Low-Resolution Models for the Interaction Dynamics of Coated Gold Nanoparticles with β2-microglobulin




Abstract
1. Introduction
2. Results and Discussion
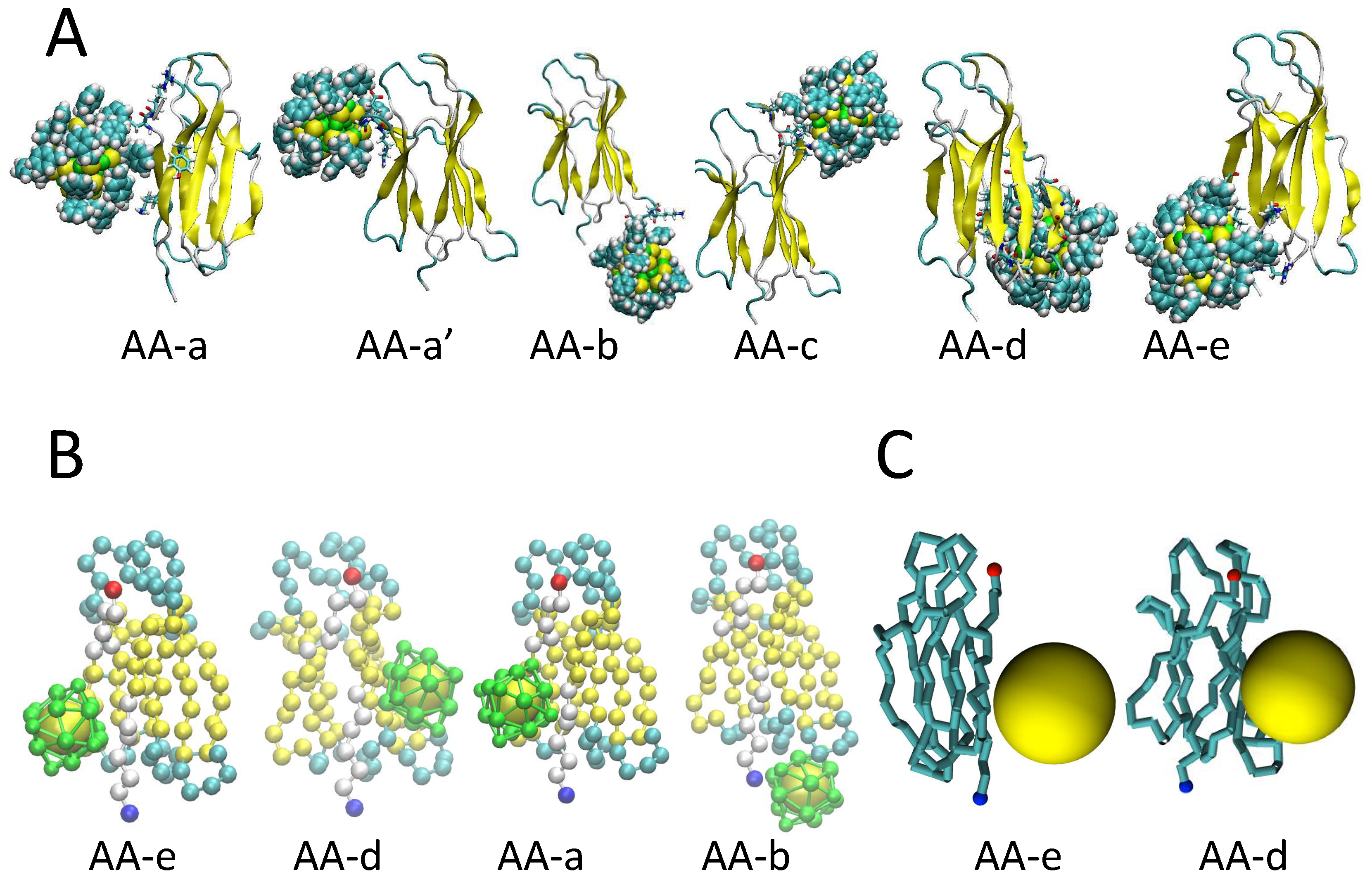
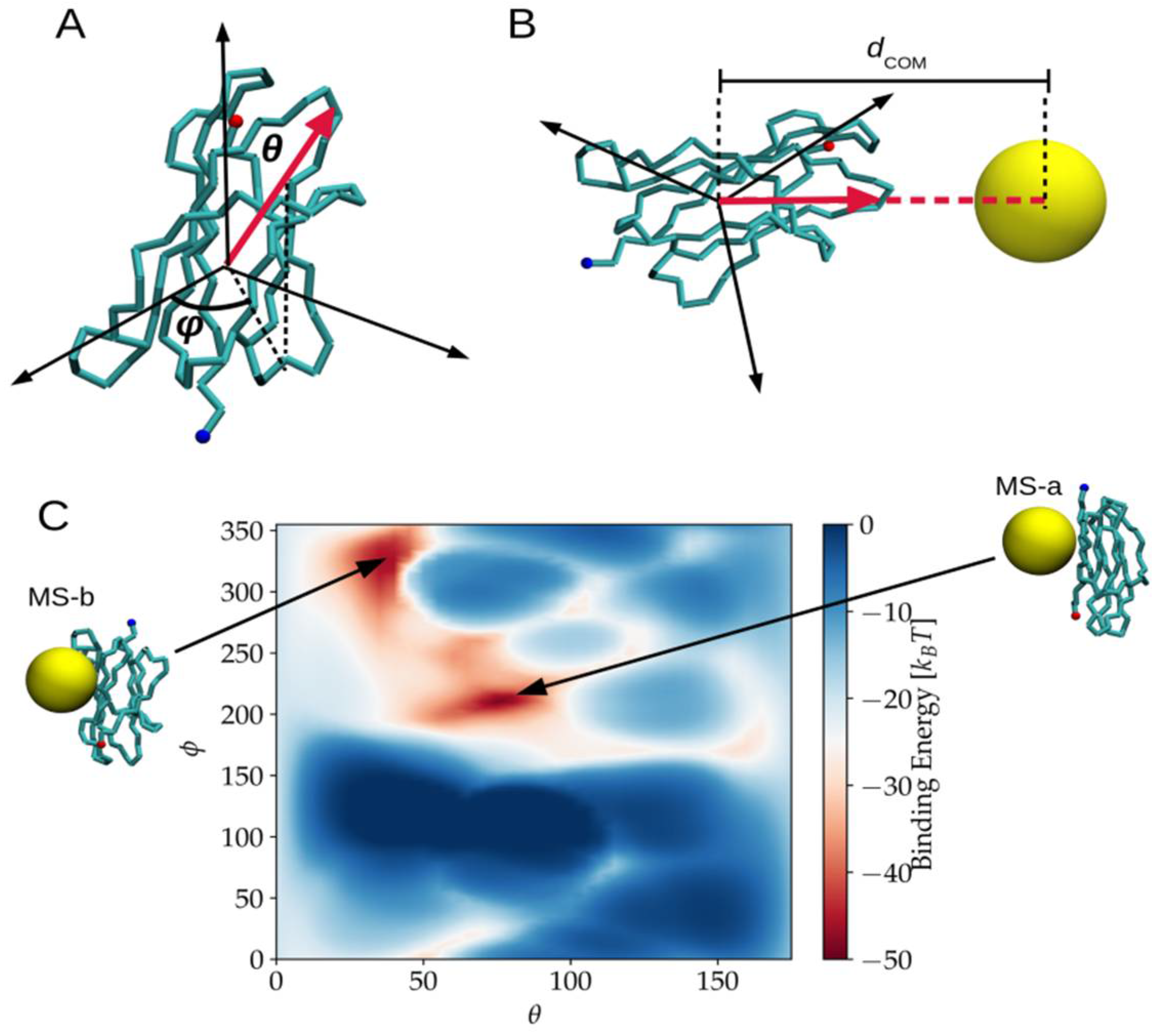
2.1. Binding Modes and Binding Energies of Couples of Proteins and Nanoparticles
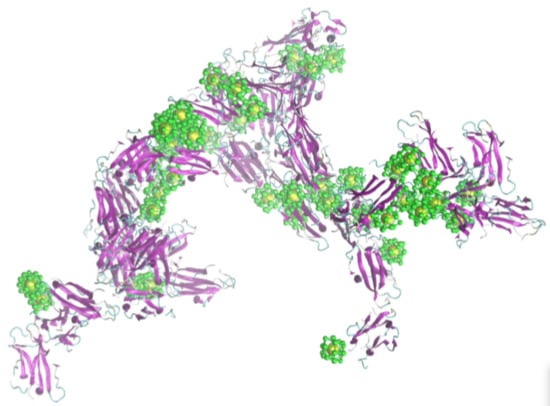
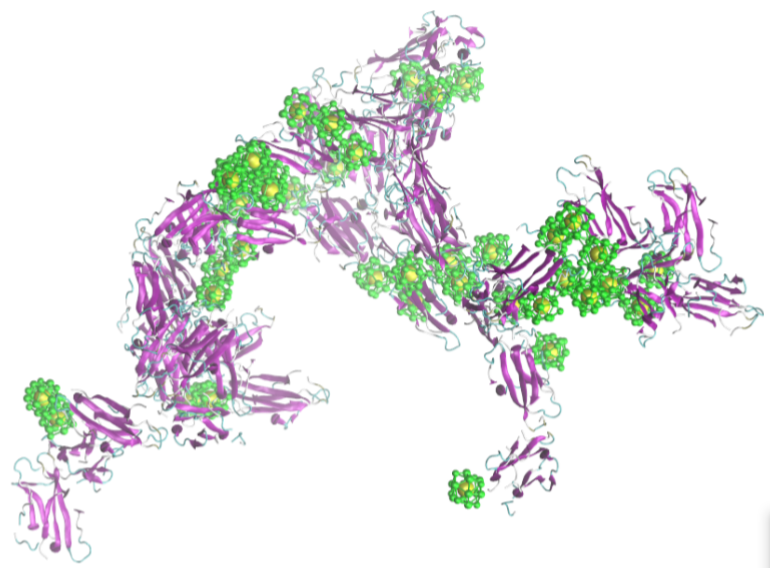
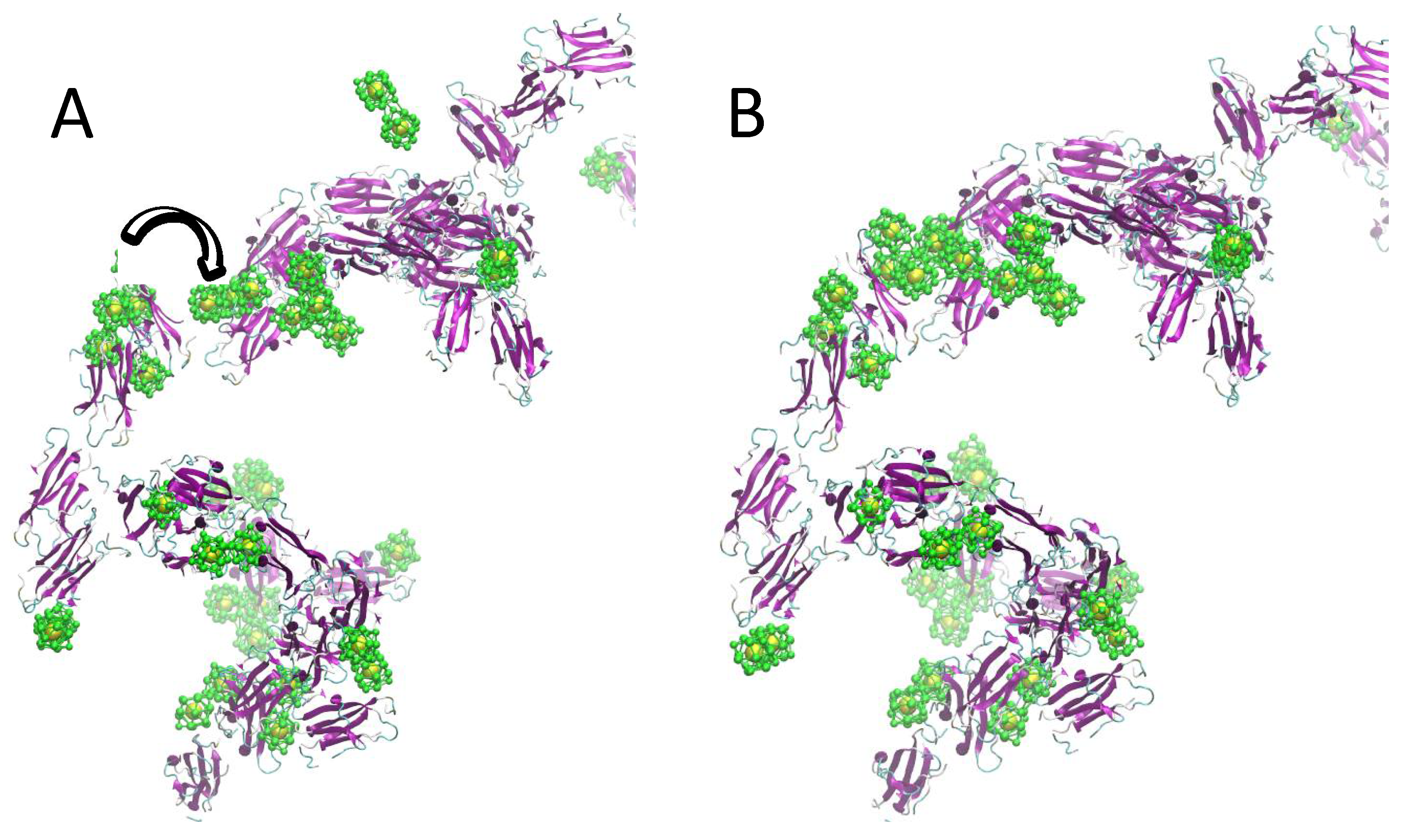
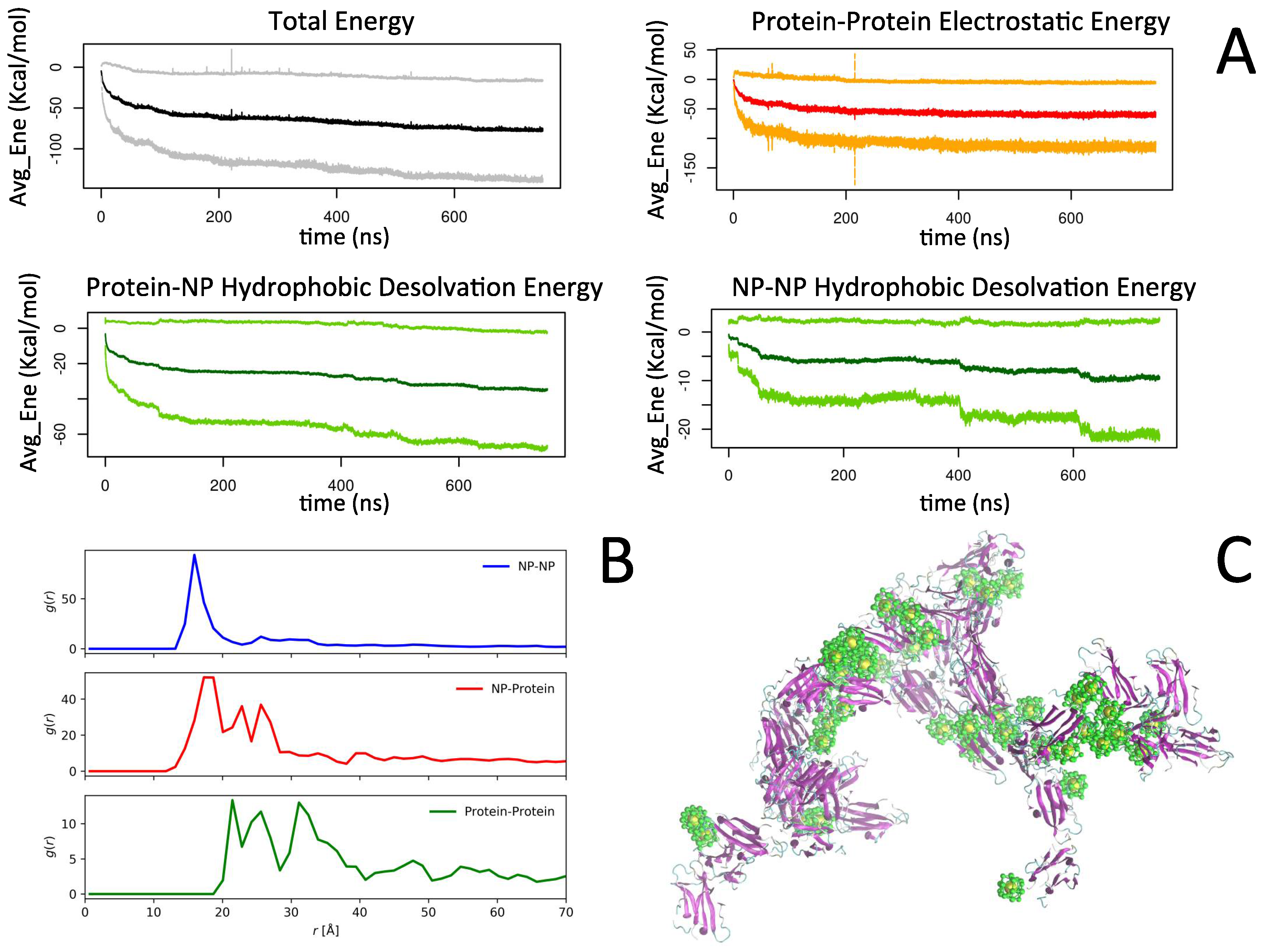
2.2. Simulation of Ensembles of Nanoparticles and Proteins
3. Materials and Methods
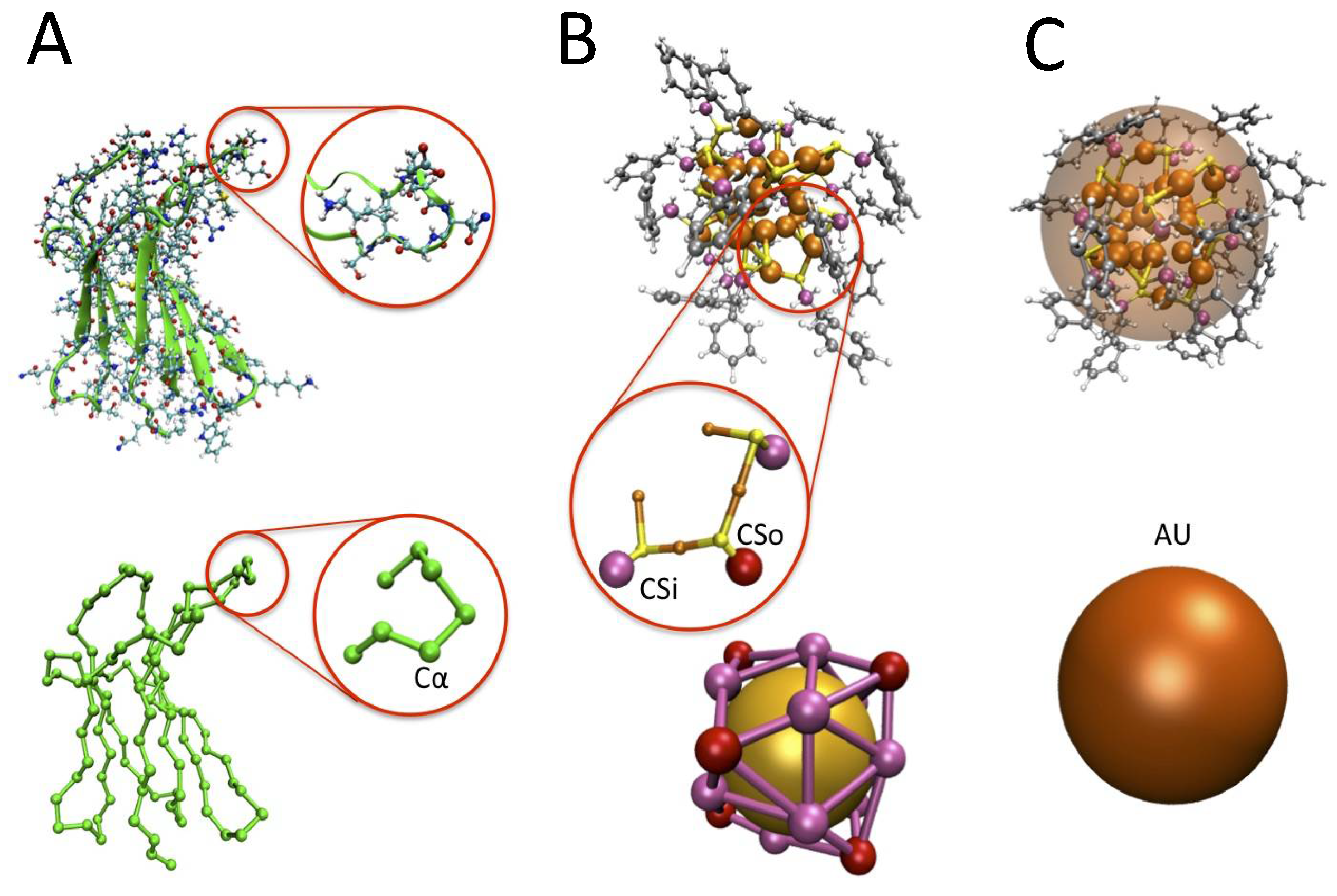
3.1. The Protein Minimalist Models
3.2. The Nanoparticle Models and Its Interaction with the Protein
3.3. Optimization of the Parameters
3.4. Simulations Methodology and Setup
3.4.1. Setup of the Atomistic Simulations
3.4.2. Setup for CG Rigid Docking Simulations
3.4.3. Dynamics MS Simulations Setup
3.4.4. NP-Protein Binding Modes and Energies Evaluation in the MS System
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Howes, P.D.; Chandrawati, R.; Stevens, M.M. Colloidal nanoparticles as advanced biological sensors. Science 2014, 346, 1247390. [Google Scholar] [CrossRef] [PubMed]
- Pelaz, B.; Alexiou, C.; Alvarez-Puebla, R.A.; Alves, F.; Andrews, A.M.; Ashraf, S.; Balogh, L.P.; Ballerini, L.; Bestetti, A.; Brendel, C.; et al. Diverse Applications of Nanomedicine. ACS Nano 2017, 11, 2313–2381. [Google Scholar] [CrossRef] [PubMed]
- Alex, S.; Tiwari, A. Functionalized gold nanoparticles: Synthesis, properties and applications—A review. J. Nanosci. Nanotechnol. 2015, 15, 1869–1894. [Google Scholar] [CrossRef] [PubMed]
- Moyano, D.F.; Rotello, V.M. Gold Nanoparticles: Testbeds for Engineered Protein—particle Interactions. Nanomedicine 2014, 9, 1905–1907. [Google Scholar] [CrossRef] [PubMed]
- Hakkinen, H. The gold-sulfur interface at the nanoscale. Nat. Chem. 2012, 4, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Brancolini, G.; Maschio, M.C.; Cantarutti, C.; Corazza, A.; Fogolari, F.; Corni, S.; Esposito, G. Citrate stabilized gold nanoparticles interfere with amyloid fibril formation: D76N and ΔN6 variants. Nanoscale 2018, 10, 4793–4806. [Google Scholar] [CrossRef] [PubMed]
- Brancolini, G.; Toroz, D.; Corni, S. Can small hydrophobic gold nanoparticles inhibit β2-microglobulin fibrillation? Nanoscale 2014, 6, 7903–7911. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef]
- Brancolini, G.; Tozzini, V. Multiscale modeling of proteins interaction with functionalized nanoparticles. Curr. Opin. Colloid Interface Sci. 2019, 41, 66–73. [Google Scholar] [CrossRef]
- Charchar, P.; Christofferson, A.J.; Todorova, N.; Yarovsky, I. Understanding and Designing the Gold-Bio Interface: Insights from Simulations. Small 2016, 12, 2395–2418. [Google Scholar] [CrossRef]
- Krupa, P.; Hałabis, A.; Zmudzinska, W.; Ołdziej, O.; Scheraga, H.A.; Liwo, A. Maximum likelihood calibration of the UNRES force field for simulation of protein structure and dynamics. J. Chem. Inf. Model 2017, 57, 2364–2377. [Google Scholar] [CrossRef] [PubMed]
- Kmiecik, S.; Gront, D.; Kolinski, M.; Wieteska, L.; Dawid, A.E.; Kolinski, A. Coarse-grained protein models and their applications. Chem. Rev. 2016, 116, 7898–7936. [Google Scholar] [CrossRef] [PubMed]
- Tozzini, V. Minimalist models for proteins: A comparative analysis. Q. Rev. Biophys. 2010, 43, 333–371. [Google Scholar] [CrossRef] [PubMed]
- Trovato, F.; Tozzini, V. Minimalist models for biopolymers: Open problems, latest advances and perspectives. AIP Conf. Proc. 2012, 1456, 187–200. [Google Scholar]
- Rocchia, A.W.; Tozzini, V. Complexes of HIV-1 integrase with HAT proteins: Multiscale models, dynamics, and hypotheses on allosteric sites of inhibition. Proteins Struct. Funct. Bioinform. 2009, 76, 946–958. [Google Scholar]
- Trylska, J.; Tozzini, V.; Chia-en, A.C.; McCammon, J.A. HIV-1 protease substrate binding and product release pathways explored with coarse-grained molecular dynamics. Biophys. J. 2007, 92, 4179–4187. [Google Scholar] [CrossRef] [PubMed]
- Trovato, F.; Nifosì, R.; Di Fenza, A.; Tozzini, V. A minimalist model of protein diffusion and interactions: The green fluorescent protein within the cytoplasm. Macromolecules 2013, 46, 8311–8322. [Google Scholar] [CrossRef]
- Vàcha, R.; Linse, S.; Lund, M. Surface Effects on Aggregation Kinetics of Amyloidogenic Peptides. J. Am. Chem. Soc. 2014, 136, 11776–11782. [Google Scholar] [CrossRef]
- Tavanti, F.; Pedone, A.; Menziani, M.C. A closer look into the ubiquitin corona on gold nanoparticles by computational studies. New. J. Chem. 2015, 39, 2474–2482. [Google Scholar] [CrossRef]
- Cantarutti, C.; Raimondi, S.; Brancolini, G.; Corazza, A.; Giorgetti, S.; Ballico, M.; Zanini, S.; Palmisano, G.; Bertoncin, P.; Marchese, L.; et al. Citrate-stabilized gold nanoparticles hinder fibrillogenesis of a pathological variant of β2-microglobulin. Nanoscale 2017, 9, 3941–3951. [Google Scholar] [CrossRef]
- Tavanti, F.; Pedone, A.; Menziani, M.C. Competitive Binding of Proteins to Gold Nanoparticles Disclosed by Molecular Dynamics Simulations. J. Phys. Chem. C 2015, 119, 22172–22180. [Google Scholar] [CrossRef]
- Cardellini, A.; Alberghini, M.; Rajan, A.G.; Misra, R.P.; Blankschtein, D.; Asinari, P. Multi-scale approach for modeling stability, aggregation, and network formation of nanoparticles suspended in aqueous solutions. Nanoscale 2019, 11, 3979–3992. [Google Scholar] [CrossRef] [PubMed]
- Brancolini, G.; Corazza, A.; Vuano, M.; Fogolari, F.; Mimmi, M.C.; Bellotti, V.; Stoppini, M.; Corni, S.; Esposito, G. Probing the influence of citrate-capped gold nanoparticles on an amyloidogenic protein. ACS Nano 2015, 9, 2600–2613. [Google Scholar] [CrossRef] [PubMed]
- Lopez, H.; Lobaskin, V. Coarse-grained model of adsorption of blood plasma proteins onto nanoparticles. J. Chem. Phys. 2015, 143, 243138. [Google Scholar] [CrossRef] [PubMed]
- Rotkiewicz, P.; Skolnick, J. Fast method for reconstruction of full-atom protein models from reduced representations. J. Comp. Chem. 2008, 29, 1460–1465. [Google Scholar] [CrossRef]
- PULCHRA—omicX. Available online: https://omictools.com/pulchra-tool (accessed on 7 August 2019).
- Spampinato, G.L.B.; Maccari, G.; Tozzini, V. Minimalist model for the dynamics of helical polypeptides: A statistic-based parameterization. J. Chem. Theory Comput. 2014, 10, 3885–3895. [Google Scholar] [CrossRef]
- Trovato, F.; Tozzini, V. Diffusion within the cytoplasm: A mesoscale model of interacting macromolecules. Biophys. J. 2014, 107, 2579–2591. [Google Scholar] [CrossRef]
- Lopez, H.; Brandt, E.G.; Mirzoev, A.; Zhurkin, D.; Lyubartsev, A.; Lobaskin, V. Multiscale Modelling of Bionano Interface. Adv. Exp. Med. Biol. 2017, 947, 173–206. [Google Scholar]
- Arkhipov, A.; Freddolino, P.L.; Schulten, K. Stability and dynamics of virus capsids described by coarse-grained modeling. Structure 2006, 14, 1767–1777. [Google Scholar] [CrossRef]
- Terakawa, T.; Takada, S. RESPAC: Method to Determine Partial Charges in Coarse-Grained Protein Model and Its Application to DNA-Binding Proteins. J. Chem. Theory Comput. 2014, 10, 711–721. [Google Scholar] [CrossRef]
- McCullagh, P.; Lake, P.T.; Mccullagh, M. Deriving Coarse-Grained Charges from All-Atom Systems: An Analytic Solution. J. Chem. Theory Comput. 2016, 12, 4390–4399. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. Gromacs: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Iori, F.; Di Felice, R.; Molinari, E.; Corni, S. GolP: An atomistic force field to describe the interaction of proteins with Au(111) surfaces in water. J. Comput. Chem. 2009, 30, 1465–1476. [Google Scholar] [CrossRef] [PubMed]
- Gabdoulline, R.R.; Wade, R.C. Simulation of the diffusional association of barnase and barstar. Biophys. J. 1997, 72, 1917–1929. [Google Scholar] [CrossRef]
- Martinez, M.; Bruce, N.J.; Romanowska, J.; Kokh, D.B.; Ozboyaci, M.; Yu, X.; Öztürk, M.A.; Richter, S.; Wade, R.C. SDA 7: A modular and parallel implementation of the simulation of diffusional association software. J. Comput. Chem. 2015, 36, 1631–1645. [Google Scholar] [CrossRef]
- Mereghetti, P.; Gabdoulline, R.R.; Wade, R.C. Brownian dynamics simulation of protein solutions: Structural and dynamical properties. Biophys. J. 2010, 99, 3782–3791. [Google Scholar] [CrossRef]
- Ortega, A.; Amorós, D.; Garcia de la Torre, J. Prediction of hydrodynamic and other solution properties of rigid proteins from atomic- and residue-level models. Biophys. J. 2011, 101, 892–898. [Google Scholar] [CrossRef]
- Anderson, J.A.; Chris, D.; Lorenz, C.D.; Travesset, A. General purpose molecular dynamics simulations fully implemented on graphics processing units. J. Comput. Phys. 2008, 227, 5342–5359. [Google Scholar] [CrossRef]
- Glaser, J.; Nguyen, T.D.; Anderson, J.A.; Lui, P.; Spiga, F.; Millan, J.A.; Morse, D.C.; Glotzer, S.C. Strong scaling of general-purpose molecular dynamics simulations on GPUs. Comput. Phys. Comm. 2015, 192, 97–107. [Google Scholar] [CrossRef]
- Nguyen, T.D.; Phillips, C.L.; Anderson, J.A.; Glotzer, S.C. Rigid body constraints realized in massively-parallel molecular dynamics on graphics processing units. Comput. Phys. Comm. 2011, 182, 2307–2313. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Label | RelPop (a) | Urep (b) | ELJ + Uds (c) | Uep (d) | Spread (e) | Contact Residues (f) |
|---|---|---|---|---|---|---|
| CG-a(AA-e) | 59 | −44.1 | −30.3 | −13.8 | 10.4 | THR4, PRO5, LYS6, LEU87, LYS91, VAL93 |
| CG-b(AA-d) | 23 | −43.6 | −35.0 | −8.5 | 9.3 | LYS6, ILE7, GLN8, TYR26, VAL27, SER28, SER55, SER57, TYR63, LEU64, LEU65 |
| CG-c(AA-b) | 12 | −45.0 | −34.6 | −10.5 | 0.6 | NTR1, ARG3, HIS31, PRO32, TRP60, SER61 |
| CG-d(AA-a) | 6 | −46.2 | −30.9 | −15.3 | 15.4 | THR4, PRO5, LYS6, VAL82, HIS84, ASN83, THR86, LEU87, GLN89, LYS91, VAL93 |
| MS-a(AA-e) | (*) | −48.8 | −28.7 | −20.1 | (**) | VAL93 LYS91 LEU87 PRO5 THR4 LYS6 ILE7 |
| MS-b(AA-d) | (*) | −45.9 | −40.9 | −5.0 | (**) | LEU64 TYR63 SER57 SER55 SER28 VAL27 TYR26 GLN8 LEU65 SER52 |
| AA-a | 28 | −48.0 | −27.4 | −20.6 | 2.2 | TYR10, LYS91, ASP96, ARG97 |
| AA-a’ | 28 | −44.5 | −27.5 | −17.1 | 15.2 | GLY43, GLU44, ARG45 |
| AA-b | 18 | −41.9 | −31.7 | −10.2 | 2.67 | LYS58, ASP59, TRP60 |
| AA-c | 16 | −42.4 | −48.3 | 5.9 | 7.9 | MET99, HIS13, PRO14, GLU16, LYS19 |
| AA-d | 4 | −47.3 | −44.5 | −2.8 | 1.8 | SER33, ASP34, ILE35, LEU54, ASP53, LEU64, GLU36, VAL37, HIS51, TYR66 |
| AA-e | 6 | −46.5 | −49.4 | 2.8 | 1.4 | THR86, LEU87, SER88, GLN89, LYS91 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brancolini, G.; Lopez, H.; Corni, S.; Tozzini, V. Low-Resolution Models for the Interaction Dynamics of Coated Gold Nanoparticles with β2-microglobulin. Int. J. Mol. Sci. 2019, 20, 3866. https://doi.org/10.3390/ijms20163866
Brancolini G, Lopez H, Corni S, Tozzini V. Low-Resolution Models for the Interaction Dynamics of Coated Gold Nanoparticles with β2-microglobulin. International Journal of Molecular Sciences. 2019; 20(16):3866. https://doi.org/10.3390/ijms20163866
Chicago/Turabian StyleBrancolini, Giorgia, Hender Lopez, Stefano Corni, and Valentina Tozzini. 2019. "Low-Resolution Models for the Interaction Dynamics of Coated Gold Nanoparticles with β2-microglobulin" International Journal of Molecular Sciences 20, no. 16: 3866. https://doi.org/10.3390/ijms20163866
APA StyleBrancolini, G., Lopez, H., Corni, S., & Tozzini, V. (2019). Low-Resolution Models for the Interaction Dynamics of Coated Gold Nanoparticles with β2-microglobulin. International Journal of Molecular Sciences, 20(16), 3866. https://doi.org/10.3390/ijms20163866