Genomics and the Acute Respiratory Distress Syndrome: Current and Future Directions


 and
and 
Abstract
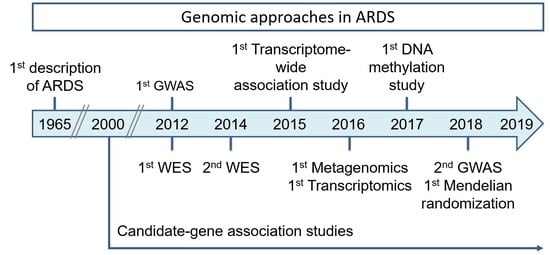
1. Definition and Epidemiology
2. Molecular Pathophysiology
3. Genetic Association Studies
4. Causal Inferences with Mendelian Randomization
5. Transcriptomics
6. Metagenomics
7. Other Incipient Genomic Approaches
8. Future Directions
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- ARDS Definition Task Force; Ranieri, V.M.; Rubenfeld, G.D.; Thompson, B.T.; Ferguson, N.D.; Caldwell, E.; Fan, E.; Camporota, L. Acute Respiratory Distress Syndrome, The Berlin Definition. JAMA 2012, 307, 2526–2533. [Google Scholar]
- Villar, J.; Blanco, J.; Kacmarek, R.M. Current incidence and outcome of the acute respiratory distress syndrome. Curr. Opin. Crit. Care 2016, 22, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology, patterns of care and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA J. Am. Med. Assoc. 2016, 315, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Aeffner, F.; Bolon, B.; Davis, I.C. Mouse models of acute respiratory distress syndrome: A Review of Analytical Approaches, Pathologic Features and Common Measurements. Toxicol. Pathol. 2015, 43, 1074–1092. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Liu, L.; Yang, Y.; Yu, W.; Li, M.; Yu, K.; Zheng, R.; Yan, J.; Wang, X.; Cai, G.; et al. A modified acute respiratory distress syndrome prediction score: A multicenter cohort study in China. J. Thorac. Dis. 2018, 10, 5764–5773. [Google Scholar] [CrossRef] [PubMed]
- Ashbaugh, D.G.; Bigelow, D.B.; Petty, T.L.; Levine, B.E. Acute Respiratory Distress in Adults Assistant in Medicine and American Thoracic Society-National Tuberculosis Association Fellow in Pulmonary Disease. Lancet 1967, 2, 319–323. [Google Scholar] [CrossRef]
- Kollef, M.H.; Schuster, D.P. Medical Progress the Acute Respiratory Distress Syndrome. Med. Prog. 1995, 332, 27–37. [Google Scholar]
- Shaw, T.D.; McAuley, D.F.; O’Kane, C.M. Emerging drugs for treating the acute respiratory distress syndrome. Expert Opin. Emerg. Drugs 2019, 24, 29–41. [Google Scholar] [CrossRef]
- Gong, M.N.; Thompson, B.T. Acute respiratory distress syndrome: Shifting the emphasis from treatment to prevention. Curr. Opin. Crit. Care 2016, 22, 21–37. [Google Scholar] [CrossRef]
- Nieman, G.F.; Gatto, L.A.; Bates, J.H.T.; Habashi, N.M. Mechanical ventilation as a therapeutic tool to reduce ards incidence. Chest 2015, 148, 1396–1404. [Google Scholar] [CrossRef]
- García-Laorden, M.I.; Lorente, J.A.; Flores, C.; Slutsky, A.S.; Villar, J. Biomarkers for the acute respiratory distress syndrome: How to make the diagnosis more precise. Ann. Transl. Med. 2017, 5, 283. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.J.; Biswas, R.S.; Mehta, H.J.; Joo, M.; Sadikot, R.T. Alternative and Natural Therapies for Acute Lung Injury and Acute Respiratory Distress Syndrome. Biomed Res. Int. 2018, 2018, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Plouët, J.; Schilling, J.; Gospodarowicz, D. Isolation and characterization of a newly identified endothelial cell mitogen produced by AtT-20 cells. EMBO J. 1989, 8, 3801–3806. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Carver-Moore, K.; Chen, H.; Dowd, M.; Lu, L.; O’Shea, K.S.; Powell-Braxton, L.; Hillan, K.J.; Moore, M.W. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 1996, 380, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Barratt, S.; Medford, A.R.; Millar, A.B. Vascular endothelial growth factor in acute lung injury and acute respiratory distress syndrome. Respiration 2014, 87, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Abadie, Y.; Bregeon, F.; Papazian, L.; Lange, F.; Chailley-Heu, B.; Thomas, P.; Duvaldestin, P.; Adnot, S.; Maitre, B.; Delclaux, C. Decreased VEGF concentration in lung tissue and vascular injury during ARDS. Eur. Respir. J. 2005, 25, 139–146. [Google Scholar] [CrossRef]
- Ourradi, K.; Blythe, T.; Jarrett, C.; Barratt, S.L.; Welsh, G.I.; Millar, A.B. VEGF isoforms have differential effects on permeability of human pulmonary microvascular endothelial cells. Respir. Res. 2017, 18, 116. [Google Scholar] [CrossRef] [PubMed]
- Becker, P.M.; Verin, A.D.; Booth, M.A.; Liu, F.; Birukova, A.; Garcia, J.G.N. Differential regulation of diverse physiological responses to VEGF in pulmonary endothelial cells. Am. J. Physiol. Cell. Mol. Physiol. 2017, 281, L1500–L1511. [Google Scholar] [CrossRef]
- Khadaroo, R.G.; Marshall, J.C. ARDS and the multiple organ dysfunction syndrome. Common mechanisms of a common systemic process. Crit. Care Clin. 2002, 18, 127–141. [Google Scholar] [CrossRef]
- Lipke, A.B.; Matute-Bello, G.; Herrero, R.; Wong, V.A.; Mongovin, S.M.; Martin, T.R. Death receptors mediate the adverse effects of febrile-range hyperthermia on the outcome of lipopolysaccharide-induced lung injury. Am. J. Physiol. Cell. Mol. Physiol. 2011, 301, L60–L70. [Google Scholar] [CrossRef]
- Herold, S.; Tabar, T.S.; Janssen, H.; Hoegner, K.; Cabanski, M.; Lewe-Schlosser, P.; Albrecht, J.; Driever, F.; Vadasz, I.; Seeger, W.; et al. Exudate macrophages attenuate lung injury by the release of IL-1 receptor antagonist in gram-negative pneumonia. Am. J. Respir. Crit. Care Med. 2011, 183, 1380–1390. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.C.; Chiu, W.C.; Yeh, C.L.; Yeh, S.L. Glutamine modulates lipopolysaccharide-induced activation of NF-κB via the Akt/mTOR pathway in lung epithelial cells. Am. J. Physiol. Cell. Mol. Physiol. 2011, 302, L174–L183. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, S.; Voulgarelis, M. Toll-Like Receptors, Tissue Injury and Tumourigenesis. Mediat. Inflamm. 2010, 2010, 1–9. [Google Scholar] [CrossRef]
- Sun, S.; Sursal, T.; Adibnia, Y.; Zhao, C.; Zheng, Y.; Li, H.; Otterbein, L.E.; Hauser, C.J.; Itagaki, K. Mitochondrial DAMPs Increase Endothelial Permeability through Neutrophil Dependent and Independent Pathways. PLoS ONE 2013, 8, e59989. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.L.; King, M.B.; Gonzalez, R.P.; Brevard, S.B.; Frotan, M.A.; Gillespie, M.N.; Simmons, J.D. Blood transfusion products contain mitochondrial DNA damage-associated molecular patterns: A potential effector of transfusion-related acute lung injury. J. Surg. Res. 2014, 191, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Tolle, L.B.; Standiford, T.J. Danger-associated molecular patterns (DAMPs) in acute lung injury. J. Pathol. 2013, 229, 145–156. [Google Scholar] [CrossRef]
- Kumar, V. Inflammation research sails through the sea of immunology to reach immunometabolism. Int. Immunopharmacol. 2019, 73, 128–145. [Google Scholar] [CrossRef]
- Ren, M.; Wang, Y.M.; Zhao, J.; Zhao, J.; Zhao, Z.M.; Zhang, T.F.; He, J.; Ren, S.P.; Peng, S.Q. Metallothioneins attenuate paraquat-induced acute lung injury in mice through the mechanisms of anti-oxidation and anti-apoptosis. Food Chem. Toxicol. 2014, 73, 140–147. [Google Scholar] [CrossRef]
- Qi, X.L.; Hao, J.; Huang, L.J.; Wu, S.; Ma, H.H.; Ye, Z.Q.; He, H.B.; Li, S.W.; Li, C.E.; Huang, X. Apoptotic mechanisms in rabbits with blast-induced acute lung injury. Acta Cir. Bras. 2018, 33, 896–903. [Google Scholar] [CrossRef]
- Fang, Y.; Gao, F.; Hao, J.; Liu, Z. MicroRNA-1246 mediates lipopolysaccharide-induced pulmonary endothelial cell apoptosis and acute lung injury by targeting angiotensin-converting enzyme 2. Am. J. Transl. Res. 2017, 9, 1287–1296. [Google Scholar]
- Flores, C.; Pino-Yanes, M.; Villar, J. A quality assessment of genetic association studies supporting susceptibility and outcome in acute lung injury. Crit. Care 2008, 12, R130. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Herrera, M.; Pino-Yanes, M.; Perez-Mendez, L.; Villar, J.; Flores, C. Assessing the quality of studies supporting genetic susceptibility and outcomes of ARDS. Front. Genet. 2014, 5, 2008–2013. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Guillén-Guío, B.; Acosta-Herrera, M.; Villar, J.; Flores, C. Genetics of Acute Respiratory Distress Syndrome. eLS 2016, 1–9. [Google Scholar] [CrossRef]
- Clark, M.F.; Baudouin, S.V. A systematic review of the quality of genetic association studies in human sepsis. Intensive Care Med. 2006, 32, 1706–1712. [Google Scholar] [CrossRef] [PubMed]
- Chanock, S.J.; Manolio, T.; Boehnke, M.; Boerwinkle, E.; Hunter, D.J.; Thomas, G.; Hirschhorn, J.N.; Abecasis, G.; Altshuler, D.; Bailey-Wilson, J.E.; et al. Replicating genotype–phenotype associations. Nature 2007, 447, 655–660. [Google Scholar] [PubMed]
- Dötsch, A.; Eisele, L.; Rabeling, M.; Rump, K.; Walstein, K.; Bick, A.; Cox, L.; Engler, A.; Bachmann, H.S.; Jöckel, K.H.; et al. Hypoxia inducible factor-2alpha and prolinhydroxylase 2 polymorphisms in patients with acute respiratory distress syndrome (ARDS). Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Rogers, A.J.; Solus, J.F.; Hunninghake, G.M.; Baron, R.M.; Meyer, N.J.; Janz, D.R.; Schwartz, D.A.; May, A.K.; Lawson, W.E.; Blackwell, T.S.; et al. MUC5B promoter polymorphism and development of ARDS. Am. J. Respir Crit. Care Med. 2018, 198, 1342–1345. [Google Scholar] [CrossRef]
- Jabaudon, M.; Berthelin, P.; Pranal, T.; Roszyk, L.; Godet, T.; Faure, J.S.; Chabanne, R.; Eisenmann, N.; Lautrette, A.; Belville, C.; et al. Receptor for advanced glycation end-products and ARDS prediction: A multicentre observational study. Sci. Rep. 2018, 8, 2603. [Google Scholar] [CrossRef]
- Wei, Y.; Wang, Z.; Su, L.; Chen, F.; Tejera, P.; Bajwa, E.K.; Wurfel, M.M.; Lin, X.; Christiani, D.C. Platelet count mediates the contribution of a genetic variant in LRRC16A to ARDS risk. Chest 2015, 147, 607–617. [Google Scholar] [CrossRef]
- Morrell, E.D.; O’Mahony, D.S.; Glavan, B.J.; Harju-Baker, S.; Nguyen, C.; Gunderson, S.; Abrahamson, A.; Radella, F. 2nd.; Rona, G.; Black, R.A.; et al. Genetic variation in MAP3K1 Associates with ventilator-free days in acute respiratory distress syndrome. Am. J. Respir. Cell Mol. Biol. 2018, 58, 117–125. [Google Scholar] [CrossRef]
- Hernandez-Pacheco, N.; Guillen-Guio, B.; Acosta-Herrera, M.; Pino-Yanes, M.; Corrales, A.; Ambrós, A.; Nogales, L.; Muriel, A.; González-Higueras, E.; Diaz-Dominguez, F.J.; et al. A vascular endothelial growth factor receptor gene variant is associated with susceptibility to acute respiratory distress syndrome. Intensive Care Med. Exp. 2018, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Cheng, B.; Ding, Y.; Wang, C.; Chen, J. Correlations of IL-17 and NF-κB gene polymorphisms with susceptibility and prognosis in acute respiratory distress syndrome in a chinese population. Biosci. Rep. 2019, 39, BSR20181987. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Liu, N.; Song, S.; Ma, Y. Relationship between β-defensin-1 gene polymorphism and susceptibility and prognosis of acute respiratory distress syndrome. Medicine 2019, 98, e14131. [Google Scholar] [CrossRef] [PubMed]
- Hinz, J.; Büttner, B.; Kriesel, F.; Steinau, M.; Frederik Popov, A.; Ghadimi, M.; Beissbarth, T.; Tzvetkov, M.; Bergmann, I.; Mansur, A. The FER rs4957796 TT genotype is associated with unfavorable 90-day survival in Caucasian patients with severe ARDS due to pneumonia. Sci. Rep. 2017, 7, 9887. [Google Scholar] [CrossRef] [PubMed]
- Reilly, J.P.; Wang, F.; Jones, T.K.; Palakshappa, J.A.; Anderson, B.J.; Shashaty, M.G.S.; Dunn, T.G.; Johansson, E.D.; Riley, T.R.; Lim, B.; et al. Plasma angiopoietin-2 as a potential causal marker in sepsis-associated ARDS development: Evidence from Mendelian randomization and mediation analysis. Intensive Care Med. 2018, 44, 1849–1858. [Google Scholar] [CrossRef]
- Frede, S.; Berchner-Pfannschmidt, U.; Fandrey, J. Regulation of Hypoxia-Inducible Factors During Inflammation. Methods Enzymol. 2007, 435, 405–419. [Google Scholar]
- Allen, R.J.; Porte, J.; Braybrooke, R.; Flores, C.; Fingerlin, T.E.; Oldham, J.M.; Guillen-Guio, B.; Ma, S.F.; Okamoto, T.; John, A.E.; et al. Genetic variants associated with susceptibility to idiopathic pulmonary fibrosis in people of European ancestry: A genome-wide association study. Lancet Respir. Med. 2017, 5, 869–880. [Google Scholar] [CrossRef]
- Bulik-Sullivan, B.; Finucane, H.K.; Anttila, V.; Gusev, A.; Day, F.R.; Loh, P.R.; ReproGen Consortium; Psychiatric Genomics Consortium; Genetic Consortium for Anorexia Nervosa of the Wellcome Trust Case Control Consortium; Duncan, L.; et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 2015, 47, 1236–1241. [Google Scholar] [CrossRef]
- Reilly, J.P.; Christie, J.D. Linking genetics to ARDS pathogenesis: The role of the platelet. Chest 2015, 147, 585–586. [Google Scholar] [CrossRef][Green Version]
- Wang, T.; Liu, Z.; Wang, Z.; Duan, M.; Li, G.; Wang, S.; Li, W.; Zhu, Z.; Wei, Y.; Christiani, D.C.; et al. Thrombocytopenia is associated with acute respiratory distress syndrome mortality: An international study. PLoS ONE 2014, 9, e94124. [Google Scholar] [CrossRef]
- Wei, Y.; Tejera, P.; Wang, Z.; Zhang, R.; Chen, F.; Su, L.; Lin, X.; Bajwa, E.K.; Thompson, B.T.; Christiani, D.C. A missense genetic variant in LRRC16A/CARMIL1 improves ARDS survival by attenuating platelet count decline. AJRCCM 2016, 195, 1353–1361. [Google Scholar]
- Acosta-Herrera, M.; Lorenzo-Diaz, F.; Pino-Yanes, M.; Corrales, A.; Valladares, F.; Klassert, T.E.; Valladares, B.; Slevogt, H.; Ma, S.F.; Villar, J.; et al. Lung transcriptomics during protective ventilatory support in sepsis-induced acute lung injury. PLoS ONE 2015, 10, e0132296. [Google Scholar]
- Shapiro, N.I.; Schuetz, P.; Yano, K.; Sorasaki, M.; Parikh, S.M.; Jones, A.E.; Trzeciak, S.; Ngo, L.; Aird, W. The association of endothelial cell signaling, severity of illness and organ dysfunction in sepsis. Crit. Care 2010, 14, R182. [Google Scholar] [CrossRef] [PubMed]
- Skibsted, S.; Jones, A.E.; Puskarich, M.A.; Arnold, R.; Sherwin, R.; Trzeciak, S.; Schuetz, P.; Aird, W.C.; Shapiro, N.I. Biomarkers of endothelial cell activation in early sepsis. Shock 2013, 39, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Christie, J.D.; Wurfel, M.M.; Feng, R.; O’Keefe, G.E.; Bradfield, J.; Ware, L.B.; Christiani, D.C.; Calfee, C.S.; Cohen, M.J.; Matthay, M.; et al. Genome wide association identifies PPFIA1 as a candidate gene for acute lung injury risk following major trauma. PLoS ONE 2012, 7, e28268. [Google Scholar] [CrossRef] [PubMed]
- Bime, C.; Pouladi, N.; Sammani, S.; Batai, K.; Casanova, N.; Zhou, T.; Kempf, C.L.; Sun, X.; Camp, S.M.; Wang, T.; et al. Genome Wide Association study in African Americans with Acute Respiratory Distress Syndrome identifies the Selectin P Ligand gene as a risk factor. Am. J. Respir. Crit. Care Med. 2018, 197, 1421–1432. [Google Scholar] [CrossRef] [PubMed]
- Rautanen, A.; Mills, T.C.; Gordon, A.C.; Hutton, P.; Steffens, M.; Nuamah, R.; Chiche, J.D.; Parks, T.; Chapman, S.J.; Davenport, E.E.; et al. Genome-wide association study of survival from sepsis due to pneumonia: An observational cohort study. Lancet Respir. Med. 2015, 3, 53–60. [Google Scholar] [CrossRef]
- Schoneweck, F.; Kuhnt, E.; Scholz, M.; Brunkhorst, F.M. Common genomic variation in the FER gene: Useful to stratify patients with sepsis due to pneumonia? Intensive Care Med. 2015, 41, 1382. [Google Scholar] [CrossRef] [PubMed]
- Scherag, A.; Schöneweck, F.; Kesselmeier, M.; Taudien, S.; Platzer, M.; Felder, M.; Sponholz, C.; Rautanen, A.; Hill, A.V.S.; Hinds, C.J.; et al. Genetic Factors of the Disease Course after Sepsis: A Genome-Wide Study for 28 Day Mortality. EBioMedicine 2016, 12, 239–246. [Google Scholar] [CrossRef]
- Wang, T.; Wang, Z.Q.; Wang, L.; Yan, L.; Wan, J.; Zhang, S.; Jiang, H.Q.; Li, W.F.; Lin, Z.F. CRISPLD2 Is Expressed at Low Levels during Septic Shock and Is Associated with Procalcitonin. PLoS ONE 2013, 8, e65743. [Google Scholar] [CrossRef]
- Muñoz-Braceras, S.; Calvo, R.; Escalante, R. TipC and the chorea-acanthocytosis protein VPS13A regulate autophagy in Dictyostelium and human HeLa cells. Autophagy 2015, 11, 918–927. [Google Scholar] [CrossRef] [PubMed]
- Goh, G.; Choi, M. Application of Whole Exome Sequencing to Identify Disease-Causing Variants in Inherited Human Diseases. Genom. Inform. 2013, 10, 214. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Emond, M.J.; Bamshad, M.J.; Barnes, K.C.; Rieder, M.J.; Nickerson, D.A.; NHLBI GO Exome Sequencing Project—ESP Lung Project Team; Christiani, D.C.; Wurfel, M.M.; Lin, X. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am. J. Hum. Genet. 2012, 91, 224–237. [Google Scholar]
- Shortt, K.; Chaudhary, S.; Grigoryev, D.; Heruth, D.P.; Venkitachalam, L.; Zhang, L.Q.; Ye, S.Q. Identification of novel single nucleotide polymorphisms associated with acute respiratory distress syndrome by exome-seq. PLoS ONE 2014, 9, e111953. [Google Scholar] [CrossRef] [PubMed]
- Terpstra, M.L.; Aman, J.; van Nieuw Amerongen, G.P.; Groeneveld, A.B.J. Plasma Biomarkers for Acute Respiratory Distress Syndrome. Crit. Care Med. 2013, 42, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Blondonnet, R.; Constantin, J.M.; Sapin, V.; Jabaudon, M. A Pathophysiologic Approach to Biomarkers in Acute Respiratory Distress Syndrome. Dis. Markers 2016, 2016, 3501373. [Google Scholar] [CrossRef] [PubMed]
- Reilly, J.P.; Christie, J.D.; Meyer, N.J. Fifty years of research in ARDS genomic contributions and opportunities. Am. J. Respir. Crit. Care Med. 2017, 196, 1113–1121. [Google Scholar] [CrossRef]
- Smith, G.D.; Hemani, G. Mendelian randomization: Genetic anchors for causal inference in epidemiological studies. Hum. Mol. Genet. 2014, 23, 89–98. [Google Scholar] [CrossRef]
- Davies, N.M.; Holmes, M.V.; Davey, S.G. Reading Mendelian randomisation studies: A guide, glossary and checklist for clinicians. BMJ 2018, 362, k601. [Google Scholar] [CrossRef]
- Bhandari, V.; Choo-Wing, R.; Lee, C.G.; Zhu, Z.; Nedrelow, J.H.; Chupp, G.L.; Zhang, X.; Matthay, M.A.; Ware, L.B.; Homer, R.J.; et al. Hyperoxia causes angiopoietin 2-mediated acute lung injury and necrotic cell death. Nat. Med. 2006, 12, 1286–1293. [Google Scholar] [CrossRef]
- Van Der Heijden, M.; Van Nieuw Amerongen, G.P.; Koolwijk, P.; Van Hinsbergh, V.W.M.; Groeneveld, A.B.J. Angiopoietin-2, permeability oedema, occurrence and severity of ALI/ARDS in septic and non-septic critically ill patients. Thorax 2008, 63, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Jesmin, S.; Gando, S.; Yanagida, Y.; Mizugaki, A.; Sultana, S.N.; Zaedi, S.; Yokota, H. The role of angiogenic factors and their soluble receptors in acute lung injury (ALI)/ acute respiratory distress syndrome (ARDS) associated with critical illness. J. Inflamm. 2013, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- Meyer, N.J.; Calfee, C.S. Novel translational approaches to the search for precision therapies for acute respiratory distress syndrome. Lancet Respir. Med. 2017, 5, 512–523. [Google Scholar] [CrossRef]
- Molyneaux, P.L.; Willis-Owen, S.A.G.; Cox, M.J.; James, P.; Cowman, S.; Loebinger, M.; Blanchard, A.; Edwards, L.M.; Stock, C.; Daccord, C.; et al. Host-microbial interactions in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2017, 195, 1640–1650. [Google Scholar] [CrossRef] [PubMed]
- Kangelaris, K.N.; Prakash, A.; Liu, K.D.; Aouizerat, B.; Woodruff, P.G.; Erle, D.J.; Rogers, A.; Seeley, E.J.; Chu, J.; Liu, T.; et al. Increased expression of neutrophil-related genes in patients with early sepsis-induced ARDS. Am. J. Physiol. Cell. Mol. Physiol. 2015, 308, L1102–L1113. [Google Scholar] [CrossRef] [PubMed]
- Grigoryev, D.N.; Cheranova, D.I.; Chaudhary, S.; Heruth, D.P.; Zhang, L.Q.; Ye, S.Q. Identification of new biomarkers for Acute Respiratory Distress Syndrome by expression-based genome-wide association study. BMC Pulm. Med. 2015, 15, 95. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lv, X.J.; Zhang, Y.J.; Lu, W.Z.; Wang, Q.; Li, S.Y.; Guo, L.; Qian, G.S.; Zhou, S.W.; Li, Y.Y. Digital gene expression analysis of transcriptomes in lipopolysaccharide-induced acute respiratory distress syndrome. Clin. Chim. Acta 2016, 453, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yan, J.; He, X.; Zhong, Q.; Zhan, C.; Li, S. Candidate genes and pathogenesis investigation for sepsis-related acute respiratory distress syndrome based on gene expression profile. Biol. Res. 2016, 49, 25. [Google Scholar] [CrossRef]
- Barker, G.F.; Manzo, N.D.; Cotich, K.L.; Shone, R.K.; Waxman, A.B. DNA damage induced by hyperoxia: Quantitation and correlation with lung injury. Am. J. Respir. Cell Mol. Biol. 2006, 35, 277–288. [Google Scholar] [CrossRef]
- Loman, N.J.; Constantinidou, C.; Christner, M.; Rohde, H.; Chan, J.Z.; Quick, J.; Weir, J.C.; Quince, C.; Smith, G.P.; Betley, J.R.; et al. A culture-independent sequence-based metagenomics approach to the investigation of an outbreak of Shiga-toxigenic Escherichia coli O104:H4. JAMA 2013, 309, 1502. [Google Scholar] [CrossRef]
- Fischer, N.; Rohde, H.; Indenbirken, D.; Günther, T.; Reumann, K.; Lütgehetmann, M.; Meyer, T.; Kluge, S.; Aepfelbacher, M.; Alawi, M.; et al. Rapid Metagenomic Diagnostics for Suspected Outbreak of Severe Pneumonia. Emerg. Infect. Dis. 2014, 20, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.D.; Xu, J.; Zhang, M.; Zhu, T.M.; Zhang, Y.H.; Sun, K.E. Microrna-21 inhibits lipopolysaccharide-induceacute lung injury by targeting nuclear factor-κb. Exp. Ther. Med. 2018, 16, 4616–4622. [Google Scholar] [PubMed]
- Tao, Z.; Yuan, Y.; Liao, Q. Alleviation of Lipopolysaccharides-Induced Acute Lung Injury by MiR-454. Cell. Physiol. Biochem. 2016, 38, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Gu, Y.; Wang, C.; Zhang, J.; Shan, S.; Gu, X.; Wang, K.; Han, Y.; Ren, T. Enforced expression of miR-125b attenuates LPS-induced acute lung injury. Immunol. Lett. 2014, 162, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Liu, S.Q.; Sun, Q.; Xie, J.F.; Xu, J.Y.; Li, Q.; Pan, C.; Liu, L.; Huang, Y.Z. Plasma microRNAs levels are different between pulmonary and extrapulmonary ARDS patients: A clinical observational study. Ann. Intensive Care 2018, 8, 23. [Google Scholar] [CrossRef]
- Han, Y.; Li, Y.; Jiang, Y. The Prognostic Value of Plasma MicroRNA-155 and MicroRNA-146a Level in Severe Sepsis and Sepsis-Induced Acute Lung Injury Patients. Clin. Lab. 2016, 62, 2355–2360. [Google Scholar] [CrossRef]
- Zhu, Z.; Liang, L.; Zhang, R.; Wei, Y.; Su, L.; Tejera, P.; Guo, Y.; Wang, Z.; Lu, Q.; Baccarelli, A.A.; et al. Whole blood microRNA markers are associated with acute respiratory distress syndrome. Intensive. Care Med. Exp. 2017, 5, 38. [Google Scholar]
- Narute, P.; Seam, N.; Tropea, M.; Logun, C.; Cai, R.; Sun, J.; Shelhamer, J.H.; Meduri, G.U.; Suffredini, A.F. Temporal Changes in MicroRNA Expression in Blood Leukocytes from Patients with the Acute Respiratory Distress Syndrome. Shock 2017, 47, 688–695. [Google Scholar] [CrossRef]
- Huffnagle, G.B.; Dickson, R.P.; Lukacs, N.W. The respiratory tract microbiome and lung inflammation: A two-way street. Mucosal Immunol. 2017, 10, 299–306. [Google Scholar] [CrossRef]
- Dickson, R.P.; Singer, B.H.; Newstead, M.W.; Falkowski, N.R.; Erb-Downward, J.R.; Standiford, T.J.; Huffnagle, G.B. Enrichment of the lung microbiome with gut bacteria in sepsis and the acute respiratory distress syndrome. Nat. Microbiol. 2016, 1, 16113. [Google Scholar] [CrossRef]
- Szilágyi, K.L.; Liu, C.; Zhang, X.; Wang, T.; Fortman, J.D.; Zhang, W.; Garcia, J.G.N. Epigenetic contribution of the myosin light chain kinase gene to the risk for acute respiratory distress syndrome. Transl. Res. 2017, 180, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.G.N.; Verin, A.D.; Herenyiova, M.; English, D. Adherent neutrophils activate endothelial myosin light chain kinase: Role in transendothelial migration. J. Appl. Physiol. 1998, 84, 1817–1821. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Agostinis, P.; Krysko, O.; Garg, A.D.; Bachert, C.; Lambrecht, B.N.; Vandenabeele, P. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011, 32, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Torralba, D.; Baixauli, F.; Villarroya-Beltri, C.; Fernández-Delgado, I.; Latorre-Pellicer, A.; Acín-Pérez, R.; Martín-Cófreces, N.B.; Jaso-Tamame, A.L.; Iborra, S.; Jorge, I.; et al. Priming of dendritic cells by DNA-containing extracellular vesicles from activated T cells through antigen-driven contacts. Nat. Commun. 2018, 9, 2658. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Song, Y. Biomarkers for patients with trauma associated acute respiratory distress syndrome. Mil. Med. Res. 2017, 4, 25. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Kyung, S.Y.; Rogers, A.J.; Gazourian, L.; Youn, S.; Massaro, A.F.; Quintana, C.; Osorio, J.C.; Wang, Z.; Zhao, Y.; et al. Circulating Mitochondrial DNA in Patients in the ICU as a Marker of Mortality: Derivation and Validation. PLoS Med. 2013, 10, e1001577. [Google Scholar] [CrossRef] [PubMed]
- Dudbridge, F. Power and Predictive Accuracy of Polygenic Risk Scores. PLoS Genet. 2013, 9, e1003348. [Google Scholar] [CrossRef]
- Martin, A.R.; Kanai, M.; Kamatani, Y.; Okada, Y.; Neale, B.M.; Daly, M.J. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet. 2019, 51, 441261. [Google Scholar] [CrossRef] [PubMed]
- Grinde, K.E.; Thornton, T.A.; Liu, S.; Shadyabç, A.H.; Chan, K.H.K.; Reiner, A.P.; Sofer, T. Generalizing polygenic risk scores from Europeans to Hispanics/Latinos. Genet. Epidemiol. 2019, 43, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Sirugo, G.; Williams, S.M.; Tishkoff, S.A. The Missing Diversity in Human Genetic Studies. Cell 2019, 177, 26–31. [Google Scholar] [CrossRef]
- Wojcik, G.L.; Graff, M.; Nishimura, K.K.; Tao, R.; Haessler, J.; Gignoux, C.R.; Highland, H.M.; Patel, Y.M.; Sorokin, E.P.; Avery, C.L.; et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature 2019, 570, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Diogo, D.; Tian, C.; Franklin, C.S.; Alanne-Kinnunen, M.; March, M.; Spencer, C.C.A.; Vangjeli, C.; Weale, M.E.; Mattsson, H.; Kilpeläinen, E.; et al. Phenome-wide association studies across large population cohorts support drug target validation. Nat. Commun. 2018, 9, 4285. [Google Scholar] [CrossRef] [PubMed]
- Denny, J.C.; Bastarache, L.; Ritchie, M.D.; Carroll, R.J.; Zink, R.; Mosley, J.D.; Field, J.R.; Pulley, J.M.; Ramirez, A.H.; Bowton, E.; et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat. Biotechnol. 2013, 31, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Skotte, L.; Jørsboe, E.; Korneliussen, T.S.; Moltke, I.; Albrechtsen, A. Ancestry-specific association mapping in admixed populations. bioRxiv 2018, 2018, 014001. [Google Scholar] [CrossRef] [PubMed]
- Locke, A.E.; Steinberg, K.M.; Chiang, C.W.K.; Service, S.K.; Havulinna, A.S.; Stell, L.; Pirinen, M.; Abel, H.J.; Chiang, C.C.; Fulton, R.S.; et al. Exome sequencing of Finnish isolates enhances rare-variant association power. Nature 2019. [Google Scholar] [CrossRef] [PubMed]
- Van Hout, C.V.; Tachmazidou, I.; Backman, J.D.; Hoffman, J.X.; Ye, B.; Pandey, A.K.; Gonzaga-Jauregui, C.; Khalid, S.; Liu, D.; Banerjee, N.; et al. Whole exome sequencing and characterization of coding variation in 49,960 individuals in the UK Biobank. bioRxiv 2019, 572347. [Google Scholar] [CrossRef]
- Meienberg, J.; Bruggmann, R.; Oexle, K.; Matyas, G. Clinical sequencing: Is WGS the better WES? Hum. Genet. 2016, 135, 359–362. [Google Scholar] [CrossRef]
- Belkadi, A.; Bolze, A.; Itan, Y.; Cobat, A.; Vincent, Q.B.; Antipenko, A.; Shang, L.; Boisson, B.; Casanova, J.L.; Abel, L. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc. Natl. Acad. Sci. USA 2015, 112, 5473–5478. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Approach | Aim | Main Advantages | Main Limitations | Phenotypes Assessed |
|---|---|---|---|---|
| Candidate-gene association study | To identify the statistical association between genetic variants for pre-specified genes of biological interest and the trait. | Simple approximation not requiring computational skills. Hypothesizes causality of the analysed variant Reduced penalty of statistical significance. | Non-reproducibility of the findings in independent studies complicating the interpretation. | Susceptibility, outcomes |
| Genome-wide association study (GWAS) | To identify the statistical association between genetic variants assessed across the genome and the trait. | Hypothesis-free approach. Allows to identify new pathogenic mechanisms, potentially leading to new therapeutic targets. Reduced proportion of false positives. | Many of the genes that are identified do not yet have a known biological implication in the trait. Large penalty of statistical significance. Large proportion of false negatives. | Susceptibility |
| Whole-exome sequencing (WES) | To identify the statistical association between genetic variants assessed across exons of all genes (exome) and the trait. | Same as indicated for GWAS. Allows analysis of rare and common genetic variants. | Blind to genetic variation occurring in the regulatory regions of genes. There is no standardization of the statistical tests. Requires advanced computational skills and dedicated infrastructure. More expensive than GWAS and candidate-gene studies for a fixed sample size. | Susceptibility, outcomes |
| Transcriptome-wide association study | To identify genomic loci associated with gene expression alterations related to the trait. | Same as indicated for GWAS. | Same as indicated for GWAS. | Susceptibility |
| Transcriptomics | To assess alterations of the gene expression and biological pathways in disease states focusing on particular targets or using array or sequencing-based approaches. | Allows to quantify and provides precise expression levels of genes simultaneously. A variant focusing on small non-coding species is possible. If sequencing-based, it allows the distinction of isoforms and allelic expression. If sequencing-based, it allows to map transcribed regions. If sequencing-based, it allows to evaluate gene expression levels in single cells. | The RNA isolation and handling require specialized materials and skills. If sequence-based, requires abundant RNA species (e.g., rRNA) to be depleted. Effects of this on the profiles are yet unknown. If sequencing-based, requires advanced computational skills and dedicated infrastructure. If sequencing-based, there is a lack of standardization of the optimal read depth. | Susceptibility and outcomes (array-based only) |
| Mendelian randomization | To assess the causality of a risk factor on a trait based on genetic predictors of the former. | Less affected by confusion or inverse causality. | Depends on many assumptions that need to be assessed for plausibility. Genetic predictors of the risk factor need to be known from previous studies. | Susceptibility |
| DNA methylation | To identify methylation levels at genomic loci associated with the trait. | Allows to quantitatively evaluate environmental exposures at DNA level. Permits the evaluation of functional effects of identified elements. | There is no standardization of the statistical tests. Necessity to control for collection tissues, environmental exposures and other relevant variables that affect the results. | Susceptibility |
| Metagenomics | To assess the collective microbial composition and function of environmental samples from genomic data. | Allows to characterize microbial communities (abundance, diversity and distribution) and deduce function without culturing. Allows to detect uncultivable microbes. With sufficient resolution, it allows to recover antibiotic resistance genes and virulence factors. | The same as indicated for DNA methylation. Requires advanced computational skills and dedicated infrastructure. | Susceptibility |
| Whole-genome sequencing | To identify the statistical association between genetic variants assessed across the genome and the trait. | Same as indicated for WES. Allows the better analysis of structural variation and variation in non-exonic regions of the genome. | There is no standardization of the statistical tests. Requires advanced computational skills and dedicated infrastructure. More expensive than WES studies for a fixed sample size. | None |
| Admixture mapping | To identify genomic regions that are associated with a trait based on ancestry markers. | Hypothesis-free approach. Reduced proportion of false positives. Reduced penalty of statistical significance. | Can only be applied in recently admixed populations and the evolutionary history must be known. Large proportion of false negatives. Identified loci at Mb resolution. There is no standardization of the statistical tests. | None |
| Polygenic risks | To stratify disease risks based on the cumulative effects of genetic variants. | Allows to stratify the risk with a single score. Allows to assess the genetic overlap among traits. | Genetic risk variants need to be known from previous studies. Difficulties in the transferability among populations. | None |
| Mitochondrial DNA levels | To assess its potential as a biomarker for a trait. | Simple approximation not requiring computational skills. May offer improvements for diagnostic or prognostic scores. Inexpensive approach. | Difficulties to reach optimal sensitivity and specificity. Strong dependency on sample collection and handling. | None |
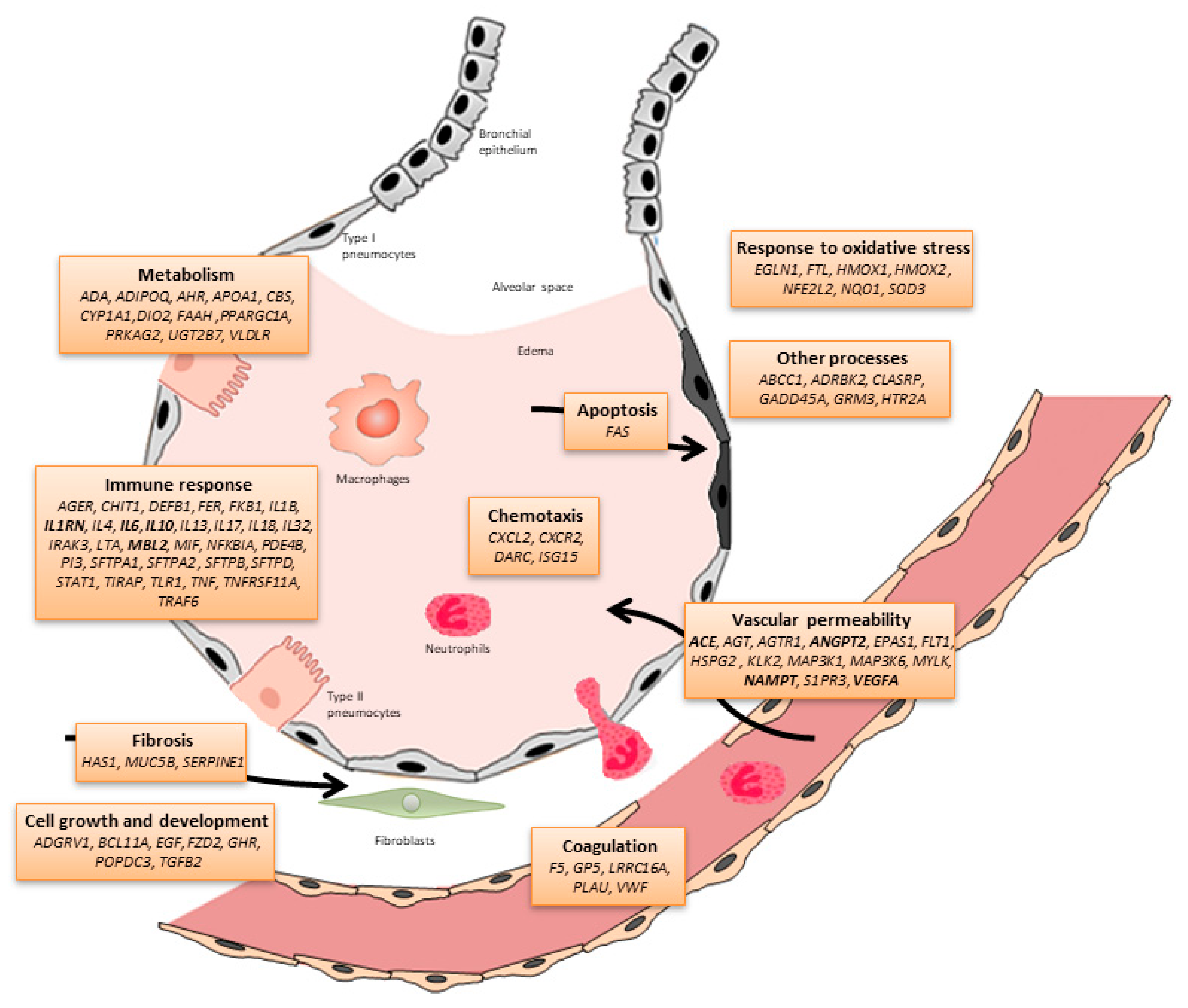
| Gene | Chr | Position (hg19) | rsID | Phenotype | Sample (Case/Control) | Population | Study | |
|---|---|---|---|---|---|---|---|---|
| Discovery | Validation | |||||||
| EGLN1 | 1 | 231542656 | rs516651 | Outcome | 264 * | -- | European | Dötsch et al. [36] |
| MUC5B | 11 | 1241221 | rs35705950 | Susceptibility | 234/669 | -- | Multi-ethnic | Rogers et al. [37] |
| AGER | 6 | 32151693 | rs2070600 | Susceptibility | 59/405 | -- | Multi-ethnic | Jabaudon et al. [38] |
| LRRC16A | 6 | 25426768 | rs9358856 | Outcome | 414 * | -- | Multi-ethnic | Wei et al. [39] |
| MAP3K1 | 5 | 56177743 | rs832582 | Outcome | 306 * | 241 * | European | Morrell et al. [40] |
| FLT1 | 13 | 28993669 | rs9513106 | Susceptibility | 225/899 | 661/234 | European | Hernandez-Pacheco et al. [41] |
| IL17 | 6 | 52185695 | rs8193036 | Susceptibility/Outcome | 210/210 | -- | East Asian | Xie et al. [42] |
| 52186235 | rs2275913 | |||||||
| DEFB1 | 8 | 6877901 | rs1800972 | Susceptibility | 300/240 | -- | European | Feng et al. [43] |
| FER | 5 | 108402140 | rs4957796 | Outcome | 27/68 | -- | European | Hinz et al. [44] |
| ANGPT2 | 8 | 6370320 | rs2442630 | Susceptibility | 178/226 | -- | European | Reilly et al. [45] |
| 6386620 | rs2442608 | |||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández-Beeftink, T.; Guillen-Guio, B.; Villar, J.; Flores, C. Genomics and the Acute Respiratory Distress Syndrome: Current and Future Directions. Int. J. Mol. Sci. 2019, 20, 4004. https://doi.org/10.3390/ijms20164004
Hernández-Beeftink T, Guillen-Guio B, Villar J, Flores C. Genomics and the Acute Respiratory Distress Syndrome: Current and Future Directions. International Journal of Molecular Sciences. 2019; 20(16):4004. https://doi.org/10.3390/ijms20164004
Chicago/Turabian StyleHernández-Beeftink, Tamara, Beatriz Guillen-Guio, Jesús Villar, and Carlos Flores. 2019. "Genomics and the Acute Respiratory Distress Syndrome: Current and Future Directions" International Journal of Molecular Sciences 20, no. 16: 4004. https://doi.org/10.3390/ijms20164004
APA StyleHernández-Beeftink, T., Guillen-Guio, B., Villar, J., & Flores, C. (2019). Genomics and the Acute Respiratory Distress Syndrome: Current and Future Directions. International Journal of Molecular Sciences, 20(16), 4004. https://doi.org/10.3390/ijms20164004