Future Aspects of CDK5 in Prostate Cancer: From Pathogenesis to Therapeutic Implications

 , , , and
, , , and 
Abstract
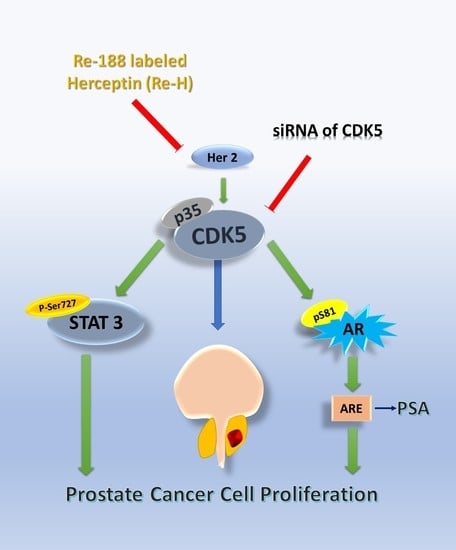
{kind=link}
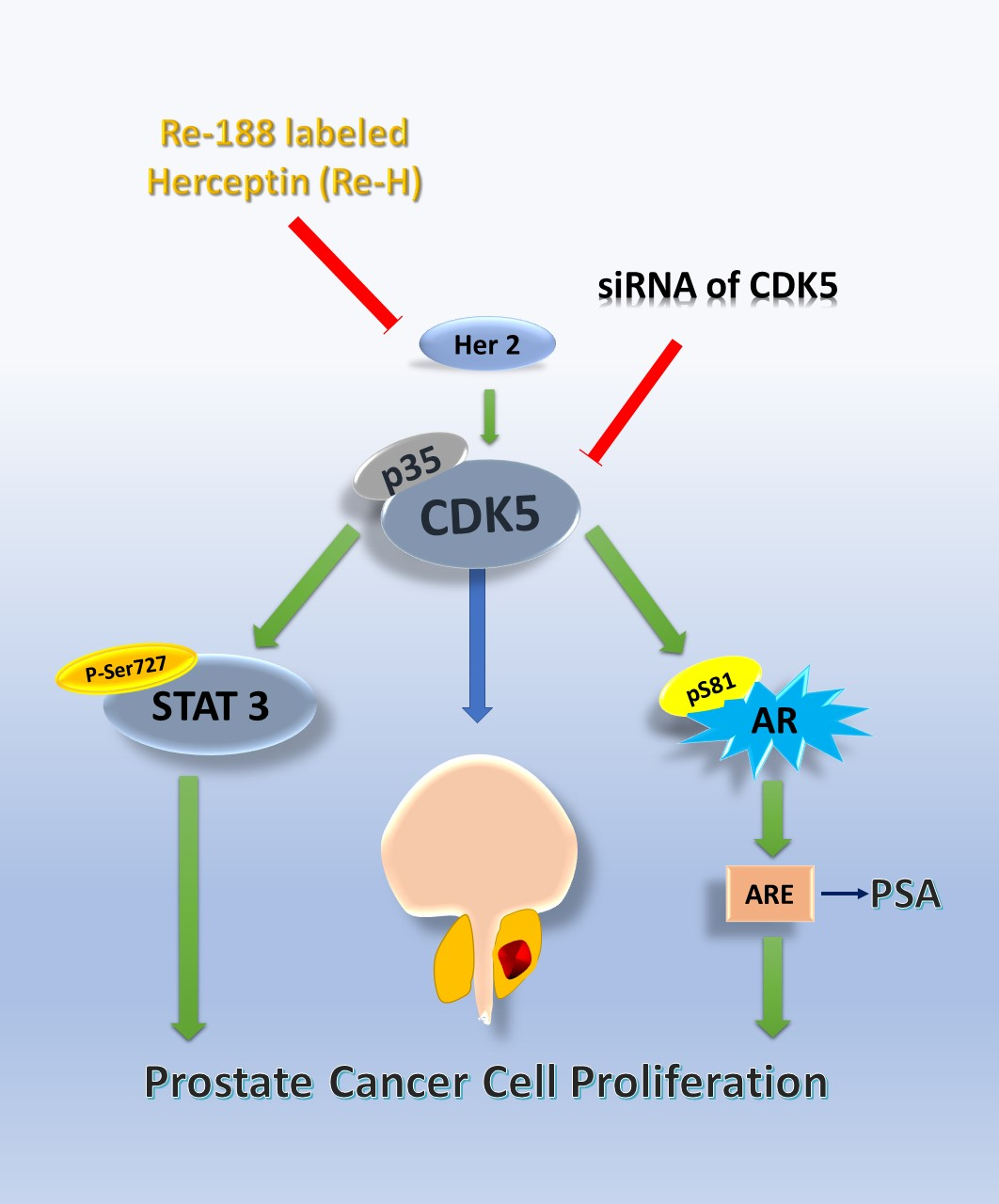
{kind=link}
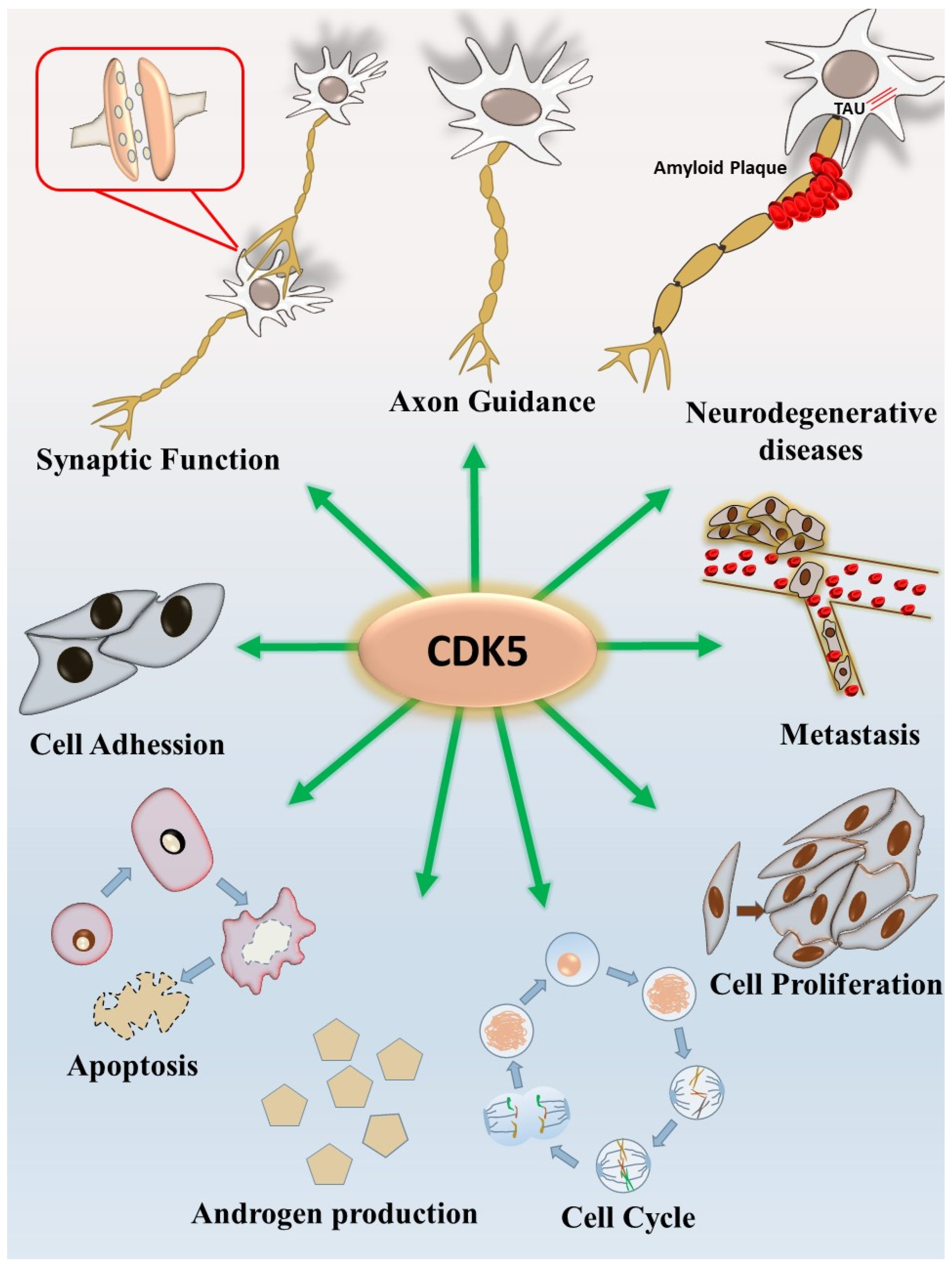
{kind=link}
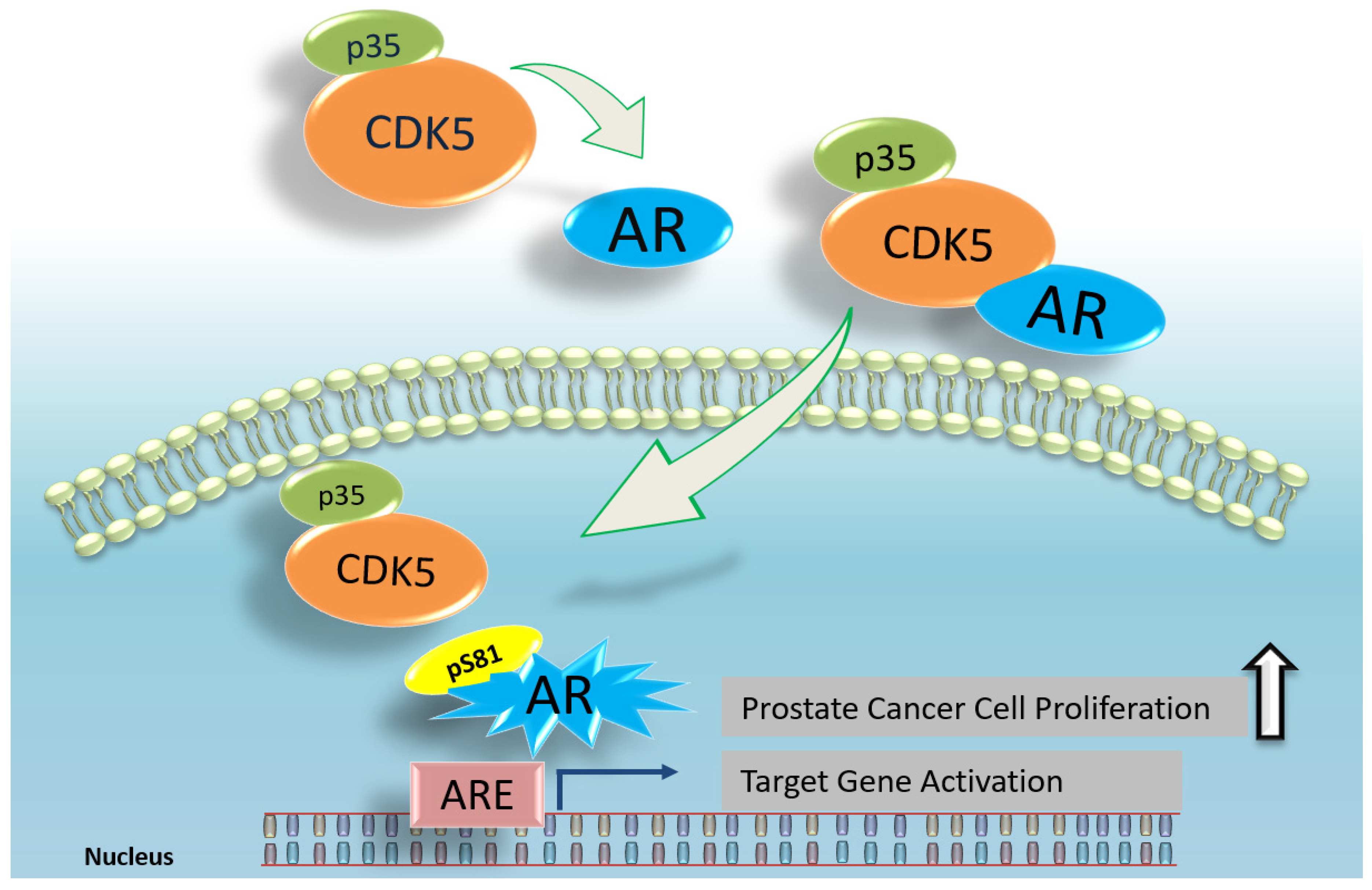
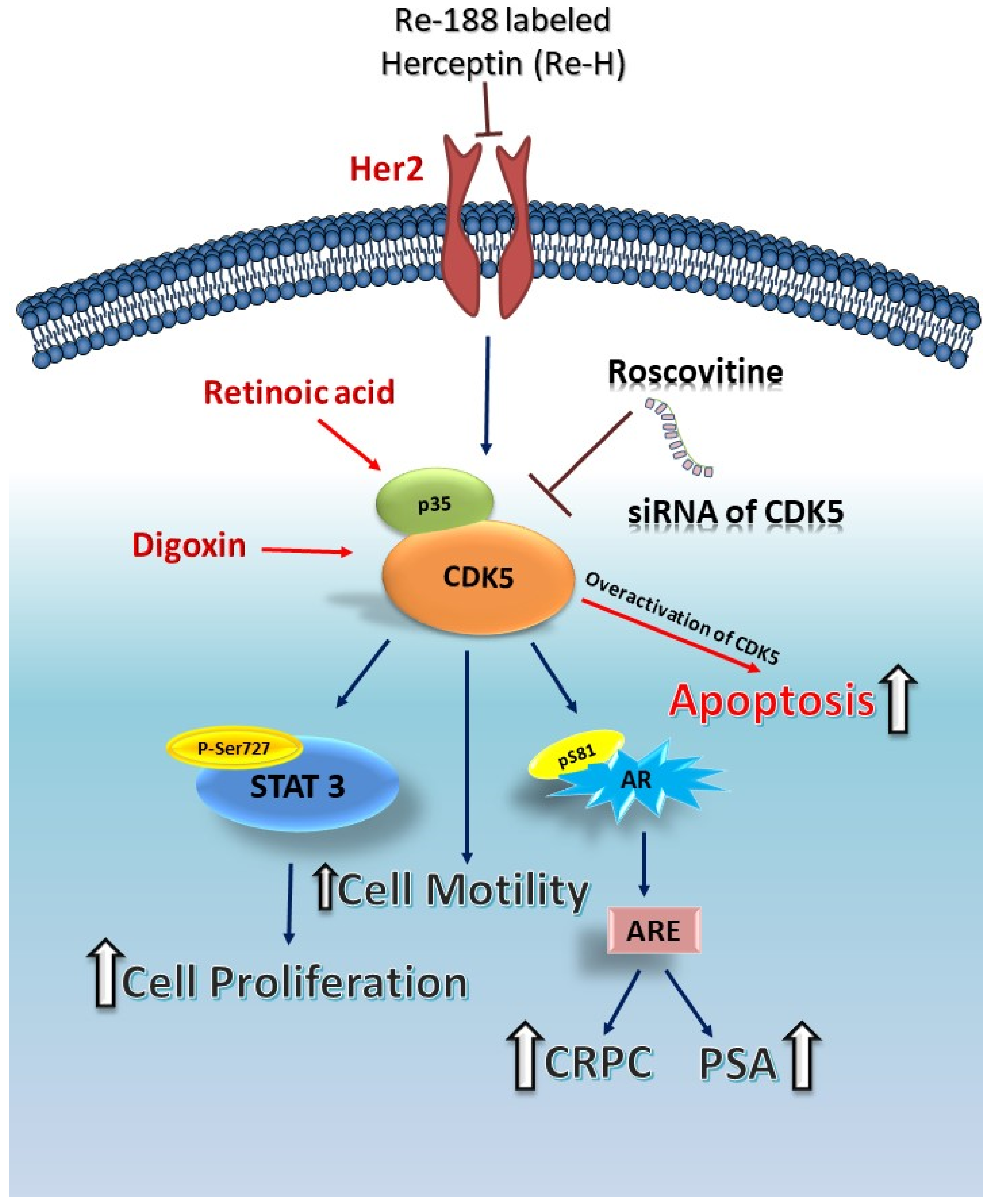{kind=link}
1. CDK5 and the Nervous System
2. CDK5 and Androgen Production
3. The Androgen Receptor and Prostate Cancer
4. CDK5 and Prostate Cancer
5. CDK5 and Apoptosis of Prostate Cancer Cells
6. CDK5 and AR in Prostate Cancer
7. CDK5-STAT3-AR in Prostate Cancer
8. The Relationship between CDK5 and Castration-Resistant Prostate Cancer
9. Conclusion and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CDK5 | Cyclin-dependent kinase 5 |
| CNS | Central nervous system |
| AR | Androgen receptor |
| siRNA | Small interfering RNAs |
| AD | Alzheimer’s disease |
| StAR | Steroidogenic acute regulatory |
| STAT3 | Signal transducer and activator of transcription 3 |
| PKC | Protein kinase C |
| DBD | DNA binding domain |
| AREs | Androgen responsive elements |
| IGF | Insulin-like growth factor |
| RA | Retinoic Acid |
| ADT | Androgen deprivation treatment |
| CRPC | Castration-resistant prostate cancer |
References
- Shupp, A.; Casimiro, M.C.; Pestell, R.G. Biological functions of CDK5 and potential CDK5 targeted clinical treatments. Oncotarget 2017, 8, 17373–17382. [Google Scholar] [CrossRef] [PubMed]
- Cortes, N.; Guzman-Martinez, L.; Andrade, V.; Gonzalez, A.; Maccioni, R.B. CDK5: A Unique CDK and Its Multiple Roles in the Nervous System. J. Alzheimers Dis. 2019. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.C.; Huang, C.Y.; Hsu, S.L.; Lin, E.; Ku, C.T.; Lin, H.; Chen, C.M. Retinoic Acid Induces Apoptosis of Prostate Cancer DU145 Cells through Cdk5 Overactivation. Evid Based Complement. Altern. Med. 2012, 2012, 580736. [Google Scholar] [CrossRef] [PubMed]
- Arif, A. Extraneuronal activities and regulatory mechanisms of the atypical cyclin-dependent kinase Cdk5. Biochem. Pharm. 2012, 84, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Tsai, L.H.; Delalle, I.; Caviness, V.S., Jr.; Chae, T.; Harlow, E. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature 1994, 371, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Minegishi, S.; Asada, A.; Miyauchi, S.; Fuchigami, T.; Saito, T.; Hisanaga, S. Membrane association facilitates degradation and cleavage of the cyclin-dependent kinase 5 activators p35 and p39. Biochemistry 2010, 49, 5482–5493. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, M.W.; Michel, T.M. Matrix metalloproteinases in autism spectrum disorders. J. Mol. Psychiatry 2013, 1, 16. [Google Scholar] [CrossRef] [PubMed]
- Hsu, F.N.; Chen, M.C.; Lin, K.C.; Peng, Y.T.; Li, P.C.; Lin, E.; Chiang, M.C.; Hsieh, J.T.; Lin, H. Cyclin-dependent kinase 5 modulates STAT3 and androgen receptor activation through phosphorylation of Ser(7)(2)(7) on STAT3 in prostate cancer cells. Am. J. Physiol Endocrinol Metab. 2013, 305, E975–E986. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.; Rossie, S. Tale of the Good and the Bad Cdk5: Remodeling of the Actin Cytoskeleton in the Brain. Mol. Neurobiol. 2018, 55, 3426–3438. [Google Scholar] [CrossRef]
- Chen, M.C.; Lin, H.; Hsu, F.N.; Huang, P.H.; Lee, G.S.; Wang, P.S. Involvement of cAMP in nerve growth factor-triggered p35/Cdk5 activation and differentiation in PC12 cells. Am. J. Physiol. Cell Physiol. 2010, 299, C516–C527. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, L.; Kordes, S.; Reinhardt, P.; Glatza, M.; Baumann, M.; Drexler, H.C.A.; Menninger, S.; Zischinsky, G.; Eickhoff, J.; Frob, C.; et al. Dual Inhibition of GSK3beta and CDK5 Protects the Cytoskeleton of Neurons from Neuroinflammatory-Mediated Degeneration In Vitro and In Vivo. Stem Cell Rep. 2019, 12, 502–517. [Google Scholar] [CrossRef] [PubMed]
- Qi, G.J.; Chen, Q.; Chen, L.J.; Shu, Y.; Bu, L.L.; Shao, X.Y.; Zhang, P.; Jiao, F.J.; Shi, J.; Tian, B. Phosphorylation of Connexin 43 by Cdk5 Modulates Neuronal Migration During Embryonic Brain Development. Mol. Neurobiol. 2016, 53, 2969–2982. [Google Scholar] [CrossRef] [PubMed]
- Diaz, A.; Jeanneret, V.; Merino, P.; McCann, P.; Yepes, M. Tissue-type plasminogen activator regulates p35-mediated Cdk5 activation in the postsynaptic terminal. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Periel, E.; Puigdellivol, M.; Brito, V.; Plattner, F.; Bibb, J.A.; Alberch, J.; Gines, S. Cdk5 Contributes to Huntington’s Disease Learning and Memory Deficits via Modulation of Brain Region-Specific Substrates. Mol. Neurobiol. 2018, 55, 6250–6268. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Ko, Y.U.; Chung, Y.; Yun, N.; Kim, M.; Kim, K.; Oh, Y.J. The acetylation of cyclin-dependent kinase 5 at lysine 33 regulates kinase activity and neurite length in hippocampal neurons. Sci. Rep. 2018, 8, 13676. [Google Scholar] [CrossRef]
- Liang, Z.; Ye, T.; Zhou, X.; Lai, K.O.; Fu, A.K.; Ip, N.Y. Cdk5 Regulates Activity-Dependent Gene Expression and Dendrite Development. J. Neurosci. 2015, 35, 15127–15134. [Google Scholar] [CrossRef]
- Liu, S.L.; Wang, C.; Jiang, T.; Tan, L.; Xing, A.; Yu, J.T. The Role of Cdk5 in Alzheimer’s Disease. Mol. Neurobiol. 2016, 53, 4328–4342. [Google Scholar] [CrossRef]
- Lin, H.; Lin, T.Y.; Juang, J.L. Abl deregulates Cdk5 kinase activity and subcellular localization in Drosophila neurodegeneration. Cell Death Differ. 2007, 14, 607–615. [Google Scholar] [CrossRef]
- Corbel, C.; Zhang, B.; Le Parc, A.; Baratte, B.; Colas, P.; Couturier, C.; Kosik, K.S.; Landrieu, I.; Le Tilly, V.; Bach, S. Tamoxifen Inhibits CDK5 Kinase Activity by Interacting with p35/p25 and Modulates the Pattern of Tau Phosphorylation. Chem. Biol. 2015, 22, 472–482. [Google Scholar] [CrossRef]
- Pozo, K.; Bibb, J.A. The Emerging Role of Cdk5 in Cancer. Trends Cancer 2016, 2, 606–618. [Google Scholar] [CrossRef]
- Wei, F.Y.; Nagashima, K.; Ohshima, T.; Saheki, Y.; Lu, Y.F.; Matsushita, M.; Yamada, Y.; Mikoshiba, K.; Seino, Y.; Matsui, H.; et al. Cdk5-dependent regulation of glucose-stimulated insulin secretion. Nat. Med. 2005, 11, 1104–1108. [Google Scholar] [CrossRef] [PubMed]
- Banks, A.S.; McAllister, F.E.; Camporez, J.P.; Zushin, P.J.; Jurczak, M.J.; Laznik-Bogoslavski, D.; Shulman, G.I.; Gygi, S.P.; Spiegelman, B.M. An ERK/Cdk5 axis controls the diabetogenic actions of PPARgamma. Nature 2015, 517, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Musa, F.R.; Takenaka, I.; Konishi, R.; Tokuda, M. Effects of luteinizing hormone, follicle-stimulating hormone, and epidermal growth factor on expression and kinase activity of cyclin-dependent kinase 5 in Leydig TM3 and Sertoli TM4 cell lines. J. Androl. 2000, 21, 392–402. [Google Scholar] [PubMed]
- Musa, F.R.; Tokuda, M.; Kuwata, Y.; Ogawa, T.; Tomizawa, K.; Konishi, R.; Takenaka, I.; Hatase, O. Expression of cyclin-dependent kinase 5 and associated cyclins in Leydig and Sertoli cells of the testis. J. Androl. 1998, 19, 657–666. [Google Scholar] [PubMed]
- Lin, H.; Chen, M.C.; Ku, C.T. Cyclin-dependent kinase 5 regulates steroidogenic acute regulatory protein and androgen production in mouse Leydig cells. Endocrinology 2009, 150, 396–403. [Google Scholar] [CrossRef]
- Harpelunde Poulsen, K.; Jorgensen, A. Role of Nodal signalling in testis development and initiation of testicular cancer. Reproduction 2019. [Google Scholar] [CrossRef]
- Roumaud, P.; Martin, L.J. Roles of leptin, adiponectin and resistin in the transcriptional regulation of steroidogenic genes contributing to decreased Leydig cells function in obesity. Horm Mol. Biol. Clin. Investig. 2015, 24, 25–45. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, F.; Ye, L.; Zirkin, B.; Chen, H. Steroidogenesis in Leydig cells: Effects of aging and environmental factors. Reproduction 2017, 154, R111–R122. [Google Scholar] [CrossRef]
- Cai, Z.; Chen, W.; Zhang, J.; Li, H. Androgen receptor: What we know and what we expect in castration-resistant prostate cancer. Int. Urol. Nephrol. 2018, 50, 1753–1764. [Google Scholar] [CrossRef]
- Zarif, J.C.; Miranti, C.K. The importance of non-nuclear AR signaling in prostate cancer progression and therapeutic resistance. Cell Signal. 2016, 28, 348–356. [Google Scholar] [CrossRef]
- Crumbaker, M.; Khoja, L.; Joshua, A.M. AR Signaling and the PI3K Pathway in Prostate Cancer. Cancers 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Davey, R.A.; Grossmann, M. Androgen Receptor Structure, Function and Biology: From Bench to Bedside. Clin. Biochem Rev. 2016, 37, 3–15. [Google Scholar] [PubMed]
- Smith, L.B.; Walker, W.H. The regulation of spermatogenesis by androgens. Semin. Cell Dev. Biol. 2014, 30, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Mayerhofer, A. Peritubular cells of the human testis: Prostaglandin E2 and more. Andrology 2019. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.; Adam, M.; Walenta, L.; Schmid, N.; Heikela, H.; Schubert, K.; Flenkenthaler, F.; Dietrich, K.G.; Gruschka, S.; Arnold, G.J.; et al. Insights into the role of androgen receptor in human testicular peritubular cells. Andrology 2018, 6, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Yang, B.; Pan, P.; Ma, Q.; Wu, Y.; Zhang, Z.; Guo, X.; Ye, J.; Gui, Y. MicroRNA-130a inhibits spermatogenesis by directly targeting androgen receptor in mouse Sertoli cells. Mol. Reprod. Dev. 2018, 85, 768–777. [Google Scholar] [CrossRef]
- Tan, M.H.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharm. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef]
- Leung, J.K.; Sadar, M.D. Non-Genomic Actions of the Androgen Receptor in Prostate Cancer. Front. Endocrinol (Lausanne) 2017, 8, 2. [Google Scholar] [CrossRef]
- Eder, I.E.; Culig, Z.; Putz, T.; Nessler-Menardi, C.; Bartsch, G.; Klocker, H. Molecular biology of the androgen receptor: From molecular understanding to the clinic. Eur. Urol. 2001, 40, 241–251. [Google Scholar] [CrossRef]
- Hsu, F.N.; Chen, M.C.; Chiang, M.C.; Lin, E.; Lee, Y.T.; Huang, P.H.; Lee, G.S.; Lin, H. Regulation of androgen receptor and prostate cancer growth by cyclin-dependent kinase 5. J. Biol. Chem. 2011, 286, 33141–33149. [Google Scholar] [CrossRef]
- Chiker, S.; Pennaneach, V.; Loew, D.; Dingli, F.; Biard, D.; Cordelieres, F.P.; Gemble, S.; Vacher, S.; Bieche, I.; Hall, J.; et al. Cdk5 promotes DNA replication stress checkpoint activation through RPA-32 phosphorylation, and impacts on metastasis free survival in breast cancer patients. Cell Cycle 2015, 14, 3066–3078. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Liu, C.; Wu, D.; Li, Z.; Li, C.; Zhang, Y. Phosphorylation of kinase insert domain receptor by cyclin-dependent kinase 5 at serine 229 is associated with invasive behavior and poor prognosis in prolactin pituitary adenomas. Oncotarget 2016, 7, 50883–50894. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pakula, H.; Xiang, D.; Li, Z. A Tale of Two Signals: AR and WNT in Development and Tumorigenesis of Prostate and Mammary Gland. Cancers 2017, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, W.J. Insulin-like growth factor-I induces androgen receptor activation in differentiating C2C12 skeletal muscle cells. Mol. Cells 2009, 28, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, W.J. Ligand-independent activation of the androgen receptor by insulin-like growth factor-I and the role of the MAPK pathway in skeletal muscle cells. Mol. Cells 2009, 28, 589–593. [Google Scholar] [CrossRef]
- Coffey, K.; Robson, C.N. Regulation of the androgen receptor by post-translational modifications. J. Endocrinol. 2012, 215, 221–237. [Google Scholar] [CrossRef]
- Daniels, G.; Pei, Z.; Logan, S.K.; Lee, P. Mini-review: Androgen receptor phosphorylation in prostate cancer. Am. J. Clin. Exp. Urol. 2013, 1, 25–29. [Google Scholar]
- Blom, N.; Gammeltoft, S.; Brunak, S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 1999, 294, 1351–1362. [Google Scholar] [CrossRef]
- Liu, S.; Yuan, Y.; Okumura, Y.; Shinkai, N.; Yamauchi, H. Camptothecin disrupts androgen receptor signaling and suppresses prostate cancer cell growth. Biochem Biophys Res. Commun. 2010, 394, 297–302. [Google Scholar] [CrossRef]
- Gordon, V.; Bhadel, S.; Wunderlich, W.; Zhang, J.; Ficarro, S.B.; Mollah, S.A.; Shabanowitz, J.; Hunt, D.F.; Xenarios, I.; Hahn, W.C.; et al. CDK9 regulates AR promoter selectivity and cell growth through serine 81 phosphorylation. Mol. Endocrinol. 2010, 24, 2267–2280. [Google Scholar] [CrossRef]
- Gioeli, D.; Ficarro, S.B.; Kwiek, J.J.; Aaronson, D.; Hancock, M.; Catling, A.D.; White, F.M.; Christian, R.E.; Settlage, R.E.; Shabanowitz, J.; et al. Androgen receptor phosphorylation. Regulation and identification of the phosphorylation sites. J. Biol. Chem. 2002, 277, 29304–29314. [Google Scholar] [CrossRef] [PubMed]
- Shigemura, K.; Isotani, S.; Wang, R.; Fujisawa, M.; Gotoh, A.; Marshall, F.F.; Zhau, H.E.; Chung, L.W. Soluble factors derived from stroma activated androgen receptor phosphorylation in human prostate LNCaP cells: Roles of ERK/MAP kinase. Prostate 2009, 69, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Juang, J.L.; Wang, P.S. Involvement of Cdk5/p25 in digoxin-triggered prostate cancer cell apoptosis. J. Biol. Chem. 2004, 279, 29302–29307. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Tucker-Burden, C.; Kaissi, E.; Newsam, A.; Duggireddy, H.; Chau, M.; Zhang, C.; Diwedi, B.; Rupji, M.; Seby, S.; et al. CDK5 Inhibition Resolves PKA/cAMP-Independent Activation of CREB1 Signaling in Glioma Stem Cells. Cell Rep. 2018, 23, 1651–1664. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Xie, S.; Liu, Y.; Shen, C.; Song, X.; Zhou, G.L.; Wang, C. CDK5 Functions as a Tumor Promoter in Human Lung Cancer. J. Cancer 2018, 9, 3950–3961. [Google Scholar] [CrossRef] [PubMed]
- Lampropoulou, E.; Logoviti, I.; Koutsioumpa, M.; Hatziapostolou, M.; Polytarchou, C.; Skandalis, S.S.; Hellman, U.; Fousteris, M.; Nikolaropoulos, S.; Choleva, E.; et al. Cyclin-dependent kinase 5 mediates pleiotrophin-induced endothelial cell migration. Sci. Rep. 2018, 8, 5893. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Chen, M.C.; Chiu, C.Y.; Song, Y.M.; Lin, S.Y. Cdk5 regulates STAT3 activation and cell proliferation in medullary thyroid carcinoma cells. J. Biol. Chem. 2007, 282, 2776–2784. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Lin, J.X.; Zhang, P.Y.; Sun, Y.Q.; Li, P.; Xie, J.W.; Wang, J.B.; Chen, Q.Y.; Cao, L.L.; Lin, Y.; et al. CDK5 suppresses the metastasis of gastric cancer cells by interacting with and regulating PP2A. Oncol. Rep. 2019, 41, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Zhou, J.; Zhang, J.; Wu, S.; Yang, X.; Zhao, X.; Li, H.; Luo, M.; Yu, Q.; Lin, G.; et al. Cyclin-dependent kinase 5 decreases in gastric cancer and its nuclear accumulation suppresses gastric tumorigenesis. Clin. Cancer Res. 2015, 21, 1419–1428. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.H.; Chen, M.C.; Peng, Y.T.; Kao, W.H.; Chang, C.H.; Wang, Y.C.; Lai, C.H.; Hsieh, J.T.; Wang, J.H.; Lee, Y.T.; et al. Cdk5 Directly Targets Nuclear p21CIP1 and Promotes Cancer Cell Growth. Cancer Res. 2016, 76, 6888–6900. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.R.; Klein, E.A. Risk factors for prostate cancer. Nat. Clin. Pr. Urol. 2009, 6, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Khurana, N.; Sikka, S.C. Interplay between SOX9, Wnt/beta-Catenin and Androgen Receptor Signaling in Castration-Resistant Prostate Cancer. Int. J. Mol. Sci. 2019, 20. [Google Scholar]
- Wadosky, K.M.; Koochekpour, S. Androgen receptor splice variants and prostate cancer: From bench to bedside. Oncotarget 2017, 8, 18550–18576. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.I. Splicing Factors Have an Essential Role in Prostate Cancer Progression and Androgen Receptor Signaling. Biomolecules 2019, 9, 131. [Google Scholar] [CrossRef] [PubMed]
- Culig, Z.; Santer, F.R. Androgen receptor signaling in prostate cancer. Cancer Metastasis Rev. 2014, 33, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef]
- Strock, C.J.; Park, J.I.; Nakakura, E.K.; Bova, G.S.; Isaacs, J.T.; Ball, D.W.; Nelkin, B.D. Cyclin-dependent kinase 5 activity controls cell motility and metastatic potential of prostate cancer cells. Cancer Res. 2006, 66, 7509–7515. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Goswami, S.; Sahai, E.; Wyckoff, J.B.; Segall, J.E.; Condeelis, J.S. Tumor cells caught in the act of invading: Their strategy for enhanced cell motility. Trends Cell Biol. 2005, 15, 138–145. [Google Scholar] [CrossRef]
- Wang, W.; Goswami, S.; Lapidus, K.; Wells, A.L.; Wyckoff, J.B.; Sahai, E.; Singer, R.H.; Segall, J.E.; Condeelis, J.S. Identification and testing of a gene expression signature of invasive carcinoma cells within primary mammary tumors. Cancer Res. 2004, 64, 8585–8594. [Google Scholar] [CrossRef]
- Sahai, E. Mechanisms of cancer cell invasion. Curr. Opin. Genet. Dev. 2005, 15, 87–96. [Google Scholar] [CrossRef]
- Dorand, R.D.; Nthale, J.; Myers, J.T.; Barkauskas, D.S.; Avril, S.; Chirieleison, S.M.; Pareek, T.K.; Abbott, D.W.; Stearns, D.S.; Letterio, J.J.; et al. Cdk5 disruption attenuates tumor PD-L1 expression and promotes antitumor immunity. Science 2016, 353, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Mandl, M.M.; Zhang, S.; Ulrich, M.; Schmoeckel, E.; Mayr, D.; Vollmar, A.M.; Liebl, J. Inhibition of Cdk5 induces cell death of tumor-initiating cells. Br. J. Cancer 2017, 116, 912–922. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, J.; Song, Y.S.; Li, Y.; Jia, Y.H.; Zhao, H.D. Cdk5 links with DNA damage response and cancer. Mol. Cancer 2017, 16, 60. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Patrick, G.N.; Zukerberg, L.; Nikolic, M.; de la Monte, S.; Dikkes, P.; Tsai, L.H. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 1999, 402, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.; Chen, M.C.; Huang, C.Y.; Hsu, S.L.; Huang, W.J.; Lin, M.S.; Wu, J.C.; Lin, H. All-trans retinoic acid induces DU145 cell cycle arrest through Cdk5 activation. Cell Physiol. Biochem. 2014, 33, 1620–1630. [Google Scholar] [CrossRef]
- Kuo, H.S.; Hsu, F.N.; Chiang, M.C.; You, S.C.; Chen, M.C.; Lo, M.J.; Lin, H. The role of Cdk5 in retinoic acid-induced apoptosis of cervical cancer cell line. Chin. J. Physiol. 2009, 52, 23–30. [Google Scholar] [CrossRef]
- Cicenas, J.; Kalyan, K.; Sorokinas, A.; Stankunas, E.; Levy, J.; Meskinyte, I.; Stankevicius, V.; Kaupinis, A.; Valius, M. Roscovitine in cancer and other diseases. Ann. Transl. Med. 2015, 3, 135. [Google Scholar]
- Gary, C.; Hajek, M.; Biktasova, A.; Bellinger, G.; Yarbrough, W.G.; Issaeva, N. Selective antitumor activity of roscovitine in head and neck cancer. Oncotarget 2016, 7, 38598–38611. [Google Scholar] [CrossRef]
- Pandey, V.; Ranjan, N.; Narne, P.; Babu, P.P. Roscovitine effectively enhances antitumor activity of temozolomide in vitro and in vivo mediated by increased autophagy and Caspase-3 dependent apoptosis. Sci. Rep. 2019, 9, 5012. [Google Scholar] [CrossRef] [PubMed]
- Ozfiliz-Kilbas, P.; Sarikaya, B.; Obakan-Yerlikaya, P.; Coker-Gurkan, A.; Arisan, E.D.; Temizci, B.; Palavan-Unsal, N. Cyclin-dependent kinase inhibitors, roscovitine and purvalanol, induce apoptosis and autophagy related to unfolded protein response in HeLa cervical cancer cells. Mol. Biol. Rep. 2018, 45, 815–828. [Google Scholar] [CrossRef] [PubMed]
- Hsu, F.N.; Kao, W.H.; Huang, P.H.; Yu, C.H.; Wang, H.Y.; Chiu, K.Y.; Yang, T.Y.; Teng, C.L.J.; Chen, K.C.; Lin, H. The Inhibitory Effect of Roscovitine on Prostate Cancer Cell Proliferation and Androgen Receptor Phosphorylation. 調適醫學 2018, 10, 34–42. [Google Scholar] [CrossRef]
- Hsu, S.L.; Chen, M.C.; Chou, Y.H.; Hwang, G.Y.; Yin, S.C. Induction of p21 (CIP1/Waf1) and activation of p34 (cdc2) involved in retinoic acid-induced apoptosis in human hepatoma Hep3B cells. Exp. Cell Res. 1999, 248, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Parkinson, D.R.; Cheson, B.D.; Friedman, M.A. Retinoids in cancer therapy. J. Clin. Oncol. 1992, 10, 839–864. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.Y.; Huang, W.J.; Kan, S.F.; Wang, P.S. Inhibitory effects of digitalis on the proliferation of androgen dependent and independent prostate cancer cells. J. Urol. 2001, 166, 1937–1942. [Google Scholar] [CrossRef]
- Amin, K.S.; Jagadeesh, S.; Baishya, G.; Rao, P.G.; Barua, N.C.; Bhattacharya, S.; Banerjee, P.P. A naturally derived small molecule disrupts ligand-dependent and ligand-independent androgen receptor signaling in human prostate cancer cells. Mol. Cancer 2014, 13, 341–352. [Google Scholar] [CrossRef]
- Hsu, F.N.; Yang, M.S.; Lin, E.; Tseng, C.F.; Lin, H. The significance of Her2 on androgen receptor protein stability in the transition of androgen requirement in prostate cancer cells. Am. J. Physiol. Endocrinol Metab. 2011, 300, E902–E908. [Google Scholar] [CrossRef]
- Coleman, K.G.; Crews, C.M. Proteolysis-Targeting Chimeras: Harnessing the Ubiquitin-Proteasome System to Induce Degradation of Specific Target Proteins. Annu. Rev. Cancer Biol. 2018, 2, 41–58. [Google Scholar] [CrossRef]
- Wade, S.L.; Auble, D.T. The Rad23 ubiquitin receptor, the proteasome and functional specificity in transcriptional control. Transcription 2010, 1, 22–26. [Google Scholar] [CrossRef]
- O’Malley, B.W. Sequentiality and processivity of nuclear receptor coregulators in regulation of target gene expression. Nucl. Recept. Signal. 2003, 1, e010. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Li, Q.; He, L.; Lim, H.Y.; Fu, X.; Cheung, N.S.; Qi, D.X.; Qi, R.Z. Microtubule association of the neuronal p35 activator of Cdk5. J. Biol. Chem. 2007, 282, 18666–18670. [Google Scholar] [CrossRef] [PubMed]
- Fu, A.K.; Fu, W.Y.; Ng, A.K.; Chien, W.W.; Ng, Y.P.; Wang, J.H.; Ip, N.Y. Cyclin-dependent kinase 5 phosphorylates signal transducer and activator of transcription 3 and regulates its transcriptional activity. Proc. Natl. Acad. Sci. USA 2004, 101, 6728–6733. [Google Scholar] [CrossRef] [PubMed]
- Cheung, Z.H.; Fu, A.K.; Ip, N.Y. Synaptic roles of Cdk5: Implications in higher cognitive functions and neurodegenerative diseases. Neuron 2006, 50, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.S.; Zhou, X.; Huang, Y.Y.; Kong, L.P.; Mei, M.; Guo, W.Y.; Zhao, M.H.; Ren, Y.; Shen, Q.; Zhang, L. Targeting STAT3/miR-21 axis inhibits epithelial-mesenchymal transition via regulating CDK5 in head and neck squamous cell carcinoma. Mol. Cancer 2015, 14, 213. [Google Scholar] [CrossRef]
- Selvendiran, K.; Koga, H.; Ueno, T.; Yoshida, T.; Maeyama, M.; Torimura, T.; Yano, H.; Kojiro, M.; Sata, M. Luteolin promotes degradation in signal transducer and activator of transcription 3 in human hepatoma cells: An implication for the antitumor potential of flavonoids. Cancer Res. 2006, 66, 4826–4834. [Google Scholar] [CrossRef]
- Courapied, S.; Sellier, H.; de Carne Trecesson, S.; Vigneron, A.; Bernard, A.C.; Gamelin, E.; Barre, B.; Coqueret, O. The cdk5 kinase regulates the STAT3 transcription factor to prevent DNA damage upon topoisomerase I inhibition. J. Biol. Chem. 2010, 285, 26765–26778. [Google Scholar] [CrossRef]
- Yeh, Y.T.; Ou-Yang, F.; Chen, I.F.; Yang, S.F.; Wang, Y.Y.; Chuang, H.Y.; Su, J.H.; Hou, M.F.; Yuan, S.S. STAT3 ser727 phosphorylation and its association with negative estrogen receptor status in breast infiltrating ductal carcinoma. Int. J. Cancer 2006, 118, 2943–2947. [Google Scholar] [CrossRef]
- Galletti, G.; Leach, B.I.; Lam, L.; Tagawa, S.T. Mechanisms of resistance to systemic therapy in metastatic castration-resistant prostate cancer. Cancer Treat. Rev. 2017, 57, 16–27. [Google Scholar] [CrossRef]
- Calcinotto, A.; Spataro, C.; Zagato, E.; Di Mitri, D.; Gil, V.; Crespo, M.; De Bernardis, G.; Losa, M.; Mirenda, M.; Pasquini, E.; et al. IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature 2018, 559, 363–369. [Google Scholar] [CrossRef]
- Perner, S.; Cronauer, M.V.; Schrader, A.J.; Klocker, H.; Culig, Z.; Baniahmad, A. Adaptive responses of androgen receptor signaling in castration-resistant prostate cancer. Oncotarget 2015, 6, 35542–35555. [Google Scholar] [CrossRef] [PubMed]
- Chi, K.N.; Bjartell, A.; Dearnaley, D.; Saad, F.; Schroder, F.H.; Sternberg, C.; Tombal, B.; Visakorpi, T. Castration-resistant prostate cancer: From new pathophysiology to new treatment targets. Eur. Urol. 2009, 56, 594–605. [Google Scholar] [CrossRef] [PubMed]
- Ferraldeschi, R.; Welti, J.; Luo, J.; Attard, G.; de Bono, J.S. Targeting the androgen receptor pathway in castration-resistant prostate cancer: Progresses and prospects. Oncogene 2014, 34, 1745. [Google Scholar] [CrossRef] [PubMed]
- Mellinghoff, I.K.; Vivanco, I.; Kwon, A.; Tran, C.; Wongvipat, J.; Sawyers, C.L. HER2/neu kinase-dependent modulation of androgen receptor function through effects on DNA binding and stability. Cancer Cell 2004, 6, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Freeman, M.R. HER2/HER3 heterodimers in prostate cancer: Whither HER1/EGFR? Cancer Cell 2004, 6, 427–428. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mountzios, G.; Kotoula, V.; Kolliou, G.A.; Papadopoulou, K.; Lazaridis, G.; Christodoulou, C.; Pentheroudakis, G.; Skondra, M.; Koutras, A.; Linardou, H.; et al. Cyclin D1 differential activation and its prognostic impact in patients with advanced breast cancer treated with trastuzumab. ESMO Open 2019, 4, e000441. [Google Scholar] [CrossRef]
- Cameron, D.; Piccart-Gebhart, M.J.; Gelber, R.D.; Procter, M.; Goldhirsch, A.; de Azambuja, E.; Castro, G., Jr.; Untch, M.; Smith, I.; Gianni, L.; et al. 11 years’ follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive early breast cancer: Final analysis of the HERceptin Adjuvant (HERA) trial. Lancet 2017, 389, 1195–1205. [Google Scholar] [CrossRef]
- Wang, H.Y.; Lin, W.Y.; Chen, M.C.; Lin, T.; Chao, C.H.; Hsu, F.N.; Lin, E.; Huang, C.Y.; Luo, T.Y.; Lin, H. Inhibitory effects of Rhenium-188-labeled Herceptin on prostate cancer cell growth: A possible radioimmunotherapy to prostate carcinoma. Int. J. Radiat. Biol. 2013, 89, 346–355. [Google Scholar] [CrossRef]
- Pareek, T.K.; Kulkarni, A.B. Cdk5: A new player in pain signaling. Cell Cycle 2006, 5, 585–588. [Google Scholar] [CrossRef]
- Ubeda, M.; Rukstalis, J.M.; Habener, J.F. Inhibition of cyclin-dependent kinase 5 activity protects pancreatic beta cells from glucotoxicity. J. Biol. Chem. 2006, 281, 28858–28864. [Google Scholar] [CrossRef]
- Lenjisa, J.L.; Tadesse, S.; Khair, N.Z.; Kumarasiri, M.; Yu, M.; Albrecht, H.; Milne, R.; Wang, S. CDK5 in oncology: Recent advances and future prospects. Future Med. Chem. 2017, 9, 1939–1962. [Google Scholar] [CrossRef] [PubMed]
- Bolin, C.; Boudra, M.T.; Fernet, M.; Vaslin, L.; Pennaneach, V.; Zaremba, T.; Biard, D.; Cordelieres, F.P.; Favaudon, V.; Megnin-Chanet, F.; et al. The impact of cyclin-dependent kinase 5 depletion on poly (ADP-ribose) polymerase activity and responses to radiation. Cell Mol. Life Sci. 2012, 69, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, B.K.; Qian, X.; Mertins, P.; Wang, D.; Papageorge, A.G.; Carr, S.A.; Lowy, D.R. CDK5 is a major regulator of the tumor suppressor DLC1. J. Cell. Biol. 2014, 207, 627–642. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oner, M.; Lin, E.; Chen, M.-C.; Hsu, F.-N.; Shazzad Hossain Prince, G.M.; Chiu, K.-Y.; Teng, C.-L.J.; Yang, T.-Y.; Wang, H.-Y.; Yue, C.-H.; et al. Future Aspects of CDK5 in Prostate Cancer: From Pathogenesis to Therapeutic Implications. Int. J. Mol. Sci. 2019, 20, 3881. https://doi.org/10.3390/ijms20163881
Oner M, Lin E, Chen M-C, Hsu F-N, Shazzad Hossain Prince GM, Chiu K-Y, Teng C-LJ, Yang T-Y, Wang H-Y, Yue C-H, et al. Future Aspects of CDK5 in Prostate Cancer: From Pathogenesis to Therapeutic Implications. International Journal of Molecular Sciences. 2019; 20(16):3881. https://doi.org/10.3390/ijms20163881
Chicago/Turabian StyleOner, Muhammet, Eugene Lin, Mei-Chih Chen, Fu-Ning Hsu, G M Shazzad Hossain Prince, Kun-Yuan Chiu, Chieh-Lin Jerry Teng, Tsung-Ying Yang, Hsin-Yi Wang, Chia-Herng Yue, and et al. 2019. "Future Aspects of CDK5 in Prostate Cancer: From Pathogenesis to Therapeutic Implications" International Journal of Molecular Sciences 20, no. 16: 3881. https://doi.org/10.3390/ijms20163881
APA StyleOner, M., Lin, E., Chen, M.-C., Hsu, F.-N., Shazzad Hossain Prince, G. M., Chiu, K.-Y., Teng, C.-L. J., Yang, T.-Y., Wang, H.-Y., Yue, C.-H., Yu, C.-H., Lai, C.-H., Hsieh, J.-T., & Lin, H. (2019). Future Aspects of CDK5 in Prostate Cancer: From Pathogenesis to Therapeutic Implications. International Journal of Molecular Sciences, 20(16), 3881. https://doi.org/10.3390/ijms20163881