A Computational Analysis of Alternative Splicing across Mammalian Tissues Reveals Circadian and Ultradian Rhythms in Splicing Events


Abstract
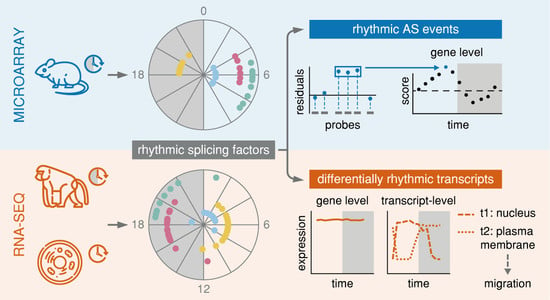
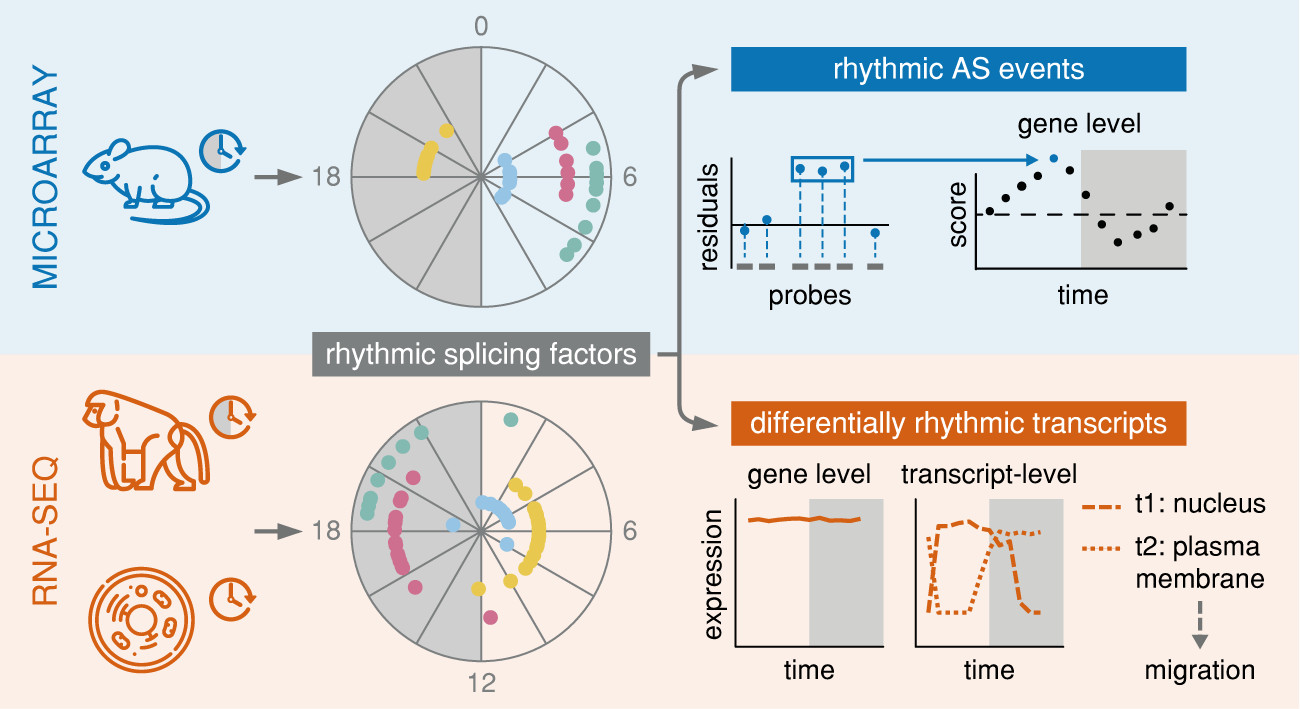{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
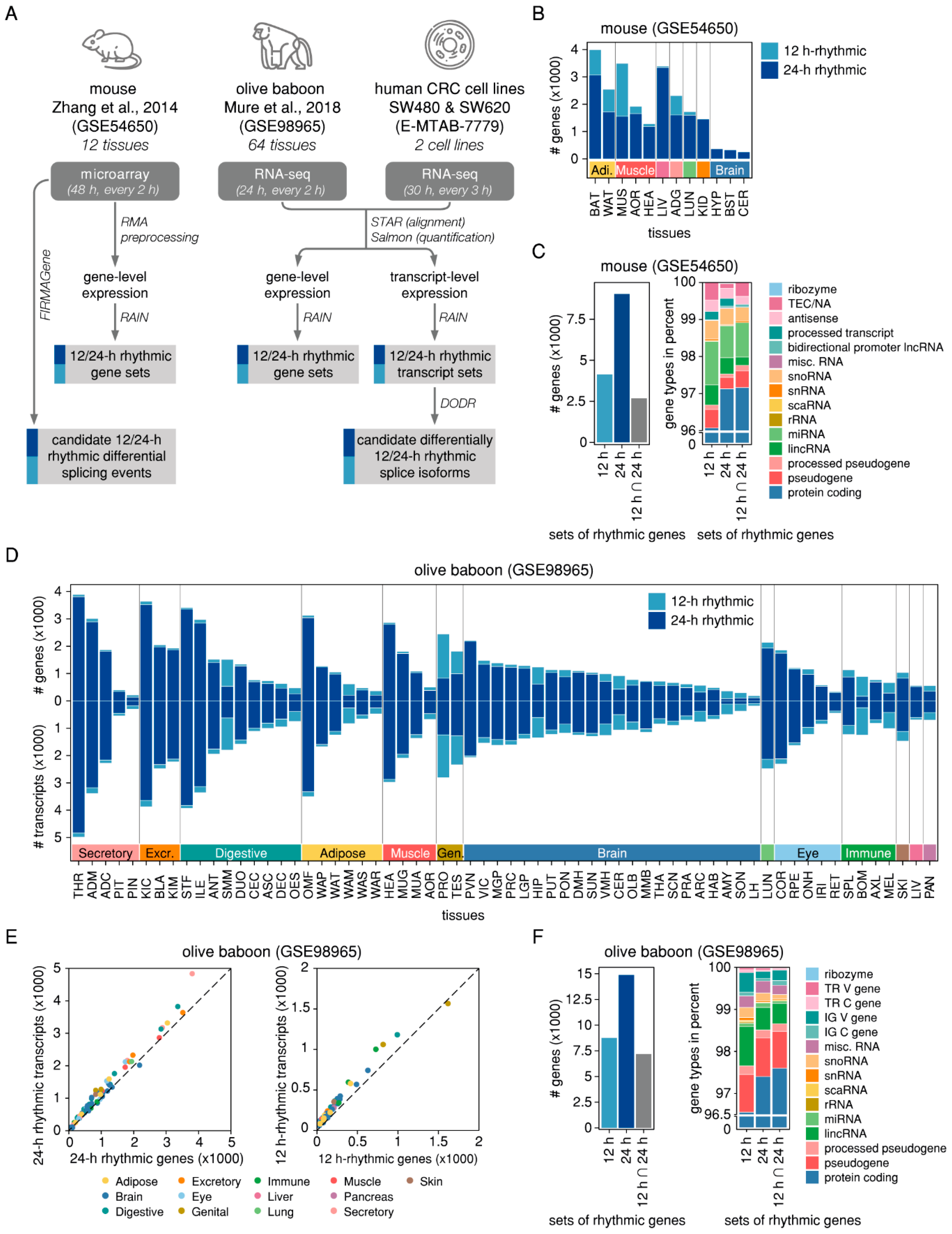
2.1. Rhythmicity Analysis of Mammalian Tissues Reveals Differences between 24-h and 12-h Rhythmic Features at the Gene- and at the Transcript-Level
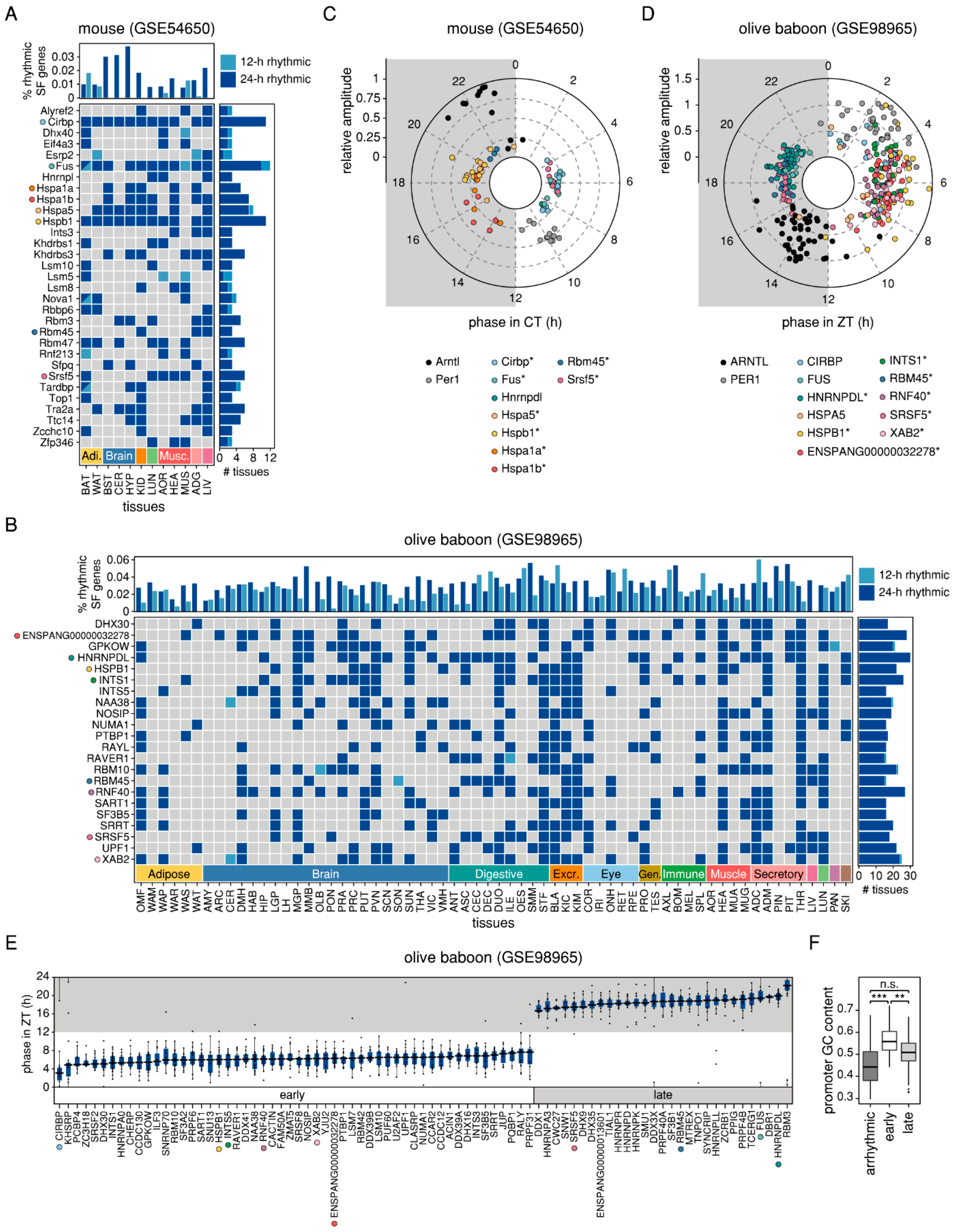
2.2. Splicing-Related Genes with 24-h Rhythmic Expression Patterns are Conserved Across Mouse and Baboon Tissues and Show Daily and Nightly Clusters of Peak Expression
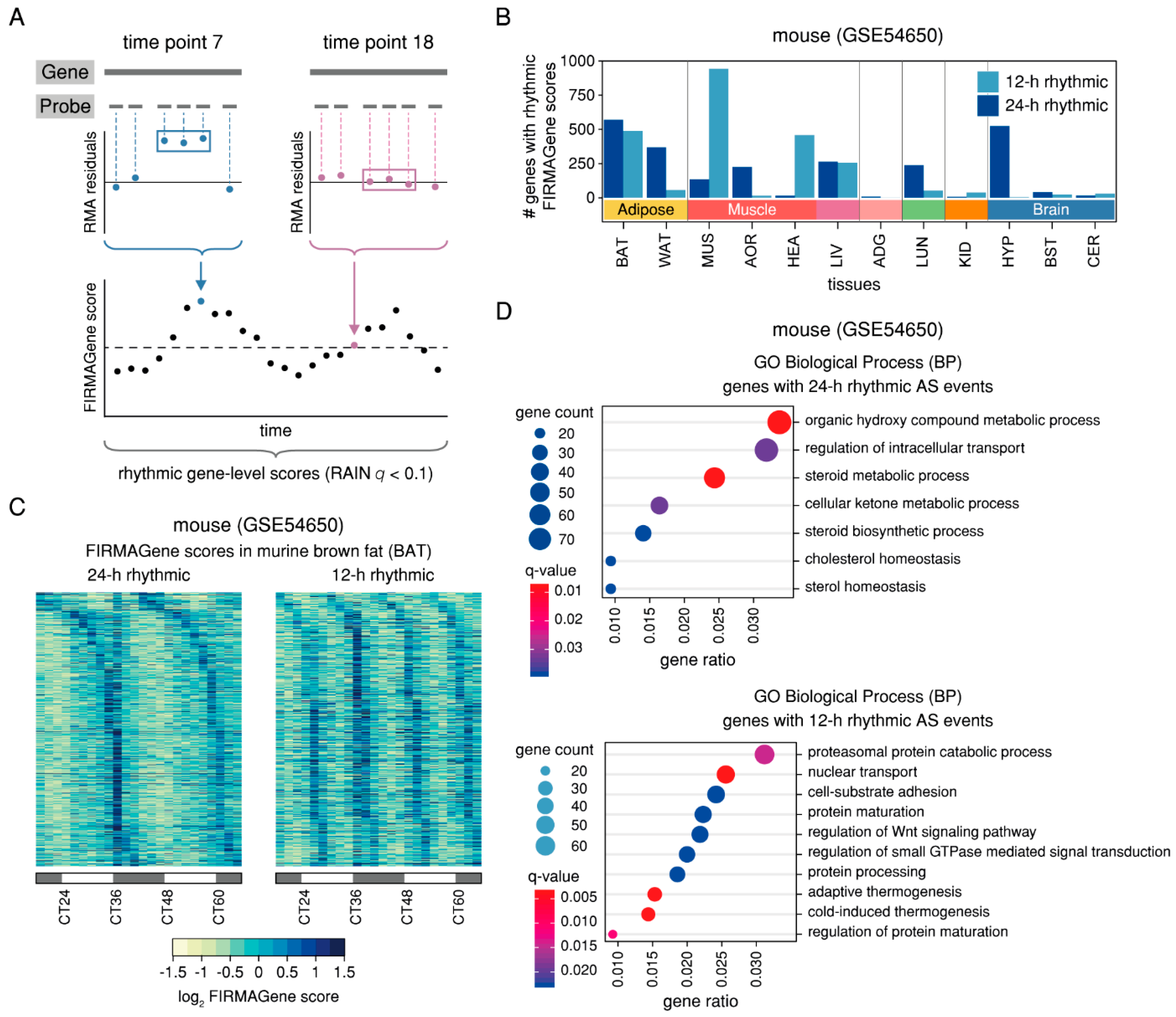
2.3. Rhythmic AS Events are Prevalent in Murine Adipose and Muscle Tissues and are Found in Genes Associated with the Circadian Clock
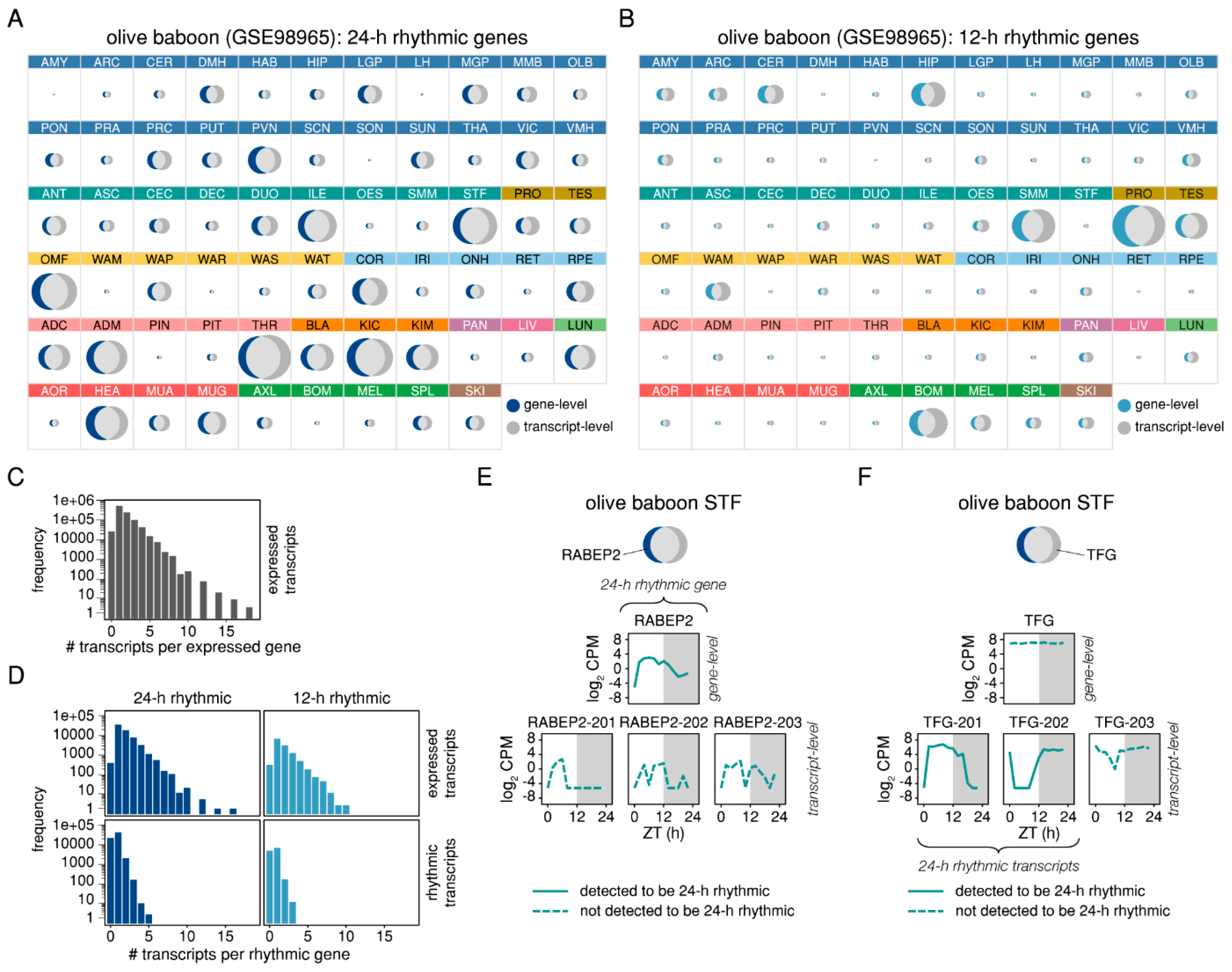
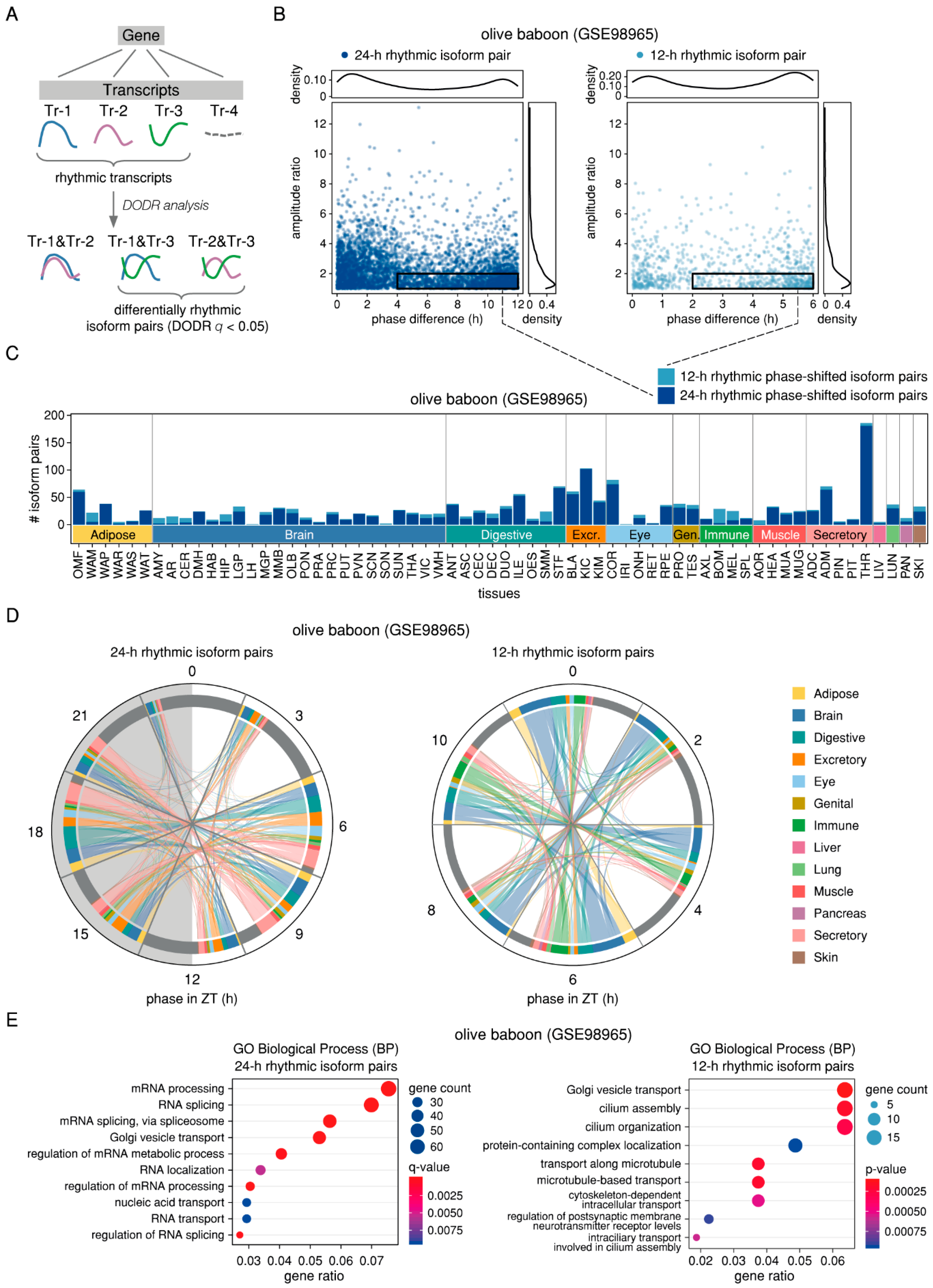
2.4. Differential 24-h Rhythmic Isoform Pairs in Baboon are Phase-Shifted Across the Daily Cycle
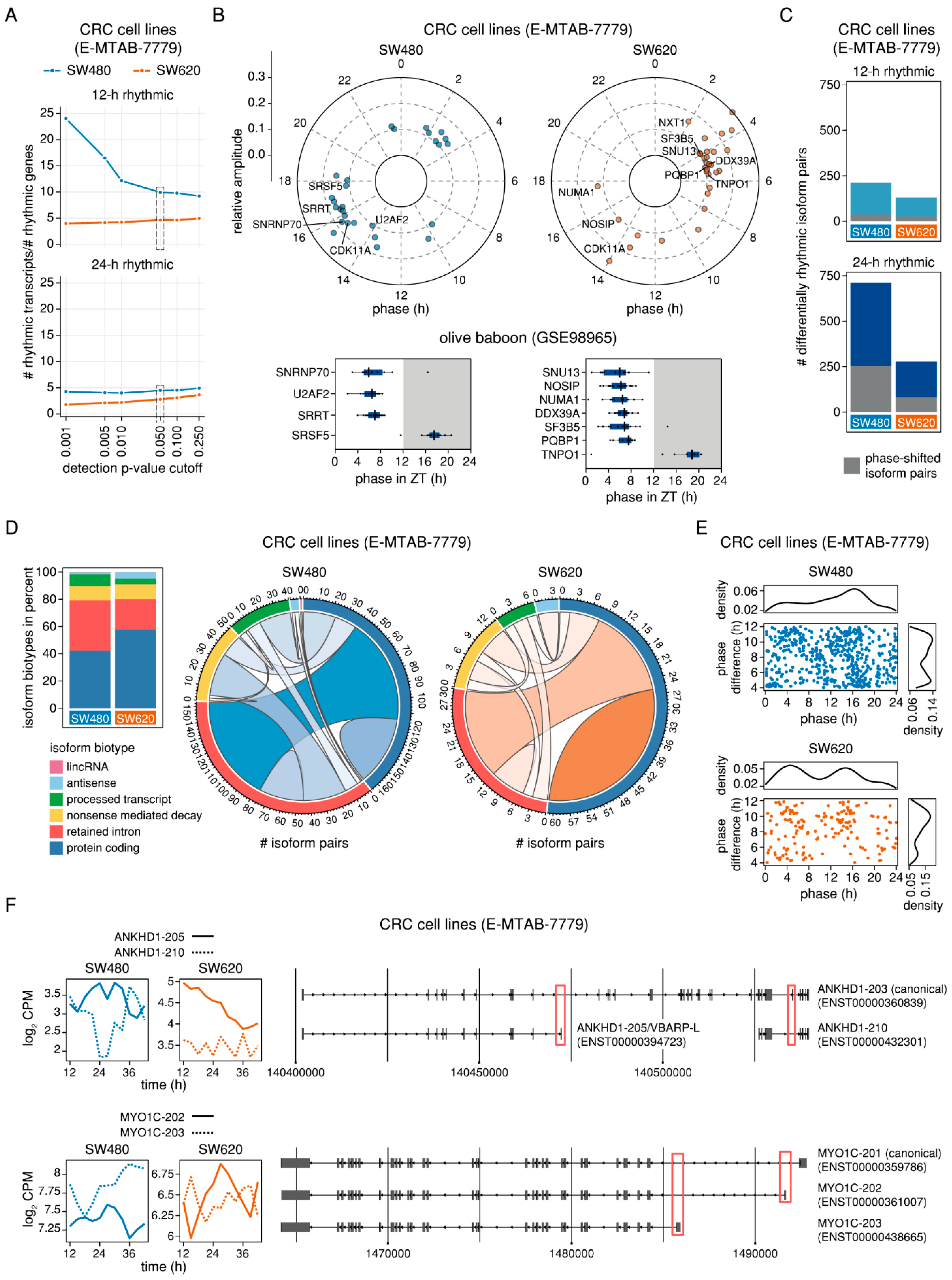
2.5. Genes with Phase-Shifted Splice Isoforms in a Human Cell Line Model of Colorectal Cancer Progression are Enriched for mRNA Splicing and Have a Putative Impact on Cell Survival and Migration
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Sample Preparation
4.3. Processing Microarray and RNA-Seq Transcriptome Data
4.4. Rhythmicity Analysis
4.5. Compilation of a List of Splicing-Related Genes
4.6. Prediction of Clock Transcription Factor Binding Sites
4.7. Detection of Rhythmic AS in Circadian Whole-Transcript Microarray Data
4.8. Detection of Differentially Rhythmic Splice Isoforms in Circadian RNA-seq Data
4.9. Gene Enrichment Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Eyras, E. The role of alternative splicing in cancer. Transcription 2017, 8, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.Y.; Alipanahi, B.; Lee, L.J.; Bretschneider, H.; Merico, D.; Yuen, R.K.; Hua, Y.; Gueroussov, S.; Najafabadi, H.S.; Hughes, T.R. The human splicing code reveals new insights into the genetic determinants of disease. Science 2015, 347, 1254806. [Google Scholar] [CrossRef] [PubMed]
- Daguenet, E.; Dujardin, G.; Valcárcel, J. The pathogenicity of splicing defects: Mechanistic insights into pre-mRNA processing inform novel therapeutic approaches. EMBO Rep. 2015, 16, 1640–1655. [Google Scholar] [CrossRef] [PubMed]
- Oltean, S.; Bates, D. Hallmarks of alternative splicing in cancer. Oncogene 2014, 33, 5311. [Google Scholar] [CrossRef]
- Carmo-Fonseca, M.; Kirchhausen, T. The timing of pre-mRNA splicing visualized in real-time. Nucleus 2014, 5, 11–14. [Google Scholar] [CrossRef][Green Version]
- McGlincy, N.J.; Valomon, A.; Chesham, J.E.; Maywood, E.S.; Hastings, M.H.; Ule, J. Regulation of alternative splicing by the circadian clock and food related cues. Genome Biol. 2012, 13, 1. [Google Scholar] [CrossRef]
- El-Athman, R.; Fuhr, L.; Relógio, A. A Systems-Level Analysis Reveals Circadian Regulation of Splicing in Colorectal Cancer. EBioMedicine 2018. [Google Scholar] [CrossRef]
- Genov, N.; Basti, A.; Abreu, M.; Relógio, A. Temporal Splicing Switches in Elements of the TNF-Pathway Identified by Computational Analysis of Transcriptome Data for Human Cell Lines. Int. J. Mol. Sci. 2019, 20, 1182. [Google Scholar] [CrossRef] [PubMed]
- Riley, L.A.; Mijares, J.R.; Zhang, X.; Esser, K.A. The Skeletal Muscle Molecular Clock Regulates Titin Splicing and Protein Expression. Biophys. J. 2018, 114, 137a. [Google Scholar] [CrossRef]
- Benegiamo, G.; Brown, S.A.; Panda, S. RNA dynamics in the control of circadian rhythm. In RNA Process; Springer: Cham, Switzerland, 2016; pp. 107–122. [Google Scholar]
- Partch, C.L.; Green, C.B.; Takahashi, J.S. Molecular architecture of the mammalian circadian clock. Trends Cell Biol. 2014, 24, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Lahens, N.F.; Ballance, H.I.; Hughes, M.E.; Hogenesch, J.B. A circadian gene expression atlas in mammals: Implications for biology and medicine. Proc. Natl. Acad. Sci. USA 2014, 111, 16219–16224. [Google Scholar] [CrossRef] [PubMed]
- Mure, L.S.; Le, H.D.; Benegiamo, G.; Chang, M.W.; Rios, L.; Jillani, N.; Ngotho, M.; Kariuki, T.; Dkhissi-Benyahya, O.; Cooper, H.M. Diurnal transcriptome atlas of a primate across major neural and peripheral tissues. Science 2018, eaao0318. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.E.; DiTacchio, L.; Hayes, K.R.; Vollmers, C.; Pulivarthy, S.; Baggs, J.E.; Panda, S.; Hogenesch, J.B. Harmonics of circadian gene transcription in mammals. PLoS Genet. 2009, 5, e1000442. [Google Scholar] [CrossRef]
- Beaulé, C.; Granados-Fuentes, D.; Marpegan, L.; Herzog, E.D. In vitro circadian rhythms: Imaging and electrophysiology. Essays Biochem. 2011, 49, 103–117. [Google Scholar] [CrossRef]
- Van der Veen, D.R.; Gerkema, M.P. Unmasking ultradian rhythms in gene expression. FASEB J. 2016, 31, 743–750. [Google Scholar] [CrossRef]
- Fuhr, L.; El-Athman, R.; Scrima, R.; Cela, O.; Carbone, A.; Knoop, H.; Li, Y.; Hoffmann, K.; Laukkanen, M.O.; Corcione, F. The Circadian Clock Regulates Metabolic Phenotype Rewiring Via HKDC1 and Modulates Tumor Progression and Drug Response in Colorectal Cancer. EBioMedicine 2018, 33, 105–121. [Google Scholar] [CrossRef]
- Wahl, M.C.; Will, C.L.; Lührmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [PubMed]
- Bozek, K.; Relógio, A.; Kielbasa, S.M.; Heine, M.; Dame, C.; Kramer, A.; Herzel, H. Regulation of clock-controlled genes in mammals. PLoS ONE 2009, 4, e4882. [Google Scholar] [CrossRef]
- Robinson, M.D.; Speed, T.P. Differential splicing using whole-transcript microarrays. BMC Bioinform. 2009, 10, 156. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.H.J.; Markus, M.A.; Mangs, A.H.; Raitskin, O.; Sperling, R.; Morris, B.J. ZRANB2 localizes to supraspliceosomes and influences the alternative splicing of multiple genes in the transcriptome. Mol. Biol. Rep. 2013, 40, 5381–5395. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, Y.; Oda, N.; Cox, N.J.; Li, X.; Orho-Melander, M.; Hara, M.; Hinokio, Y.; Lindner, T.H.; Mashima, H.; Schwarz, P.E. Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat. Genet. 2000, 26, 163. [Google Scholar] [CrossRef] [PubMed]
- Thaben, P.F.; Westermark, P.O. Differential rhythmicity: Detecting altered rhythmicity in biological data. Bioinformatics 2016, 32, 2800–2808. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Jia, R. Splicing factor poly (rC)-binding protein 1 is a novel and distinctive tumor suppressor. J. Cell. Physiol. 2019, 234, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Dang, H.; Takai, A.; Forgues, M.; Pomyen, Y.; Mou, H.; Xue, W.; Ray, D.; Ha, K.C.; Morris, Q.D.; Hughes, T.R. Oncogenic activation of the RNA binding protein NELFE and MYC signaling in hepatocellular carcinoma. Cancer Cell 2017, 32, 101–114.e108. [Google Scholar] [CrossRef]
- Lujan, D.A.; Ochoa, J.L.; Hartley, R.S. Cold-inducible RNA binding protein in cancer and inflammation. Wiley Interdiscip. Rev. 2018, 9, e1462. [Google Scholar] [CrossRef]
- Adams, D.R.; Ron, D.; Kiely, P.A. RACK1, A multifaceted scaffolding protein: Structure and function. Cell Commun. Signal. 2011, 9, 22. [Google Scholar] [CrossRef]
- Bhuiyan, S.A.; Ly, S.; Phan, M.; Huntington, B.; Hogan, E.; Liu, C.C.; Liu, J.; Pavlidis, P. Systematic evaluation of isoform function in literature reports of alternative splicing. BMC Genom. 2018, 19, 637. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Manley, J.L. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes Dev. 2010, 24, 2343–2364. [Google Scholar] [CrossRef] [PubMed]
- Kaida, D.; Schneider-Poetsch, T.; Yoshida, M. Splicing in oncogenesis and tumor suppression. Cancer Sci. 2012, 103, 1611–1616. [Google Scholar] [CrossRef] [PubMed]
- Seiler, M.; Peng, S.; Agrawal, A.A.; Palacino, J.; Teng, T.; Zhu, P.; Smith, P.G.; Caesar-Johnson, S.J.; Demchok, J.A.; Felau, I. Somatic mutational landscape of splicing factor genes and their functional consequences across 33 cancer types. Cell Rep. 2018, 23, 282–296. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, M.-P.; Armero, V.E.; Allaire, A.; Boudreault, S.; Martenon-Brodeur, C.; Durand, M.; Lapointe, E.; Thibault, P.; Tremblay-Létourneau, M.; Perreault, J.-P. Global profiling of alternative RNA splicing events provides insights into molecular differences between various types of hepatocellular carcinoma. BMC Genom. 2016, 17, 683. [Google Scholar] [CrossRef] [PubMed]
- Tabaglio, T.; Low, D.H.; Teo, W.K.L.; Goy, P.A.; Cywoniuk, P.; Wollmann, H.; Ho, J.; Tan, D.; Aw, J.; Pavesi, A. MBNL1 alternative splicing isoforms play opposing roles in cancer. Life Sci. Alliance 2018, 1, e201800157. [Google Scholar] [CrossRef] [PubMed]
- Miles, M.C.; Janket, M.L.; Wheeler, E.D.; Chattopadhyay, A.; Majumder, B.; DeRicco, J.; Schafer, E.A.; Ayyavoo, V. Molecular and functional characterization of a novel splice variant of ANKHD1 that lacks the KH domain and its role in cell survival and apoptosisc. FEBS J. 2005, 272, 4091–4102. [Google Scholar] [CrossRef] [PubMed]
- Zattelman, L.; Regev, R.; Ušaj, M.; Reinke, P.Y.; Giese, S.; Samson, A.O.; Taft, M.H.; Manstein, D.J.; Henn, A. N-terminal splicing extensions of the human MYO1C gene fine-tune the kinetics of the three full-length myosin IC isoforms. J. Biol. Chem. 2017, 292, 17804–17818. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, J.S. Transcriptional architecture of the mammalian circadian clock. Nat. Rev. Genet. 2017, 18, 164. [Google Scholar] [CrossRef]
- Matys, V.; Fricke, E.; Geffers, R.; Gößling, E.; Haubrock, M.; Hehl, R.; Hornischer, K.; Karas, D.; Kel, A.E.; Kel-Margoulis, O.V. TRANSFAC®: Transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003, 31, 374–378. [Google Scholar] [CrossRef]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’ayan, A. The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhang, T.; Wang, H.; Wang, T.; Qin, M.; Bao, P.; Wang, R.; Liu, Y.; Chang, H.-C.; Yan, J. Neurodegeneration-associated FUS is a novel regulator of circadian gene expression. Transl. Neurodegener. 2018, 7, 24. [Google Scholar] [CrossRef]
- Rogelj, B.; Easton, L.E.; Bogu, G.K.; Stanton, L.W.; Rot, G.; Curk, T.; Zupan, B.; Sugimoto, Y.; Modic, M.; Haberman, N. Widespread binding of FUS along nascent RNA regulates alternative splicing in the brain. Sci. Rep. 2012, 2, 603. [Google Scholar] [CrossRef] [PubMed]
- Arrigo, A.-P.; Gibert, B. Protein interactomes of three stress inducible small heat shock proteins: HspB1, HspB5 and HspB8. Int. J. Hyperth. 2013, 29, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Higashitsuji, H.; Higashitsuji, H.; Liu, Y.; Itoh, K.; Sakurai, T.; Kojima, T.; Kandori, S.; Nishiyama, H.; Fukumoto, M. TRPV4-dependent induction of a novel mammalian cold-inducible protein SRSF5 as well as CIRP and RBM3. Sci. Rep. 2017, 7, 2295. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Alavez, M.; Alboni, S.; Conti, B. Sex-and age-specific differences in core body temperature of C57Bl/6 mice. Age 2011, 33, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Gotic, I.; Omidi, S.; Fleury-Olela, F.; Molina, N.; Naef, F.; Schibler, U. Temperature regulates splicing efficiency of the cold-inducible RNA-binding protein gene Cirbp. Genes Dev. 2016, 30, 2005–2017. [Google Scholar] [CrossRef] [PubMed]
- Gotic, I.; Schibler, U. Posttranscriptional mechanisms controlling diurnal gene expression cycles by body temperature rhythms. RNA Biol. 2017, 14, 1294–1298. [Google Scholar] [CrossRef]
- Preußner, M.; Heyd, F. Temperature-controlled Rhythmic Gene Expression in Endothermic Mammals: All Diurnal Rhythms are Equal, but Some are Circadian. BioEssays 2018, 40, 1700216. [Google Scholar] [CrossRef]
- Yurtsever, T.; Streit, F.; Foo, J.C.; Trifonova, S.; Kumsta, R.; Muller, C.P.; Turner, J.D.; Meyer, J.; Schote, A.B. Temporal dynamics of cortisol-associated changes in mRNA expression of glucocorticoid responsive genes FKBP5, GILZ, SDPR, PER1, PER2 and PER3 in healthy humans. Psychoneuroendocrinology 2019, 102, 63–67. [Google Scholar] [CrossRef]
- Preußner, M.; Goldammer, G.; Neumann, A.; Haltenhof, T.; Rautenstrauch, P.; Müller-McNicoll, M.; Heyd, F. Body temperature cycles control rhythmic alternative splicing in mammals. Mol. Cell 2017, 67, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Buoso, E.; Galasso, M.; Ronfani, M.; Serafini, M.M.; Lanni, C.; Corsini, E.; Racchi, M. Role of spliceosome proteins in the regulation of glucocorticoid receptor isoforms by cortisol and dehydroepiandrosterone. Pharmacol. Res. 2017, 120, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Neumann, A.; Schindler, M.; Olofsson, D.; Wilhelmi, I.; Schürmann, A.; Heyd, F. Genome-wide identification of alternative splicing events that regulate protein transport across the secretory pathway. J Cell Sci. 2019, jcs230201. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Garva, R.; Pickard, A.; Yeung, C.-Y.C.; Mallikarjun, V.; Swift, J.; Holmes, D.F.; Calverley, B.; Lu, Y.; Adamson, A. Circadian control of the secretory pathway is a central mechanism in tissue homeostasis. bioRxiv 2019, 304014. [Google Scholar]
- Hatami, R.; Sieuwerts, A.M.; Izadmehr, S.; Yao, Z.; Qiao, R.F.; Papa, L.; Look, M.P.; Smid, M.; Ohlssen, J.; Levine, A.C. KLF6-SV1 drives breast cancer metastasis and is associated with poor survival. Sci. Transl. Med. 2013, 5, ra112–ra169. [Google Scholar] [CrossRef] [PubMed]
- Narla, G.; DiFeo, A.; Fernandez, Y.; Dhanasekaran, S.; Huang, F.; Sangodkar, J.; Hod, E.; Leake, D.; Friedman, S.L.; Hall, S.J. KLF6-SV1 overexpression accelerates human and mouse prostate cancer progression and metastasis. J. Clin. Investig. 2008, 118, 2711–2721. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.A.; Ripperger, J.; Kadener, S.; Fleury-Olela, F.; Vilbois, F.; Rosbash, M.; Schibler, U. PERIOD1-associated proteins modulate the negative limb of the mammalian circadian oscillator. Science 2005, 308, 693–696. [Google Scholar] [CrossRef]
- Kowalska, E.; Ripperger, J.A.; Hoegger, D.C.; Bruegger, P.; Buch, T.; Birchler, T.; Mueller, A.; Albrecht, U.; Contaldo, C.; Brown, S.A. NONO couples the circadian clock to the cell cycle. Proc. Natl. Acad. Sci. 2013, 110, 1592–1599. [Google Scholar] [CrossRef]
- House, R.P.; Talwar, S.; Hazard, E.S.; Hill, E.G.; Palanisamy, V. RNA-binding protein CELF1 promotes tumor growth and alters gene expression in oral squamous cell carcinoma. Oncotarget 2015, 6, 43620. [Google Scholar] [CrossRef]
- Cifdaloz, M.; Osterloh, L.; Graña, O.; Riveiro-Falkenbach, E.; Ximénez-Embún, P.; Muñoz, J.; Tejedo, C.; Calvo, T.G.; Karras, P.; Olmeda, D. Systems analysis identifies melanoma-enriched pro-oncogenic networks controlled by the RNA binding protein CELF1. Nat. Commun. 2017, 8, 2249. [Google Scholar] [CrossRef]
- Traina, F.; Favaro, P.M.; de Souza Medina, S.; Duarte, A.d.S.S.; Winnischofer, S.M.B.; Costa, F.F.; Saad, S.T. ANKHD1, ankyrin repeat and KH domain containing 1, is overexpressed in acute leukemias and is associated with SHP2 in K562 cells. Biochim. Biophys. Acta (BBA). 2006, 1762, 828–834. [Google Scholar] [CrossRef]
- Maly, I.V.; Domaradzki, T.M.; Gosy, V.A.; Hofmann, W.A. Myosin isoform expressed in metastatic prostate cancer stimulates cell invasion. Sci. Rep. 2017, 7, 8476. [Google Scholar] [CrossRef] [PubMed]
- Venit, T.; Kalendová, A.; Petr, M.; Dzijak, R.; Pastorek, L.; Rohožková, J.; Malohlava, J.; Hozák, P. Nuclear myosin I regulates cell membrane tension. Sci. Rep. 2016, 6, 30864. [Google Scholar] [CrossRef]
- Sefer, E.; Kleyman, M.; Bar-Joseph, Z. Tradeoffs between dense and replicate sampling strategies for high-throughput time series experiments. Cell Syst. 2016, 3, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, C.-C.; Kang, S.; Li, J.-R.; Tseng, Y.-T.; Zhou, X.J. Pushing the annotation of cellular activities to a higher resolution: Predicting functions at the isoform level. Methods 2016, 93, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Bolstad, B.M.; Collin, F.; Cope, L.M.; Hobbs, B.; Speed, T.P. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003, 31, e15. [Google Scholar] [CrossRef] [PubMed]
- Bolstad, B.M.; Irizarry, R.A.; Åstrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef]
- Carvalho, B.S.; Irizarry, R.A. A framework for oligonucleotide microarray preprocessing. Bioinformatics 2010, 26, 2363–2367. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/. (accessed on 15 Augst 2019).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417. [Google Scholar] [CrossRef] [PubMed]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Thaben, P.F.; Westermark, P.O. Detecting rhythms in time series with RAIN. J. Biol. Rhythm. 2014, 29, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Lück, S.; Thurley, K.; Thaben, P.F.; Westermark, P.O. Rhythmic degradation explains and unifies circadian transcriptome and proteome data. Cell Rep. 2014, 9, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Agostinelli, C.; Lund, U. R Package Circular: Circular Statistics. 2017. Available online: https://rdrr.io/cran/circular/. (accessed on 15 Augst 2019).
- Larsson, J. Eulerr: Area-Proportional Euler and Venn Diagrams with Ellipses. 2018. Available online: https://rdrr.io/cran/eulerr/. (accessed on 15 Augst 2019).
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Girón, C.G. Ensembl 2018. Nucleic Acids Res. 2017, 46, D754–D761. [Google Scholar] [CrossRef]
- Quinlan, A.R. BEDTools: The Swiss-army tool for genome feature analysis. Curr. Protoc. Bioinform. 2014, 47, 11–12. [Google Scholar] [CrossRef]
- McLeay, R.C.; Bailey, T.L. Motif Enrichment Analysis: A unified framework and an evaluation on ChIP data. BMC Bioinform. 2010, 11, 165. [Google Scholar] [CrossRef]
- Bengtsson, H.; Simpson, K.; Bullard, J.; Hansen, K. Aroma. Affymetrix: A Generic Framework in R for Analyzing Small to Very Large Affymetrix Data Sets in Bounded Memory. 2018. Technical Report. Available online: https://www.google.com.hk/url?sa=t&rct=j&q=&esrc=s&source=web&cd=3&ved=2ahUKEwjPmfqxz4TkAhWlGaYKHa9zD2cQFjACegQIBBAC&url=https%3A%2F%2Fpdfs.semanticscholar.org%2F71ee%2F9edadec02aed109c1e9142f31ef49d5e81e2.pdf&usg=AOvVaw0r8_vMftA8RH-d5U7Jndsphttps://rdrr.io/cran/eulerr/ (accessed on 15 Augst 2019).
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics: A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Athman, R.; Knezevic, D.; Fuhr, L.; Relógio, A. A Computational Analysis of Alternative Splicing across Mammalian Tissues Reveals Circadian and Ultradian Rhythms in Splicing Events. Int. J. Mol. Sci. 2019, 20, 3977. https://doi.org/10.3390/ijms20163977
El-Athman R, Knezevic D, Fuhr L, Relógio A. A Computational Analysis of Alternative Splicing across Mammalian Tissues Reveals Circadian and Ultradian Rhythms in Splicing Events. International Journal of Molecular Sciences. 2019; 20(16):3977. https://doi.org/10.3390/ijms20163977
Chicago/Turabian StyleEl-Athman, Rukeia, Dora Knezevic, Luise Fuhr, and Angela Relógio. 2019. "A Computational Analysis of Alternative Splicing across Mammalian Tissues Reveals Circadian and Ultradian Rhythms in Splicing Events" International Journal of Molecular Sciences 20, no. 16: 3977. https://doi.org/10.3390/ijms20163977
APA StyleEl-Athman, R., Knezevic, D., Fuhr, L., & Relógio, A. (2019). A Computational Analysis of Alternative Splicing across Mammalian Tissues Reveals Circadian and Ultradian Rhythms in Splicing Events. International Journal of Molecular Sciences, 20(16), 3977. https://doi.org/10.3390/ijms20163977