Exploring the Phylogeography of Ancient Platycladus orientalis in China by Specific-Locus Amplified Fragment Sequencing

 ,
,
Abstract
1. Introduction
2. Results
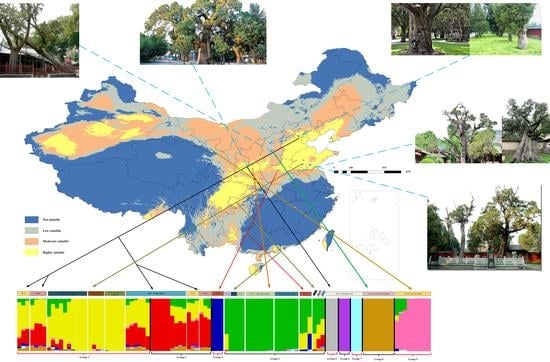
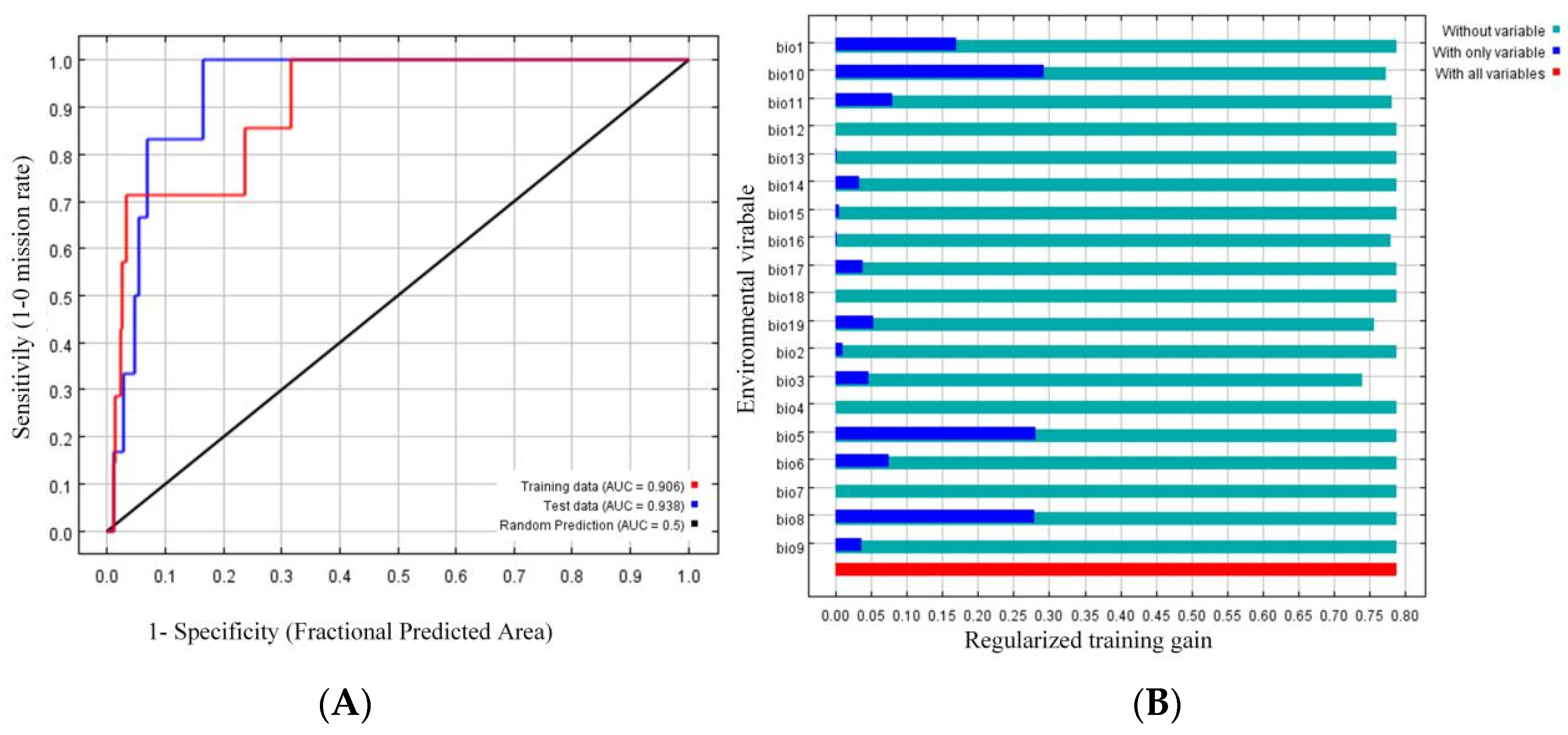
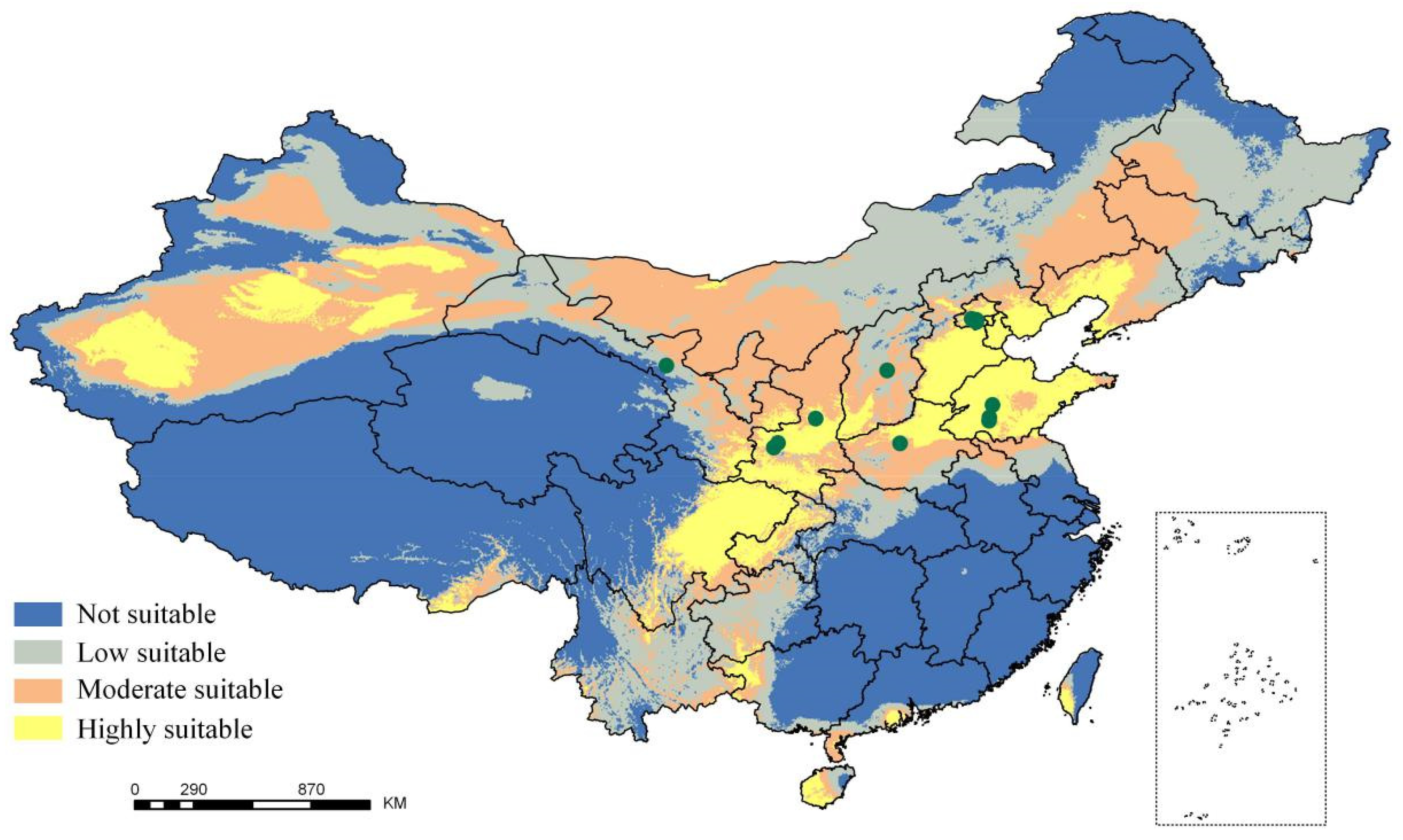
2.1. Potential Tree Distribution and Effects of Climatic Factors
2.2. Statistical Analysis of the Sequencing Data
2.3. SLAF Tags and SNP Sites
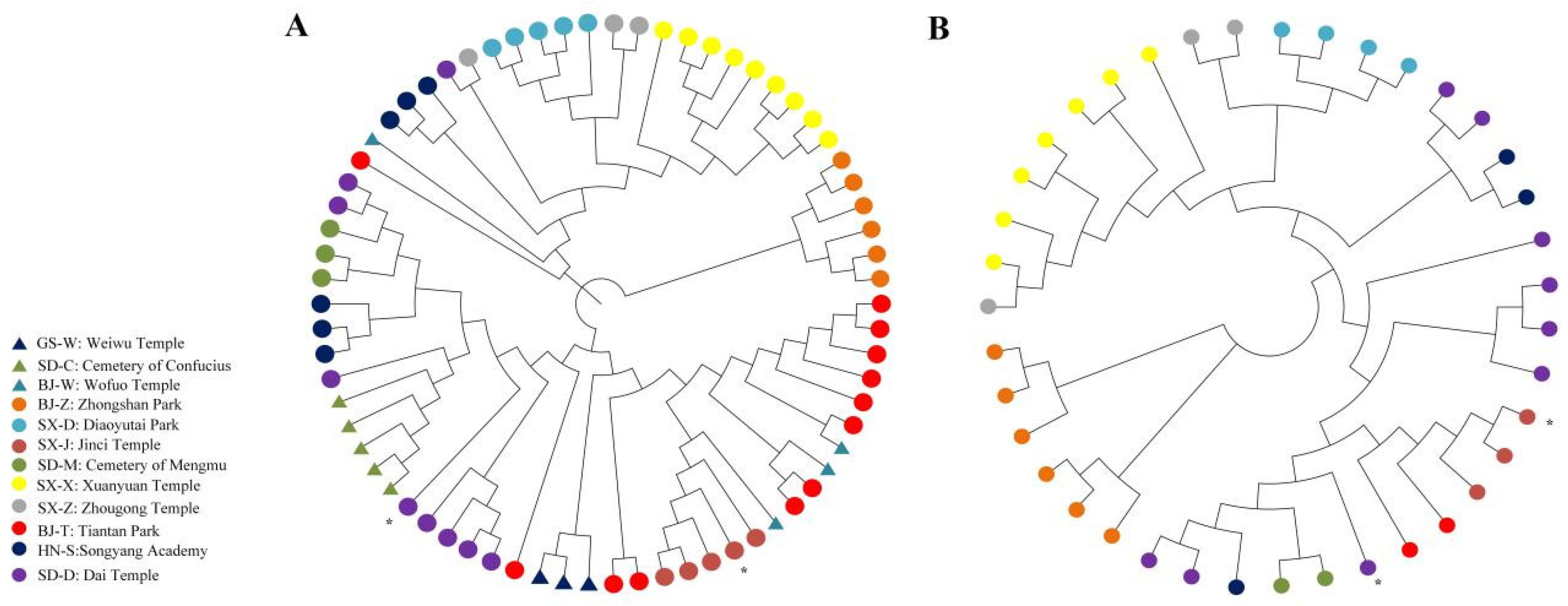
2.4. Genome-Wide Estimates of Genetic Diversity
3. Discussion
4. Materials and Methods
4.1. Sample Preparation
4.2. Potential Distribution Predictions
4.3. SLAF Library Construction and Sequencing
4.4. Sequencing Data Processing and SNP Calling
4.5. Phylogenetic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jiang, P.; Shi, J.; Niu, P.X.; Lu, Y. Effect on activities of defensive enzymes and MDA content in leaves of Platycladus orientalis under naturally decreasing temperature. J. Shihezi Univ. 2009, 27, 30–33, (In Chinese with English Abstract). [Google Scholar]
- Li, X.P.; He, Y.P.; Wu, X.J.; Ren, Q.F. Water stress experiments of Platycladus orientalis and Pinus tablaeformis young trees. For. Res. 2011, 24, 91–96, (In Chinese with English Abstract). [Google Scholar]
- Chang, E.; Shi, S.; Liu, J.; Cheng, T.; Xue, L.; Yang, X.; Yang, W.; Lan, Q.; Jiang, Z. Selection of reference genes for quantitative gene expression studies in Platycladus orientalis (Cupressaceae) using real-time PCR. PLoS ONE 2012, 7, e33278. [Google Scholar] [CrossRef] [PubMed]
- Lanner, R.M.; Connor, K.F. Does bristlecone pine senescence? Exp. Gerontol. 2001, 36, 675–685. [Google Scholar] [CrossRef]
- Flanary, B.E.; Kletetschka, G. Analysis of telomere length and telomerase activity in tree species of various lifespans, and with age in the bristlecone pine Pinus longaeva. Rejuvenation Res. 2006, 9, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Klimešová, J.; Nobis, M.P.; Herben, T. Senescence, ageing and death of the whole plant: morphological prerequisites and constraints of plant immortality. Electron. Des. Eng. 2015, 206, 1–8. [Google Scholar] [CrossRef]
- Thomas, H. Senescence, ageing and death of the whole plant. New Phytol. 2013, 197, 696–711. [Google Scholar] [CrossRef] [PubMed]
- Sillett, S.C.; Van Pelt, R.; Carroll, A.L.; Kramer, R.D.; Ambrose, A.R.; Trask, D. How do tree structure and old age affect growth potential of California redwoods? Ecol. Monogr. 2015, 85, 181–212. [Google Scholar] [CrossRef]
- Bowen, B.W.; Bass, A.L.; Soares, L.; Toonen, R.J. Conservation implications of complex population structure: Lessons from the loggerhead turtle (Caretta caretta). Mol. Ecol. 2005, 14, 2389–2402. [Google Scholar] [CrossRef]
- McRae, B.H.; Beier, P.; Dewald, L.E.; Huynh, L.Y.; Keim, P. Habitat barriers limit gene flow and illuminate historical events in a wide-ranging carnivore, the American puma. Mol. Ecol. 2005, 14, 1965–1977. [Google Scholar] [CrossRef]
- Hendricks, S.; Epstein, B.; Schönfeld, B.; Wiench, C.; Hamede, R.; Jones, M.; Storfer, A.; Hohenlohe, P. Conservation implications of limited genetic diversity and population structure in Tasmanian devils (Sarcophilus harrisii). Conserv. Genet. 2017, 18, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Hohenlohe, P.A.; Bassham, S.; Etter, P.D.; Stiffler, N.; Johnson, E.A.; Cresko, W.A. Population genomics of parallel adaptation in Threespine Stickleback using sequenced RAD tags. PLoS Genet. 2010, 6, e1000862. [Google Scholar] [CrossRef] [PubMed]
- Bonin, A. Population genomics: A new generation of genome scans to bridge the gap with functional genomics. Mol. Ecol. 2008, 17, 3583–3584. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, F.S.; Parkhill, J. Population genomics: Modeling the new and a renaissance of the old. Curr. Opin. Microbiol. 2008, 11, 439–441. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.P.; Zhao, X.; Liu, D.Y.; Li, Y.; Lightfoot, D.A.; Yang, Z.; Zhao, L.; Zhou, G.; Wang, Z.; Huang, L.; et al. Domestication footprints anchor genomic regions of agronomic importance in soybeans. New Phytol. 2016, 209, 871–884. [Google Scholar] [CrossRef] [PubMed]
- Curto, M.; Schachtler, C.; Puppo, P.; Meimberg, H. Using a new RAD-sequencing approach to study the evolution of Micromeria in the Canary islands. Mol. Phylogenet. Evol. 2018, 119, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Stobie, C.S.; Oosthuizen, C.J.; Cunningham, M.J.; Bloomer, P. Exploring the phylogeography of a hexaploid freshwater fish by RAD sequencing. Ecol. Evol. 2018, 8, 2326–2342. [Google Scholar] [CrossRef] [PubMed]
- Humble, E.; Dasmahapatra, K.K.; Martinez-Barrio, A.; Gregório, I.; Forcada, J.; Polikeit, A.C.; Goldsworthy, S.D.; Goebel, M.E.; Kalinowski, J.; Wolf, J.B.; et al. RAD Sequencing and a hybrid antarctic fur seal genome assembly reveal rapidly decaying linkage disequilibrium, global population structure and evidence for inbreeding. G3 Genes Genom. Genet. 2018, 8, 2709–2722. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liu, D.; Zhang, X.; Li, W.; Liu, H.; Hong, W.; Jiang, C.; Guan, N.; Ma, C.; Zeng, H.; et al. SLAF-seq: An efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS ONE 2013, 8, e58700. [Google Scholar] [CrossRef]
- Li, M.; Tian, S.; Jin, L.; Zhou, G.; Li, Y.; Zhang, Y.; Wang, T.; Yeung, C.K.L.; Chen, L.; Ma, J.; et al. Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nat. Genet. 2013, 45, 1431–1438. [Google Scholar] [CrossRef]
- Wang, Y.S.; Xing, S.Y.; Tang, H.X.; Feng, D.Q. Genetic diversity of Platycladus orientalis provenances. Sci. Silvae Sin. 2011, 47, 91–96. [Google Scholar]
- Liu, J.F.; Xiao, W.F. RAPD analysis of the genetic diversity of a critically endangered plant, Thuja sutchuenensis (Cupressaceae). Acta Agric. Univ. Jiangxiensis 2008, 30, 68–72, (In Chinese with English Abstract). [Google Scholar]
- Vanagas, G. Receiver operating characteristic curves and comparison of cardiac surgery risk stratification systems. Interact. Cardiovasc. Thorac. Surg. 2004, 3, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Deng, N.; Liu, C.X.; Sun, C.Y.; Ma, F.; Luo, J.; Tian, Y.X. Distribution patterns of Gnetum, L. species in China under climate change. Bangl. J. Bot. 2018, 47, 615–622. [Google Scholar]
- Peterson, A.T.; Cohoon, K.P. Sensitivity of distributional prediction algorithms to geographic data completeness. Ecol. Model. 1999, 117, 159–164. [Google Scholar] [CrossRef]
- Birol, I.; Raymond, A.; Jackman, S.D.; Pleasance, S.; Coope, R.; Taylor, G.A.; Yuen, M.M.S.; Keeling, C.I.; Brand, D.; Vandervalk, B.P.; et al. Assembling the 20 Gb white spruce (Picea glauca) genome from whole-genome shotgun sequencing data. Bioinformatics 2013, 29, 1492–1497. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, R. Molecular Markers and Selection for complex traits in plants: Learning from the last 20 years. Crop. Sci. 2015, 48, 1649–1664. [Google Scholar] [CrossRef]
- Elliotl, H.; Marke, S.; Jeanluc, J. Genomic Selection for Crop Improvement. Crop. Sci. 2017, 49, 1–12. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, G. 1000 Genome project data processing subgroup The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Boil. Evol. 1987, 4, 406. [Google Scholar]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Boil. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Meirmans, P.G.; Van Tienderen, P.H. genotype and genodive: Two programs for the analysis of genetic diversity of asexual organisms. Mol. Ecol. Notes 2004, 4, 792–794. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Sample Number | Total Reads | Q30 (%) | GC Content (%) | SLAF Number | Average Depth | Polymorphic SLAF | SNP Number | Heterozygosity (%) |
|---|---|---|---|---|---|---|---|---|---|
| SD-D | 12 | 5,851,890 | 82.63 | 34.47 | 352,123 | 11.44 | 314,150 | 3,069,621 | 0.43 |
| BJ-T | 16 | 7,163,133 | 82.56 | 34.39 | 456,407 | 10.84 | 406,430 | 3,912,571 | 0.36 |
| SD-M | 4 | 1,893,816 | 82.62 | 34.50 | 118,467 | 10.83 | 105,589 | 1,036,869 | 0.36 |
| BJ-W | 8 | 3,058,893 | 82.65 | 34.54 | 231,879 | 8.80 | 206,623 | 1,963,840 | 0.35 |
| HN-S | 6 | 2,307,247 | 82.66 | 34.49 | 170,807 | 9.36 | 152,372 | 1,426,753 | 0.35 |
| SX-D | 6 | 2,514,465 | 82.97 | 34.23 | 164,998 | 11.02 | 147,496 | 1,358,212 | 0.38 |
| GS-W | 9 | 3,300,962 | 83.00 | 34.53 | 247,813 | 9.28 | 220,406 | 2,015,626 | 0.28 |
| SX-Z | 2 | 57,506 | 83.25 | 34.25 | 57,506 | 19.34 | 51,351 | 485,378 | 0.68 |
| SD-C | 5 | 201,0243 | 82.42 | 34.31 | 145,908 | 9.67 | 130,346 | 1,231,667 | 0.34 |
| BJ-D | 6 | 2,356,156 | 82.81 | 34.38 | 175,280 | 9.37 | 156,352 | 1,478,408 | 0.35 |
| BJ-Z | 9 | 3,885,951 | 82.80 | 34.25 | 256,074 | 10.45 | 228,777 | 2,170,506 | 0.24 |
| SX-J | 8 | 3,270,113 | 82.85 | 34.21 | 236,456 | 9.85 | 209,765 | 1,974,304 | 0.34 |
| SX-X | 9 | 3,676,621 | 83.10 | 34.78 | 252,694 | 9.37 | 224,749 | 2,093,102 | 0.20 |
| Population | SX-D | BJ-D | GS-W | SX-J | SD-C | SD-M | SX-X | SD-D | BJ-T | BJ-W | HN-S | BJ-Z | SX-Z |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SX-D | 0.302 | 0.330 | 0.315 | 0.312 | 0.309 | 0.261 | 0.296 | 0.302 | 0.306 | 0.309 | 0.338 | 0.261 | |
| BJ-D | 0.109 | 0.286 | 0.237 | 0.253 | 0.254 | 0.291 | 0.254 | 0.237 | 0.244 | 0.266 | 0.330 | 0.298 | |
| GS-W | 0.163 | 0.125 | 0.287 | 0.294 | 0.291 | 0.323 | 0.294 | 0.285 | 0.294 | 0.307 | 0.365 | 0.324 | |
| SX-J | 0.334 | 0.265 | 0.353 | 0.262 | 0.265 | 0.300 | 0.262 | 0.234 | 0.235 | 0.277 | 0.345 | 0.306 | |
| SD-C | 0.144 | 0.076 | 0.160 | 0.314 | 0.229 | 0.302 | 0.241 | 0.255 | 0.264 | 0.259 | 0.330 | 0.310 | |
| SD-M | 0.143 | 0.078 | 0.155 | 0.317 | 0.074 | 0.297 | 0.245 | 0.258 | 0.264 | 0.262 | 0.325 | 0.298 | |
| SX-X | 0.067 | 0.091 | 0.150 | 0.318 | 0.127 | 0.124 | 0.286 | 0.291 | 0.294 | 0.299 | 0.337 | 0.257 | |
| SD-D | 0.095 | 0.041 | 0.121 | 0.278 | 0.051 | 0.058 | 0.077 | 0.258 | 0.263 | 0.262 | 0.328 | 0.290 | |
| BJ-T | 0.107 | 0.026 | 0.122 | 0.259 | 0.076 | 0.081 | 0.089 | 0.044 | 0.242 | 0.272 | 0.336 | 0.301 | |
| BJ-W | 0.105 | 0.028 | 0.123 | 0.254 | 0.079 | 0.080 | 0.084 | 0.042 | 0.024 | 0.279 | 0.340 | 0.301 | |
| HN-S | 0.166 | 0.117 | 0.197 | 0.354 | 0.138 | 0.142 | 0.151 | 0.105 | 0.122 | 0.123 | 0.339 | 0.298 | |
| BJ-Z | 0.171 | 0.157 | 0.222 | 0.386 | 0.182 | 0.180 | 0.163 | 0.146 | 0.159 | 0.157 | 0.217 | 0.336 | |
| SX-Z | 0.122 | 0.100 | 0.071 | 0.328 | 0.135 | 0.134 | 0.108 | 0.094 | 0.097 | 0.098 | 0.164 | 0.190 |
| Population Code | Location | Number of Individuals | Latitude (N) | Longitude (E) | Average Age (Years) |
|---|---|---|---|---|---|
| BJ-T | Tiantan Park, Beijing Municipality | 16 | 39°52 | 116°24′ | 650 |
| BJ-D | Ditan Park, Beijing Municipality | 6 | 39°57′ | 116°24′ | 191.7 |
| BJ-W | Wofuo Temple, Beijing Municipality | 8 | 40°0′ | 116°12′ | 437.5 |
| BJ-Z | Zhongshan Park, Beijing Municipality | 9 | 39°54′ | 116°23′ | 1000 |
| SD-M | Cemetery of Mengmu, Qufu City, Shandong Province | 4 | 35°29 | 116°58′ | 800 |
| SD-D | Dai Temple, Tai’an City, Shandong Province | 12 | 36°11′ | 117°07′ | 1341.7 |
| SD-C | Cemetery of Confucius, Qufu City, Shandong Province | 5 | 35°37′ | 116°59′ | 500 |
| HN-S | Songyang Academy, Zhengzhou City, Henan Province | 6 | 34°28′ | 113°01′ | 1250 |
| SX-Z | Zhougong Temple, Baoji City, Shaanxi Province | 2 | 34°29′ | 107°36′ | 1300 |
| SX-D | Diaoyutai Park, Baoji City, Shanxi Province | 6 | 34°16 | 107°25′ | 991.7 |
| SX-J | Jinci Temple, Taiyuan City, Shanxi Province | 8 | 37°42′ | 112°26′ | 912.5 |
| SX-X | Xuanyuan Temple, Yan’an City, Shanxi Province | 9 | 35°35′ | 109°16′ | 1744.4 |
| GS-W | Weiwu Temple, Wuwei City, Gansu Province | 9 | 37°55′ | 102°38′ | 381.1 |
| Code | Description |
|---|---|
| Bio1 | Annual Mean Temperature |
| Bio2 | Mean Diurnal Range (Mean of monthly (max.temp.–min.temp.)) |
| Bio3 | Isothermality (BIO2/BIO7) (* 100) |
| Bio4 | Temperature Seasonality (standard deviation *100) |
| Bio5 | Max Temperature of Warmest Month |
| Bio6 | Min Temperature of Coldest Month |
| Bio7 | Temperature Annual Range (BIO5–BIO6) |
| Bio8 | Mean Temperature of Wettest Quarter |
| Bio9 | Mean Temperature of Driest Quarter |
| Bio10 | Mean Temperature of Warmest Quarter |
| Bio11 | Mean Temperature of Coldest Quarter |
| Bio12 | Annual Precipitation |
| Bio13 | Precipitation of Wettest Month |
| Bio14 | Precipitation of Driest Month |
| Bio15 | Precipitation Seasonality (Coefficient of Variation) |
| Bio16 | Precipitation of Wettest Quarter |
| Bio17 | Precipitation of Driest Quarter |
| Bio18 | Precipitation of Warmest Quarter |
| Bio19 | Precipitation of Coldest Quarter |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, E.; Tian, Y.; Wang, C.; Deng, N.; Jiang, Z.; Liu, C. Exploring the Phylogeography of Ancient Platycladus orientalis in China by Specific-Locus Amplified Fragment Sequencing. Int. J. Mol. Sci. 2019, 20, 3871. https://doi.org/10.3390/ijms20163871
Chang E, Tian Y, Wang C, Deng N, Jiang Z, Liu C. Exploring the Phylogeography of Ancient Platycladus orientalis in China by Specific-Locus Amplified Fragment Sequencing. International Journal of Molecular Sciences. 2019; 20(16):3871. https://doi.org/10.3390/ijms20163871
Chicago/Turabian StyleChang, Ermei, Yuxin Tian, Caiyun Wang, Nan Deng, Zeping Jiang, and Caixia Liu. 2019. "Exploring the Phylogeography of Ancient Platycladus orientalis in China by Specific-Locus Amplified Fragment Sequencing" International Journal of Molecular Sciences 20, no. 16: 3871. https://doi.org/10.3390/ijms20163871
APA StyleChang, E., Tian, Y., Wang, C., Deng, N., Jiang, Z., & Liu, C. (2019). Exploring the Phylogeography of Ancient Platycladus orientalis in China by Specific-Locus Amplified Fragment Sequencing. International Journal of Molecular Sciences, 20(16), 3871. https://doi.org/10.3390/ijms20163871