Leu239 are equivalent to TRAAK residues Leu151, Leu236, Ile279, and Leu283, respectively). As observed on previously reported structures of K
2P channels, TASK-3 fenestrations are cavities formed by hydrophobic residues [
41,
42,
43], and the hydrophobic regions of the ligands can be included in these cavities. In our report, ligands
DR16 and
DR16.1 establish hydrophobic interactions with Leu239 and Ile235 at the fenestrations. Also,
DR16 interacts hydrophobically with Val115 and through a hydrogen bond with Leu 197 (
Figure 3D). With Leu197,
DR16.1 established hydrophobic interactions (
Figure 4B). The mutagenesis study previously suggested could reveal whether these new drugs interfere with the opening/closed mechanism at the fenestrations of TASK channels in a similar way as bupivacaine could do it in TASK-1 [
19].
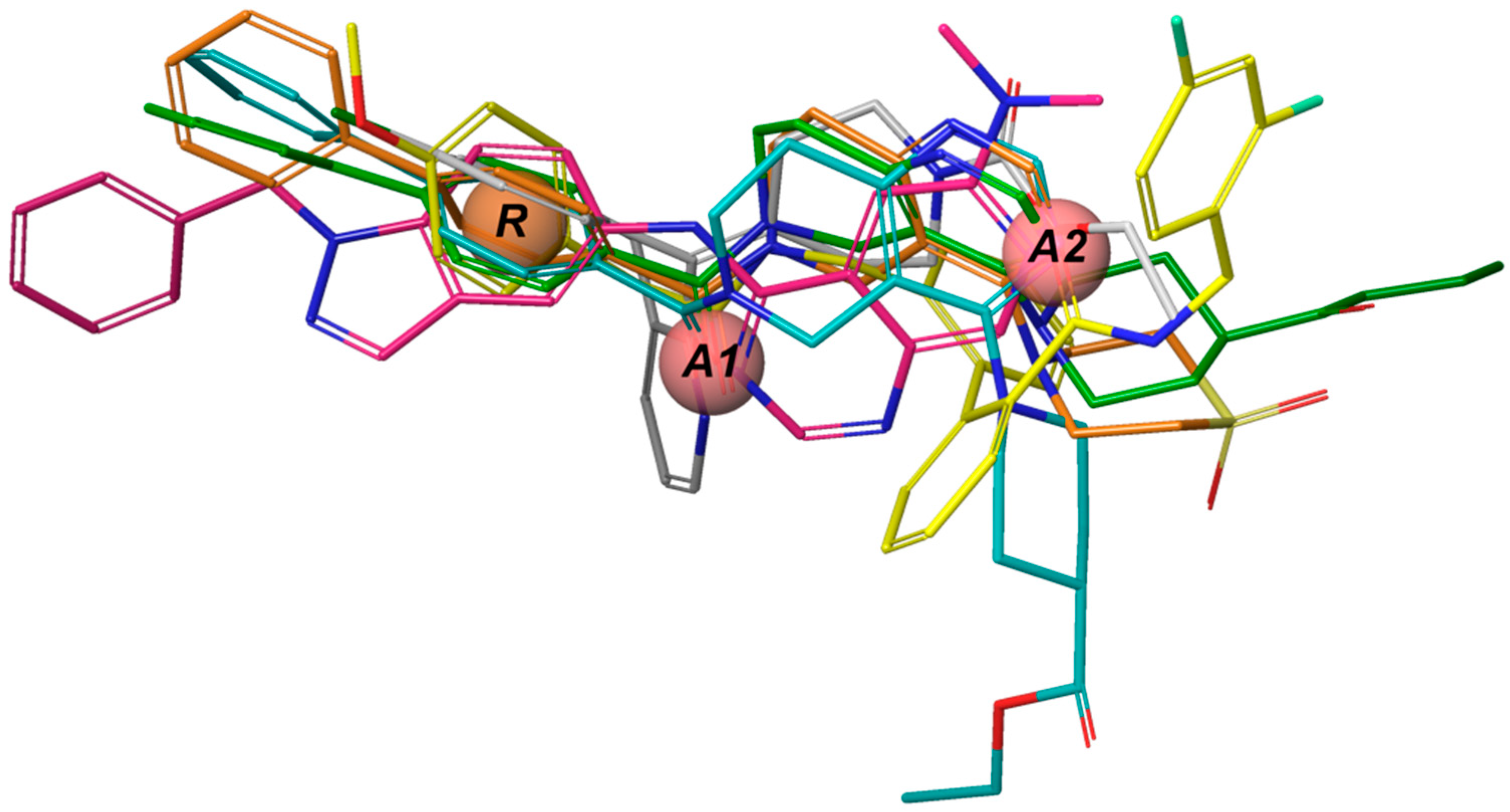
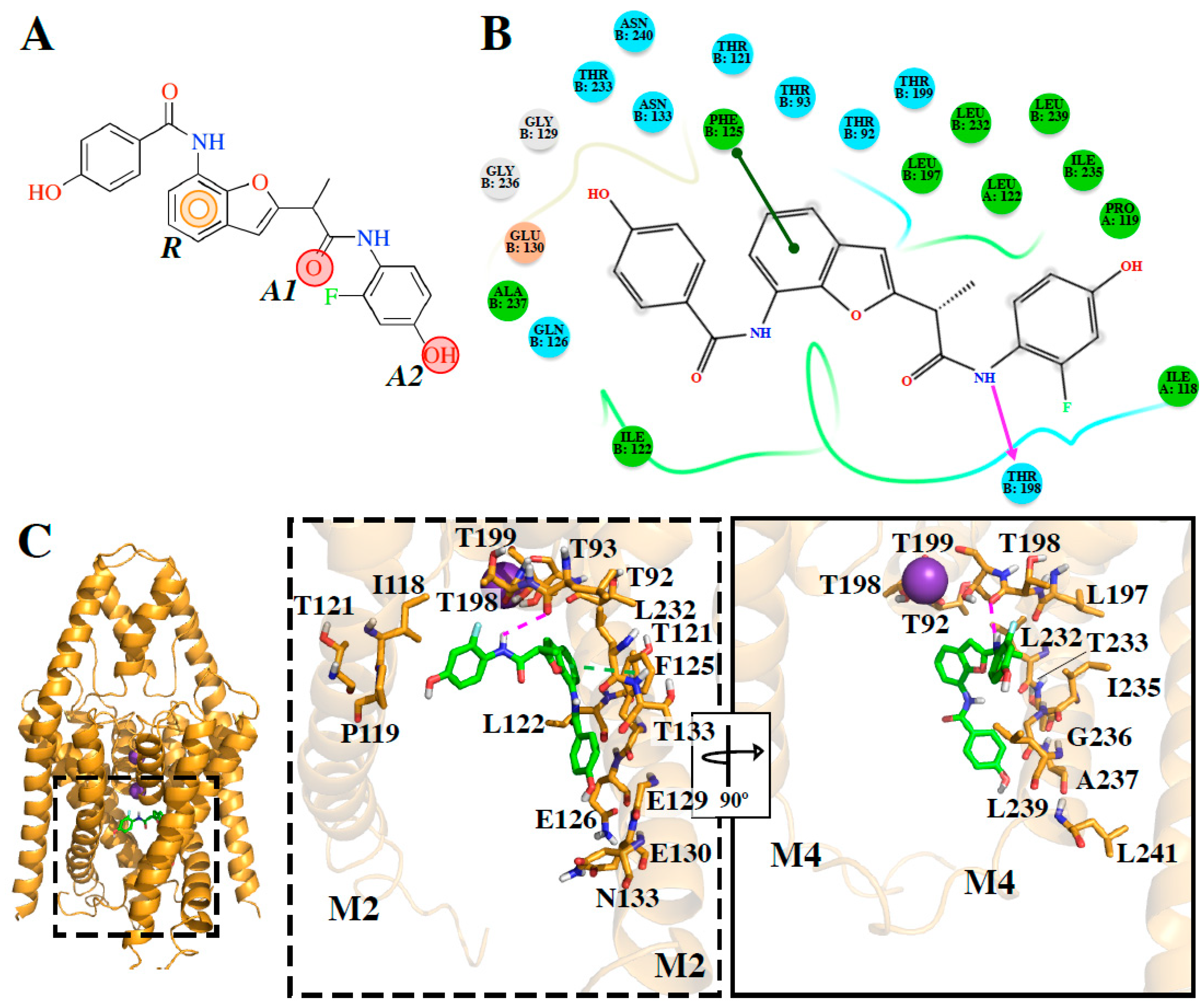
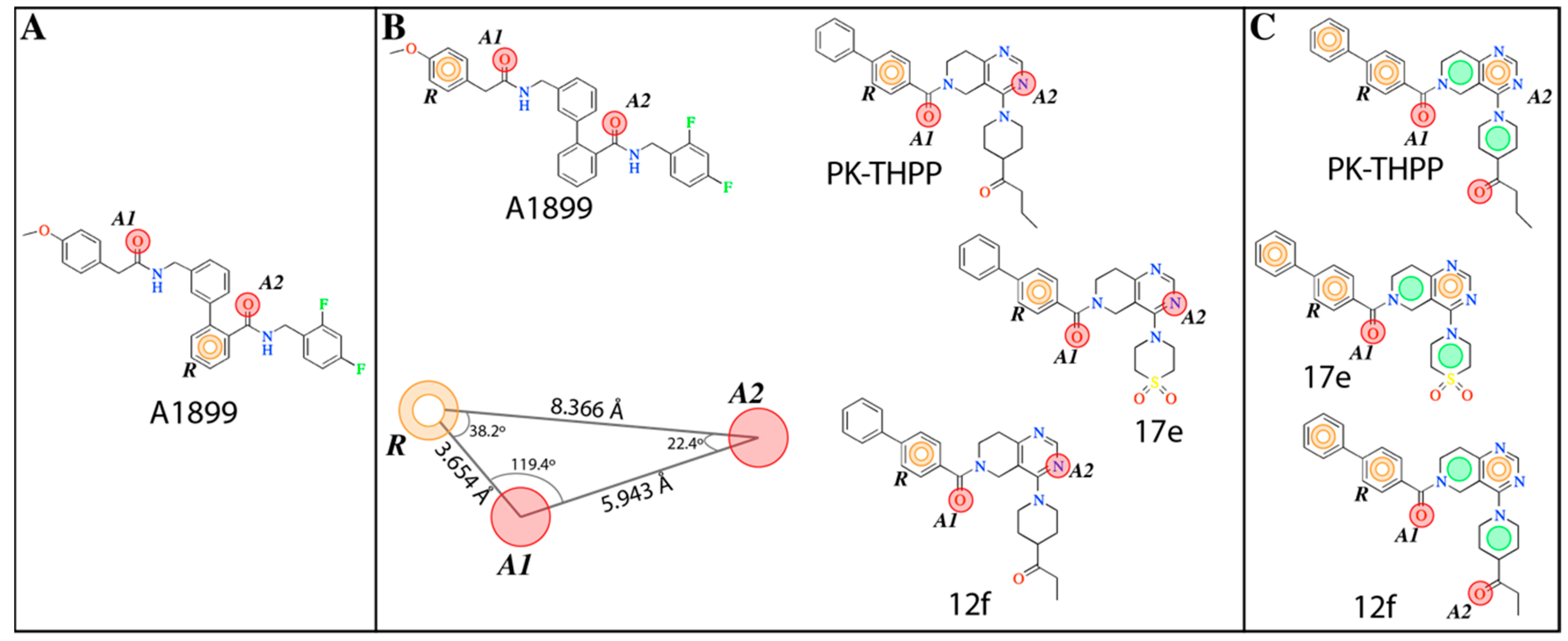
The
e-Pharmacophore model derived from the 12 blockers of
Table 2 presents two hydrogen bond acceptors (
A1 and
A2) and one aromatic ring (
R) (
Figure 1). These results are in concordance with those previously reported by our group [
35] since the common pharmacophore identified for TASK-1 and Kv1.5 blockers is similar to our model. However, it differs in the position of the aromatic ring (
Figure 6A,B). Also,
A1 and
R groups are contained in the shared seven-point pharmacophore
RRAHRHA of the 5,6,7,8 tetrahydropyrido[4,3-d]pyrimidine derivatives (
Figure 6B,C). Comparing the three pharmacophores, they will differ because they define the interaction patterns of three different groups of bioactive molecules that interact with TASK channels: Those that interact simultaneously with TASK-1 and K
V1.5 channels (
Figure 6A) [
34], those that interact with high affinity with TASK-3 channels (
Figure 6C) [
20,
24], and the pharmacophore reported and used here for TASK-3 blockers with different affinities (
Figure 6B). This comparison reflects that a pharmacophore is, definitely, a quantitative measure of molecular similarity. However, some features must be shared between these three groups of molecules, for example, the hydrogen bond acceptor groups that can establish interactions with the threonines of the selectivity filter of TASK channels [
20,
30].
After pharmacophore identification,
e-Pharmacophore-based virtual screening (
e-PBVS) was performed to select which compounds of the ZINC database (>22 million) fitted the model requirements; then, a docking-based HTVS following by a re-docking process with a more precise function and binding free energy calculations (MM-GBSA) were done to finally obtain 19 ligands (DR1–DR19 in
Table 4 and
Figure S4). These hits contain at least two rigid aromatic units connected by amide or ester groups (except DR7), which act as linkers. They also have H-bond acceptor groups and hydrophobic groups that could interact with the hydrophobic residues of the TASK-3 binding site. These common chemical features between the obtained hits and the previously reported blockers allowed us to perform the experimental evaluation of the identified compounds.
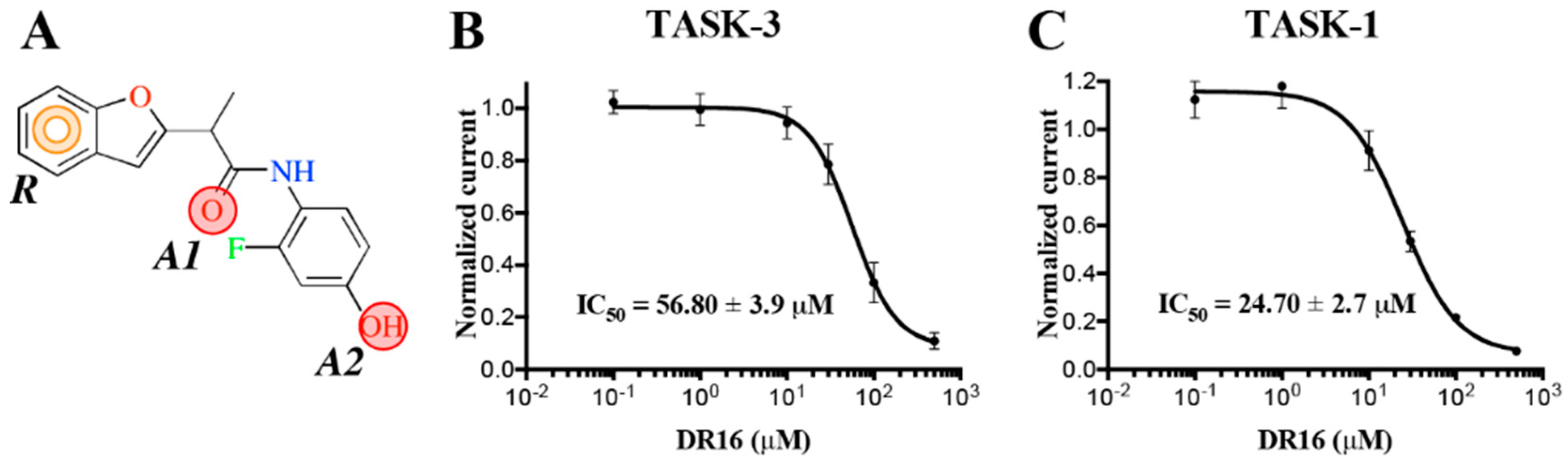
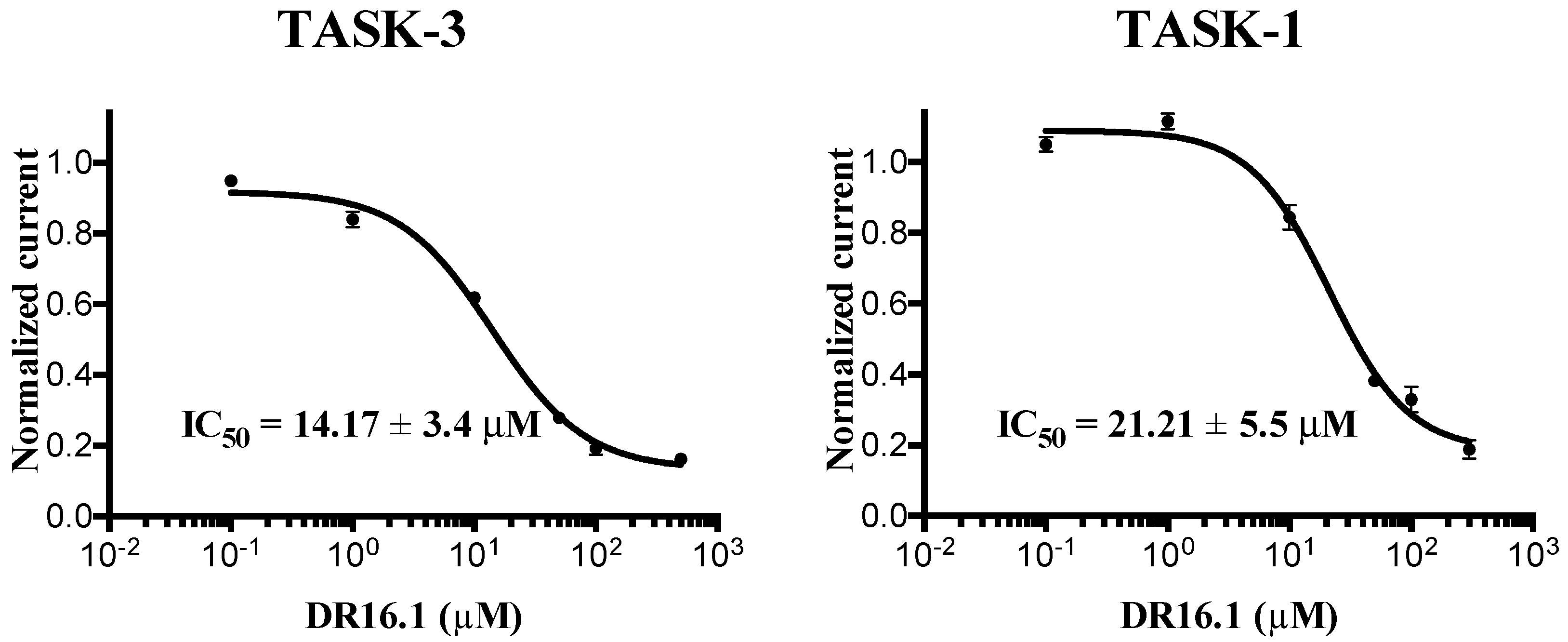
The experimental activity evaluation was performed using patch clamp. We obtained one lead ligand from the 19 tested:
DR16 (IC
50 = 56.8 ± 3.9 μM); this compound binds TASK-3 with a 1000-fold lower affinity with respect to the most active compounds of the THPP series (compound 23) [
20,
25], the most active compound reported to date. However, its activity is in the same IC
50 range with respect to other reported blockers, such as dihyro-β-erythromidine, doxapram, GW2974, L-703,606, loratadine, mevastatin, mibefradil, and octoclothepin (
Table 2). Our results are in concordance with those reported by several authors where novel modulators have been identified through virtual screening and/or molecular docking simulations [
25,
44,
45,
46,
47,
48,
49,
50,
51], with a successful result in the prediction of compounds with the same biological activity range of the reported modulators used to construct the pharmacophore model. Using
DR16, we designed a derivative (
DR16.1) converging the common pharmacophore identified in TASK-3 blockers (
Figure 1) and present in
DR16 (
Figure 2) as well. This novel inhibitor has an IC
50 = 14.17 ± 3.4 and 21.21 ± 5.5 µM against TASK-3 and TASK-1, respectively. Both inhibitors,
DR16 and
DR16.1, presented ADME/tox (Administration, Distribution, Metabolism, Excretion and Toxicity) properties in accepted ranges for druggability, which suggests that scaffold modifications of these molecules can lead to drug-like compounds. Thus, these two compounds are part of the reduced number of K
2P channel modulators reported until today and they are novel scaffolds that could be chemically optimized in the future to get more potent TASK modulators.
DR16.1 activity is four times better than
DR16 activity. The putative binding site of
DR16.1 (
Figure 4B) exhibits 11 different residues from the putative binding site of
DR16. Within them, more than 50% are residues of the binding site of high affinity compounds, A1899 and PK-THPP (
Table S2). Another factor that could increase the activity of
DR16.1 in TASK-3 could be the hydrogen bond established with Thr198, which is not present in
DR16. Thr198 is at the S4 site of the selectivity filter and we observed recently that the marked difference in the potency of compounds of THPP series (blocking TASK-3 in the nanomolar range), with respect to some less potent compounds of the series (inhibiting TASK-3 channels in the micromolar range), is due to the presence of a hydrogen bond interaction with a threonine of the selectivity filter [
20]. Hydrogen bond interactions with threonines of the selectivity filter are also essential for A1899 binding in the TASK-1 channel [
18].
The experimental activity also reveals that
DR16.1 increased its activity four times in TASK-3 but not in TASK-1 with respect to
DR16. We hypothesize that this is due to the interactions that appear in
DR16.1 with residues of the binding site of high-affinity TASK-3 compounds, such as PK-THPP, that are not present in
DR16 (i.e., Gln126, Ala237,
Table S2).
The results presented in this work allow us to conclude that interactions of the identified compounds could be established in the TASK-3 inner cavity and the fenestrations. However, a mutagenesis approach is certainly needed to discern the role of both cavities in the binding sites.
DR16 shows a binding similar to norfluoxetine and bupivacaine, which are located in the fenestrations of TREK-2 [
16] and TASK-1 [
19], respectively.
DR16.1 orientation is similar to THPP in TASK-3 [
20] or A1899 in TASK-1 [
18].

 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











