Structural and Functional Properties of the Capsid Protein of Dengue and Related Flavivirus

 , ,
, ,  ,
, 

Abstract
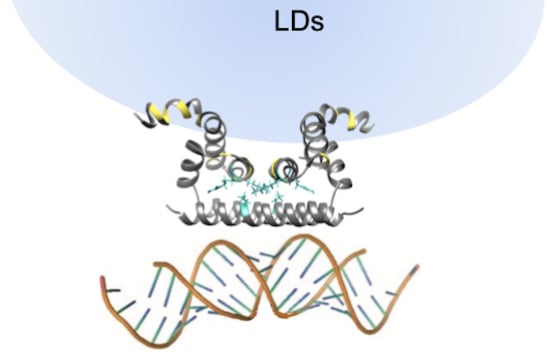
1. Introduction
2. Results
2.1. Analysis of Amino Acid Sequence Conservation Among Flavivirus C proteins
2.2. Analysis of the Flavirus C Protein Sequences Hydrophobicity and Secondary Structure Propensity
2.3. Analysis of the Flavivirus C Protein Tertiary Structure Propensity
2.4. Analysis of Dengue Virus (DENV) C Protein Rotational Correlation Time
2.5. Analysis of DENV C Conformational Stability
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Flavivirus C Proteins Primary, Secondary and Tertiary Structural Predictions
4.3. Structure Comparison Between DENV C and Influenza NS1
4.4. DENV C Recombinant Protein Production and Purification
4.5. Time-Resolved Fluorescence Anisotropy
4.6. Rotational Correlation Time Corrections
4.7. Temperature Denaturation Measurements via Circular Dichroism (CD) Spectroscopy
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALFV | Alfuy virus |
| APOE | Apolipoprotein E |
| AROAV | Aroa virus |
| BAGV | Bagaza virus |
| C protein | Capsid protein |
| CD | Circular dichroism |
| DENV | Dengue virus |
| ICTV | International Committee on Taxonomy of Viruses |
| IDP | Intrinsically disordered protein |
| IFN | Interferon |
| IGUV | Iguape virus |
| ILHV | Ilheus virus |
| JEV | Japanese encephalitis virus |
| KEDV | Kedougou virus |
| KOKV | Kokobera virus |
| LDs | Lipid droplets |
| MVEV | Murray Valley encephalitis virus |
| NS1 | Non-structural protein 1 from influenza virus A |
| PDB | Protein Data Bank |
| pep14-23 | Inhibitor peptide pep14-23 (amino acid sequence NMLKRARNRV) |
| PLIN3 | Perilipin 3 |
| ROCV | Rocio virus |
| SLEV | Saint Louis encephalitis virus |
| SPOV | Spondweni virus |
| USUV | Usutu virus |
| VLDL | Very low-density lipoproteins |
| WNV | West Nile virus |
| WNV-K | WNV serotype Kunjin |
| YFV | Yellow fever virus |
| ZIKV | Zika virus |
References
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Sanofi Pasteur. Available online: https://www.sanofipasteur.com/en/media-room/press-releases/dengvaxia-vaccine-approved-for-prevention-of-dengue-in-europe (accessed on 30 January 2019).
- Durbin, A.P. A dengue vaccine. Cell 2016, 166, 1. [Google Scholar] [CrossRef] [PubMed]
- Villar, L.; Dayan, G.H.; Arredondo-Garcia, J.L.; Rivera, D.M.; Cunha, R.; Deseda, C.; Reynales, H.; Costa, M.S.; Morales-Ramirez, J.O.; Carrasquilla, G.; et al. Efficacy of a tetravalent dengue vaccine in children in Latin America. N. Engl. J. Med. 2015, 372, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Takeda. Available online: https://www.takeda.com/newsroom/newsreleases/2019/takedas-dengue-vaccine-candidate-meets-primary-endpoint-in-pivotal-phase-3-efficacy-trial/ (accessed on 4 February 2019).
- ICTV Taxonomy. Available online: https://talk.ictvonline.org/taxonomy/ (accessed on 17 April 2019).
- Grard, G.; Moureau, G.; Charrel, R.N.; Holmes, E.C.; Gould, E.A.; de Lamballerie, X. Genomics and evolution of Aedes-borne flaviviruses. J. Gen. Virol. 2019, 91, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Schubert, A.M.; Putonti, C. Infection, genetics and evolution of the sequence composition of flaviviruses. Infect. Genet. Evol. 2010, 10, 129–136. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Calisher, C.H.; Gould, E.A. Taxonomy of the virus family Flaviviridae. Adv. Virus Res. 2003, 59, 1–19. [Google Scholar] [PubMed]
- Mukhopadhyay, S.; Kuhn, R.J.; Rossmann, M.G. A structural perspective of the flavivirus life cycle. Nat. Rev. Microbiol. 2005, 3, 13–22. [Google Scholar] [CrossRef]
- Kuhn, R.J.; Zhang, W.; Rossmann, M.G.; Pletnev, S.V.; Corver, J.; Lenches, E.; Jones, C.T.; Mukhopadhyay, S.; Chipman, P.R.; Strauss, E.G.; et al. Structure of dengue virus: Implications for flavivirus organization, maturation, and fusion. Cell 2002, 108, 717–725. [Google Scholar] [CrossRef]
- Ma, L.; Jones, C.T.; Groesch, T.D.; Kuhn, R.J.; Post, C.B. Solution structure of dengue virus capsid protein reveals another fold. Proc. Natl. Acad. Sci. USA 2004, 101, 3414–3419. [Google Scholar] [CrossRef]
- Faustino, A.F.; Barbosa, G.M.; Silva, M.; Castanho, M.A.R.B.; da Poian, A.T.; Cabrita, E.J.; Santos, N.C.; Almeida, F.C.L.; Martins, I.C. Fast NMR method to probe solvent accessibility and disordered regions in proteins. Sci. Rep. 2019, 9, 1647. [Google Scholar] [CrossRef]
- Martins, I.C.; Gomes-Neto, F.; Faustino, A.F.; Carvalho, F.A.; Carneiro, F.A.; Bozza, P.T.; Mohana-Borges, R.; Castanho, M.A.R.B.; Almeida, F.C.L.; Santos, N.C.; et al. The disordered N-terminal region of dengue virus capsid protein contains a lipid-droplet-binding motif. Biochem. J. 2012, 444, 405–415. [Google Scholar] [CrossRef]
- Faustino, A.F.; Guerra, G.M.; Huber, R.G.; Hollmann, A.; Domingues, M.M.; Barbosa, G.M.; Enguita, F.J.; Bond, P.J.; Castanho, M.A.R.B.; da Poian, A.T.; et al. Understanding Dengue virus capsid protein disordered N-terminus and pep14-23-based inhibition. ACS Chem. Biol. 2015, 10, 517–526. [Google Scholar] [CrossRef]
- Jones, C.T.; Ma, L.; Burgner, J.W.; Groesch, T.D.; Post, C.B.; Kuhn, R.J. Flavivirus capsid is a dimeric alpha-helical protein. J. Virol. 2003, 77, 7143–7149. [Google Scholar] [CrossRef] [PubMed]
- Van Gorp, E.C.M.; Suharti, C.; Mairuhu, A.T.A.; Dolmans, W.M.V.; van der Ven, J.; Demacker, P.N.M.; van der Meer, J.W.M. Changes in the plasma lipid profile as a potential predictor of clinical outcome in dengue hemorrhagic fever. Clin. Infect. Dis. 2002, 34, 1150–1153. [Google Scholar] [CrossRef]
- Samsa, M.M.; Mondotte, J.A.; Iglesias, N.G.; Assuncao-Miranda, I.; Barbosa-Lima, G.; da Poian, A.T.; Bozza, P.T.; Gamarnik, A. V Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog. 2009, 5, e1000632. [Google Scholar] [CrossRef]
- Suvarna, J.C.; Rane, P.P. Serum lipid profile: A predictor of clinical outcome in dengue infection. Trop. Med. Int. Heal. 2009, 14, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, F.A.; Carneiro, F.A.; Martins, I.C.; Assunção-Miranda, I.; Faustino, A.F.; Pereira, R.M.; Bozza, P.T.; Castanho, M.A.R.B.; Mohana-Borges, R.; da Poian, A.T.; et al. Dengue virus capsid protein binding to hepatic lipid droplets (LD) is potassium ion dependent and is mediated by LD surface proteins. J. Virol. 2012, 86, 2096–2108. [Google Scholar] [CrossRef]
- Faustino, A.F.; Carvalho, F.A.; Martins, I.C.; Castanho, M.A.R.B.; Mohana-Borges, R.; Almeida, F.C.L.; da Poian, A.T.; Santos, N.C. Dengue virus capsid protein interacts specifically with very low-density lipoproteins. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 247–255. [Google Scholar] [CrossRef]
- Faustino, A.F.; Martins, I.C.; Carvalho, F.A.; Castanho, M.A.R.B.; Maurer-Stroh, S.; Santos, N.C. Understanding dengue virus capsid protein interaction with key biological targets. Sci. Rep. 2015, 5, 10592. [Google Scholar] [CrossRef]
- Martins, A.S.; Carvalho, F.A.; Faustino, A.F.; Martins, I.C.; Santos, N.C. West Nile virus capsid protein interacts with biologically relevant host lipid systems. Front. Cell. Infect. Microbiol. 2019, 9, 8. [Google Scholar] [CrossRef]
- Martins, A.S.; Martins, I.C.; Santos, N.C. Methods for lipid droplet biophysical characterization in Flaviviridae infections. Front. Microbiol. 2018, 9, 1951. [Google Scholar] [CrossRef] [PubMed]
- Shang, Z.; Song, H.; Shi, Y.; Qi, J.; Gao, G.F. Crystal structure of the capsid protein from Zika virus. J. Mol. Biol. 2018, 430, 948–962. [Google Scholar] [CrossRef] [PubMed]
- Kyte, J.; Doolittle, R.F. A Simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef]
- Deléage, G.; Roux, B. An algorithm for protein secondary structure prediction based on class prediction. Protein Eng. 1987, 1, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 8, 1–8. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Arbor, A.; Arbor, A. The I-TASSER suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2011, 5, 725–738. [Google Scholar] [CrossRef]
- Dokland, T.; Walsh, M.; Mackenzie, J.M.; Khromykh, A.A.; Ee, K.-H.; Wang, S. West Nile virus core protein; tetramer structure and ribbon formation. Structure 2004, 12, 1157–1163. [Google Scholar] [CrossRef]
- Zhan, C.; Zhao, L.; Chen, X.; Lu, W.; Lu, W. Total chemical synthesis of dengue 2 virus capsid protein via native chemical ligation: Role of the conserved salt-bridge. Bioorg. Med. Chem. 2013, 21, 3443–3449. [Google Scholar] [CrossRef][Green Version]
- Morando, M.A.; Barbosa, G.M.; Cruz-Oliveira, C.; da Poian, A.T.; Almeida, F.C.L. Dynamics of Zika virus capsid protein in solution: The properties and exposure of the hydrophobic cleft are controlled by the α-helix 1 sequence. Biochemistry 2019, 58, 2488–2498. [Google Scholar] [CrossRef]
- Kumar, S.; Ravi, V.K.; Swaminathan, R. How do surfactants and DTT affect the size, dynamics, activity and growth of soluble lysozyme aggregates? Biochem. J. 2008, 415, 275–288. [Google Scholar] [CrossRef]
- Lakowicz, J. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer Science, LLC: Berlin/Heidelberg, Germany, 2006; ISBN 9780387312781. [Google Scholar]
- Rossi, P.; Yuanpeng, G.V.T.S.; James, J.H.; Anklin, C.; Conover, K.; Hamilton, K.; Xiao, R. A microscale protein NMR sample screening pipeline. J. Biomol. NMR 2010, 46, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.H.; Urquidi, J.; Singh, S.; Robinson, G.W. Thermal offset viscosities of liquid H2O, D2O, and T2O. J. Phys. Chem. B 1999, 103, 1991–1994. [Google Scholar] [CrossRef]
- Martins, I.C.; Almeida, F.C.L.; Santos, N.C.; da Poian, A.T. DENV–Derived Peptides and Methods for the Inhibition of Flavivirus Replication. International Patent Publication Nr WO/2012/159187, 26 May 2011. [Google Scholar]
- Ivanyi-Nagy, R.; Lavergne, J.; Gabus, C.; Ficheux, D.; Darlix, J.; Inserm, L.; Supe, E.N. RNA chaperoning and intrinsic disorder in the core proteins of Flaviviridae. Nucleic Acids Res. 2008, 36, 712–725. [Google Scholar] [CrossRef]
- Ivanyi-Nagy, R.; Darlix, J. Core protein-mediated 5–3 annealing of the West Nile virus genomic RNA in vitro. Virus Res. 2012, 167, 226–235. [Google Scholar] [CrossRef]
- Kumar, M.; Gromiha, M.M.; Raghava, G.P.S. SVM based prediction of RNA-binding proteins using binding residues and evolutionary information. J. Mol. Recognit. 2011, 24, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Järvelin, A.I.; Noerenberg, M.; Davis, I.; Castello, A. The new (dis)order in RNA regulation. Cell Commun. Signal. 2016, 14, 9. [Google Scholar] [CrossRef]
- Shavinskaya, A.; Boulant, S.; Penin, F.; McLauchlan, J.; Bartenschlager, R. The lipid droplet binding domain of hepatitis C virus core protein is a major determinant for efficient virus assembly. J. Biol. Chem. 2007, 282, 37158–37169. [Google Scholar] [CrossRef]
- Cheng, A.; Wong, S.M.; Yuan, Y.A. Structural basis for dsRNA recognition by NS1 protein of influenza A virus. Cell Res. 2009, 19, 187–195. [Google Scholar] [CrossRef]
- Fernandez-Sesma, A.; Marukian, S.; Ebersole, B.J.; Kaminski, D.; Park, M.S.; Yuen, T.; Sealfon, S.C.; Garcia-Sastre, A.; Moran, T.M. Influenza virus evades innate and adaptive immunity via the NS1 protein. J. Virol. 2006, 80, 6295–6304. [Google Scholar] [CrossRef]
- Wang, S.H.; Syu, W.J.; Huang, K.J.; Lei, H.Y.; Yao, C.W.; King, C.C.; Hu, S.T. Intracellular localization and determination of a nuclear localization signal of the core protein of dengue virus. J. Gen. Virol. 2002, 83, 3093–3102. [Google Scholar] [CrossRef] [PubMed]
- Kobe, B. Autoinhibition by an internal nuclear localization signal revealed by the crystal structure of mammalian importin α. Nat. Struct. Biol. 1999, 6, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Catimel, B.; Teh, T.; Fontes, M.R.M.; Jennings, I.G.; Jans, D.A.; Howlett, G.J.; Nice, E.C.; Kobe, B. Biophysical characterization of interactions involving importin-α during nuclear import. J. Biol. Chem. 2001, 276, 34189–34198. [Google Scholar] [CrossRef] [PubMed]
- Marfori, M.; Mynott, A.; Ellis, J.J.; Mehdi, A.M.; Saunders, N.F.W.; Curmi, P.M.; Forwood, J.K.; Boden, M.; Kobe, B. Molecular basis for specificity of nuclear import and prediction of nuclear localization. Biochim. Biophys. Acta 2011, 1813, 1562–1577. [Google Scholar] [CrossRef] [PubMed]
- Fontes, M.R.M.; Teh, T.; Kobe, B. Structural basis of recognition of monopartite and bipartite nuclear localization sequences by mammalian importin-α. J. Mol. Biol. 2000, 297, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Marfori, M.; Lonhienne, T.G.; Forwood, J.K.; Kobe, B. Structural basis of high-affinity nuclear localization signal interactions with importin-alpha. Traffic 2012, 13, 532–548. [Google Scholar] [CrossRef] [PubMed]
- Tadano, M.; Makino, Y.; Fukunaga, T.; Okuno, Y.; Fukai, K. Detection of dengue 4 virus core protein in the nucleus I. A monoclonal antibody to dengue 4 virus reacts with the antigen in the nucleus and cytoplasm. J. Gen. Virol. 1989, 70, 1409–1415. [Google Scholar] [CrossRef] [PubMed]
- Makino, Y.; Tadano, M.; Anzai, T.; Ma, S.P.; Yasuda, S.; Žagar, E. Detection of dengue 4 virus core protein in the nucleus II. Antibody against dengue 4 core protein produced by a recombinant baculovirus reacts with the antigen in the nucleus. J. Gen. Virol. 1989, 70, 1417–1425. [Google Scholar] [CrossRef] [PubMed]
- Wagstaff, K.M.; Sivakumaran, H.; Heaton, S.M.; Harrich, D.; Jans, D.A. Ivermectin is a specific inhibitor of importin α/β-mediated nuclear import able to inhibit replication of HIV-1 and dengue virus. Biochem. J. 2012, 443, 851–856. [Google Scholar] [CrossRef]
- Bergmann, M.; Garcia-Sastre, A.; Carnero, E.; Pehamberger, H.; Wolff, K.; Palese, P.; Muster, T. Influenza virus NS1 protein counteracts PKR-mediated inhibition of replication. J. Virol. 2000, 74, 6203–6206. [Google Scholar] [CrossRef]
- Kochs, G.; Garcia-Sastre, A.; Martinez-Sobrido, L. Multiple anti-interferon actions of the influenza A virus NS1 protein. J. Virol. 2007, 81, 7011–7021. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Madoz, J.R.; Bernal-Rubio, D.; Kaminski, D.; Boyd, K.; Fernandez-Sesma, A. Dengue virus inhibits the production of type I interferon in primary human dendritic cells. J. Virol. 2010, 84, 4845–4850. [Google Scholar] [CrossRef] [PubMed]
- Min, J.Y.; Krug, R.M. The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: Inhibiting the 2′–5′ oligo (A) synthetase/RNase L pathway. Proc. Natl. Acad. Sci. USA 2006, 103, 7100–7105. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsically disordered proteins and their “mysterious” (meta)physics. Front. Phys. 2019, 7, 10. [Google Scholar] [CrossRef]
- Na, J.H.; Lee, W.K.; Yu, Y.G. How do we study the dynamic structure of unstructured proteins: A case study on nopp140 as an example of a large, intrinsically disordered protein. Int. J. Mol. Sci. 2018, 19, 381. [Google Scholar] [CrossRef]
- Uversky, V.N. Introduction to intrinsically disordered proteins (IDPs). Chem. Rev. 2014, 114, 6557–6560. [Google Scholar] [CrossRef] [PubMed]
- Minde, D.P.; Halff, E.F.; Tans, S. Designing disorder: Tales of the unexpected tails. Intrinsically Disord. Proteins 2013, 1, e26790. [Google Scholar] [CrossRef]
- Krystkowiak, I.; Manguy, J.; Davey, N.E. PSSMSearch: A server for modeling, visualization, proteome-wide discovery and annotation of protein motif specificity determinants. Nucleic Acids Res. 2018, 46, W235–W241. [Google Scholar] [CrossRef]
- Bera, A.K.; Kuhn, R.J.; Smith, J.L. Functional characterization of cis and trans activity of the Flavivirus NS2B-NS3 protease. J. Biol. Chem. 2007, 282, 12883–12892. [Google Scholar] [CrossRef]
- Niyomrattanakit, P.; Yahorava, S.; Mutule, I.; Mutulis, F.; Petrovska, R.; Prusis, P.; Katzenmeier, G.; Wikberg, J.E. Probing the substrate specificity of the dengue virus type 2 NS3 serine protease by using internally quenched fluorescent peptides. Biochem. J. 2006, 397, 203–211. [Google Scholar] [CrossRef]
- Sievers, F.; Higgins, D.G. Clustal omega. Curr. Protoc. Bioinform. 2014, 13, 1–16. [Google Scholar]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; Thompson, J.D.; Higgins, D.G.; Mcwilliam, H.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Irie, K.; Mohan, P.; Sasaguri, Y.; Putnak, R.; Padmanabhan, R. Sequence analysis of cloned dengue virus type 2 genome (New Guinea-C strain). Gene 1989, 75, 197–211. [Google Scholar] [CrossRef]
- Smith, P.; van Gunsteren, W. Translational and rotational diffusion of proteins. J. Mol. Biol. 1994, 236, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Mok, Y.; de Prat Gay, G.; Butler, P.; Bycroft, M. Equilibrium dissociation and unfolding. Protein Sci. 1996, 5, 310–319. [Google Scholar] [CrossRef]
- Rumfeldt, J.; Galvagnion, C.; Vassall, K.; Meiering, E. Conformational stability and folding mechanisms of dimeric proteins. Prog. Biophys. Mol. Biol. 2008, 98, 61–84. [Google Scholar] [CrossRef]
- Neet, K.E.; Timm, D.E. Conformational stability of dimeric proteins: Quantitative studies by equilibrium denaturation. Protein Sci. 1994, 3, 2167–2174. [Google Scholar] [CrossRef]
- Allen, D.L.; Pielak, G.J. Baseline length and automated fitting of denaturation data. Protein Sci. 1998, 7, 1262–1263. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Cluster A | Cluster B | Cluster C | Cluster D | Excluded |
|---|---|---|---|---|---|
| ALFV C | 1 | 0 | 1 | 0 | 3 |
| AROAV C | 1 | 1 | 1 | 0 | 2 |
| BAGV C | 1 | 0 | 2 | 1 | 1 |
| DENV C | 1 | 2 | 1 | 0 | 1 |
| IGUV C | 1 | 2 | 1 | 0 | 1 |
| ILHV C | 0 | 2 | 2 | 0 | 1 |
| JEV C | 1 | 1 | 0 | 1 | 2 |
| KEDV C | 0 | 1 | 1 | 1 | 2 |
| KOKV C | 1 | 0 | 1 | 1 | 2 |
| MVEV C | 0 | 2 | 1 | 0 | 2 |
| ROCV C | 1 | 0 | 1 | 2 | 1 |
| SLEV C | 1 | 0 | 2 | 0 | 2 |
| SPOV C | 1 | 2 | 1 | 0 | 1 |
| USUV C | 1 | 0 | 2 | 0 | 2 |
| WNV C | 1 | 0 | 1 | 1 | 2 |
| ZIKV C | 0 | 1 | 0 | 1 | 3 |
| Total | 12 | 14 | 18 | 8 | 28 |
| Parameter | pH 6.0 | pH 7.5 |
|---|---|---|
| τ1 (ns) * | 0.209 (± 3.9%) | 0.520 (± 4.0%) |
| τ2 (ns) | 3.106 (± 0.4%) | 3.108 (± 0.9%) |
| τ3 (ns) * | 6.328 (± 0.4%) | 6.506 (± 0.4%) |
| α1 * | 0.275 (± 0.7%) | 0.178 (± 3.4%) |
| α2 * | 0.315 (± 0.9%) | 0.385 (± 0.4%) |
| α3 * | 0.410 (± 0.4%) | 0.437 (± 0.4%) |
| τc (ns) * | 16.46 (± 2.9%) | 16.41 (± 3.4%) |
| r0 | 0.130 (± 0.8%) | 0.131 (± 1.1%) |
| τc (ns) at T | T (°C) | τc (ns) at 25 °C in H2O | Method | Source |
|---|---|---|---|---|
| 16.4 ± 0.5 | 22 | 15.2 ± 0.5 | Time-resolved fluorescence anisotropy | This work |
| 13.0 | 27 | 13.4 | Overall NMR relaxation analysis | Jones et al., 2003 [16] |
| 14.1 | 20 | 12.0 | τc (ns) ≈ 0.6×MW (kDa) | Rossi et al., 2010 [36] |
| Parameter | pH 6.0 | pH 7.5 |
|---|---|---|
| (°C) | 70.02 ± 0.63 | 69.03 ± 0.65 |
| (°C) | 88.26 ± 0.80 | 88.80 ± 0.83 |
| (kJ mol−1) | 612 ± 26 | 564 ± 23 |
| (kJ mol−1 K−1) | 1.693 ± 0.073 | 1.557 ± 0.065 |
| T (°C) | η in H2O (cP) | η in 10% D2O (cP) | ||
|---|---|---|---|---|
| 20 | 1.002 | 1.027 | 0.8736 | 0.8523 |
| 22 | 0.955 | 0.978 | 0.9231 | 0.9012 |
| 25 | 0.890 | 0.911 | 1 | 0.9770 |
| 27 | 0.851 | 0.871 | 1.0530 | 1.0293 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faustino, A.F.; Martins, A.S.; Karguth, N.; Artilheiro, V.; Enguita, F.J.; Ricardo, J.C.; Santos, N.C.; Martins, I.C. Structural and Functional Properties of the Capsid Protein of Dengue and Related Flavivirus. Int. J. Mol. Sci. 2019, 20, 3870. https://doi.org/10.3390/ijms20163870
Faustino AF, Martins AS, Karguth N, Artilheiro V, Enguita FJ, Ricardo JC, Santos NC, Martins IC. Structural and Functional Properties of the Capsid Protein of Dengue and Related Flavivirus. International Journal of Molecular Sciences. 2019; 20(16):3870. https://doi.org/10.3390/ijms20163870
Chicago/Turabian StyleFaustino, André F., Ana S. Martins, Nina Karguth, Vanessa Artilheiro, Francisco J. Enguita, Joana C. Ricardo, Nuno C. Santos, and Ivo C. Martins. 2019. "Structural and Functional Properties of the Capsid Protein of Dengue and Related Flavivirus" International Journal of Molecular Sciences 20, no. 16: 3870. https://doi.org/10.3390/ijms20163870
APA StyleFaustino, A. F., Martins, A. S., Karguth, N., Artilheiro, V., Enguita, F. J., Ricardo, J. C., Santos, N. C., & Martins, I. C. (2019). Structural and Functional Properties of the Capsid Protein of Dengue and Related Flavivirus. International Journal of Molecular Sciences, 20(16), 3870. https://doi.org/10.3390/ijms20163870