1. Introduction
Sushi Domain Containing 2 (SUSD2) has been studied in cancer, neurodevelopment, and as a marker for mesenchymal stem cells. In cancer, SUSD2 has been described as both protumor [
1,
2] and antitumor [
3,
4] depending on the type of cancer. The mouse homolog of SUSD2, Susd2/SVS-1, has been implicated in the regulation of neurite outgrowth during development [
5]. SUSD2 has also been identified as the antigen for the W5C5 antibody, which is used as a marker for the isolation of mesenchymal stem cells from human tonsil and bone marrow [
6,
7]. While SUSD2 has been studied in diverse biological systems, there are currently few studies on the biochemical properties of the protein. Knowledge of the structure and post-translational processing of SUSD2 may prove useful in determining the functions of the protein in association with health and disease.
Our studies have been focused on the role of SUSD2 in breast cancer. Previous work has shown that 80% of patient breast tumors have moderate to high staining for SUSD2, and healthy tissues have minimal SUSD2 staining [
1,
8]. In a syngeneic mouse model, tumors with Susd2 had increased angiogenesis and decreased T cells in the tumor microenvironment [
1]. These results suggested that SUSD2 is a promising candidate for targeted therapy in breast cancer. SUSD2′s pathogenicity in breast cancer may be mediated through Galectin-1 (Gal-1). SUSD2 interacts with Gal-1 and is necessary for Gal-1 surface presentation [
1]. Gal-1 contributes to tumor immune evasion by inducing apoptosis of activated T cells [
9]. Kovacs-Solyom et al. demonstrated that Gal-1 surface presentation is required to induce T cell apoptosis [
10]. Therefore, SUSD2-mediated surface presentation of Gal-1 may be contributing to tumor immune evasion. To disrupt this process as a potential treatment for breast cancer, a more detailed understanding of cell surface presentation of Gal-1 by SUSD2 is required.
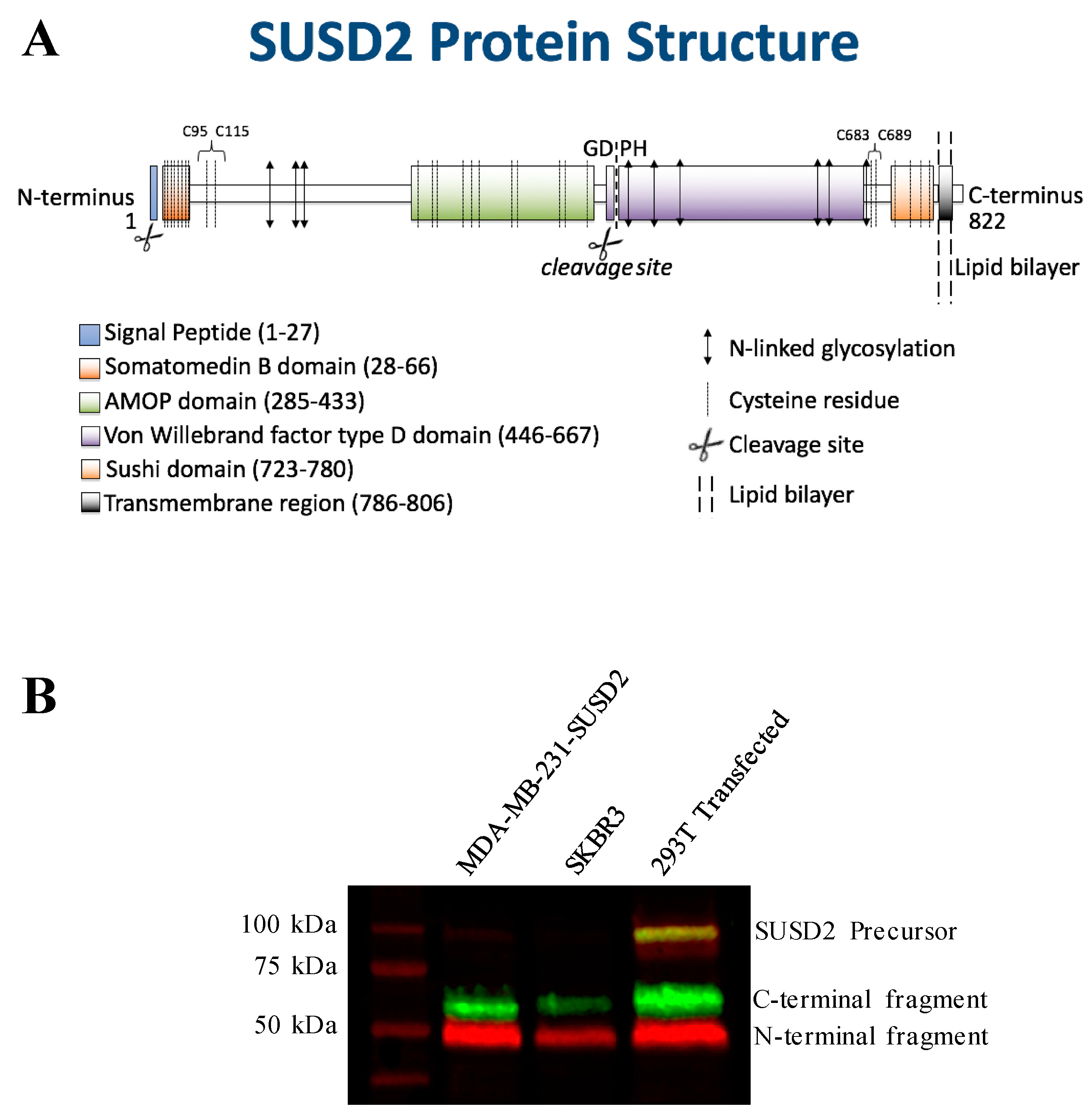
SUSD2 is an 822-amino acid type 1 transmembrane protein, and it is composed of somatomedin b, adhesion associated domain in MUC4 and other proteins (AMOP), von Willebrand factor type D (VWFD) and sushi domains (
Figure 1A). These domains have been implicated in cell adhesion in other proteins, and consistently, SUSD2 has been shown to play a role in cell adhesion functions [
5,
11]. SUSD2 shares the AMOP and VWFD domains with Mucin 4 (MUC4). A phylogenetic tree generated using AMOP and VWFD domain sequences suggests that SUSD2 and MUC4 developed from a common ancestral gene [
12]. The VWFD domain contains a glycine-aspartic acid-proline-histidine (GDPH) amino acid sequence that is conserved in many MUC family members and results in protein cleavage between the aspartic acid and proline residues of the GDPH sequence [
13,
14,
15]. Being a paralog to MUC4, we hypothesized that SUSD2 may have similar processing.
The predicted molecular weight of SUSD2 is 90.4 kDa. However, SUSD2 has nine predicted glycosylation sites (
Figure 1A), which would increase the size of the protein. We previously demonstrated by western immunoblot analysis using an anti-SUSD2 antibody that two bands were detected. The larger 110-kDa band was most likely a glycosylated form of SUSD2, and a 60-kDa band suggested that SUSD2 was post-translationally cleaved into two fragments [
1]. Since the anti-SUSD2 antibody used as a probe only recognized the C-terminal domain of the protein, only one of the two cleaved fragments was observed. Details of post-translational processing of SUSD2 may reveal critical steps that could be targeted therapeutically to inhibit the function of SUSD2 in breast cancer. Therefore, we investigated the mechanism by which SUSD2 was cleaved and whether the fragments remained associated. Here we report the identification of key post-translational processing steps for SUSD2 that are critical for surface localization of SUSD2 and consequently, Gal-1.
3. Discussion
SUSD2 is a paralog to MUC4 and contains a VWFD domain which is conserved in many MUC family members [
13,
14,
15]. Identical to MUC4, our data demonstrate that SUSD2 is cleaved between the aspartic acid and proline residues of the GDPH sequence in the VWFD domain (
Figure 1A and
Figure 2B). Although several MUC family members are cleaved at the conserved GDPH site, the cleavage may have different functional consequences depending on the specific protein. A summary of the similarities and differences of SUSD2, MUC4, MUC5AC, and MUC2 processing can be found in
Table 1. For example, when cleavage of the GDPH site in MUC5AC was inhibited, the uncleaved MUC5AC protein traveled through the secretory pathway [
13]. However, when SUSD2 GDPH cleavage was inhibited, SUSD2 was sequestered in the ER and was unable to traffic to the plasma membrane (
Figure 4A and
Figure 5B). Similar to SUSD2, MUC4-GDPH cleavage was required for its secretory pathway trafficking, as precursor MUC4 was degraded if it could not be cleaved [
18].
SUSD2 shares the GDPH sequence in common with MUC4, MUC5AC, and MUC2. Each of these proteins is cleaved at its GDPH sequence. However, the consequences of this cleavage are different in these proteins.
Table 1 highlights notable similarities and differences between SUSD2 and three MUC Family proteins. References are shown in parentheses.
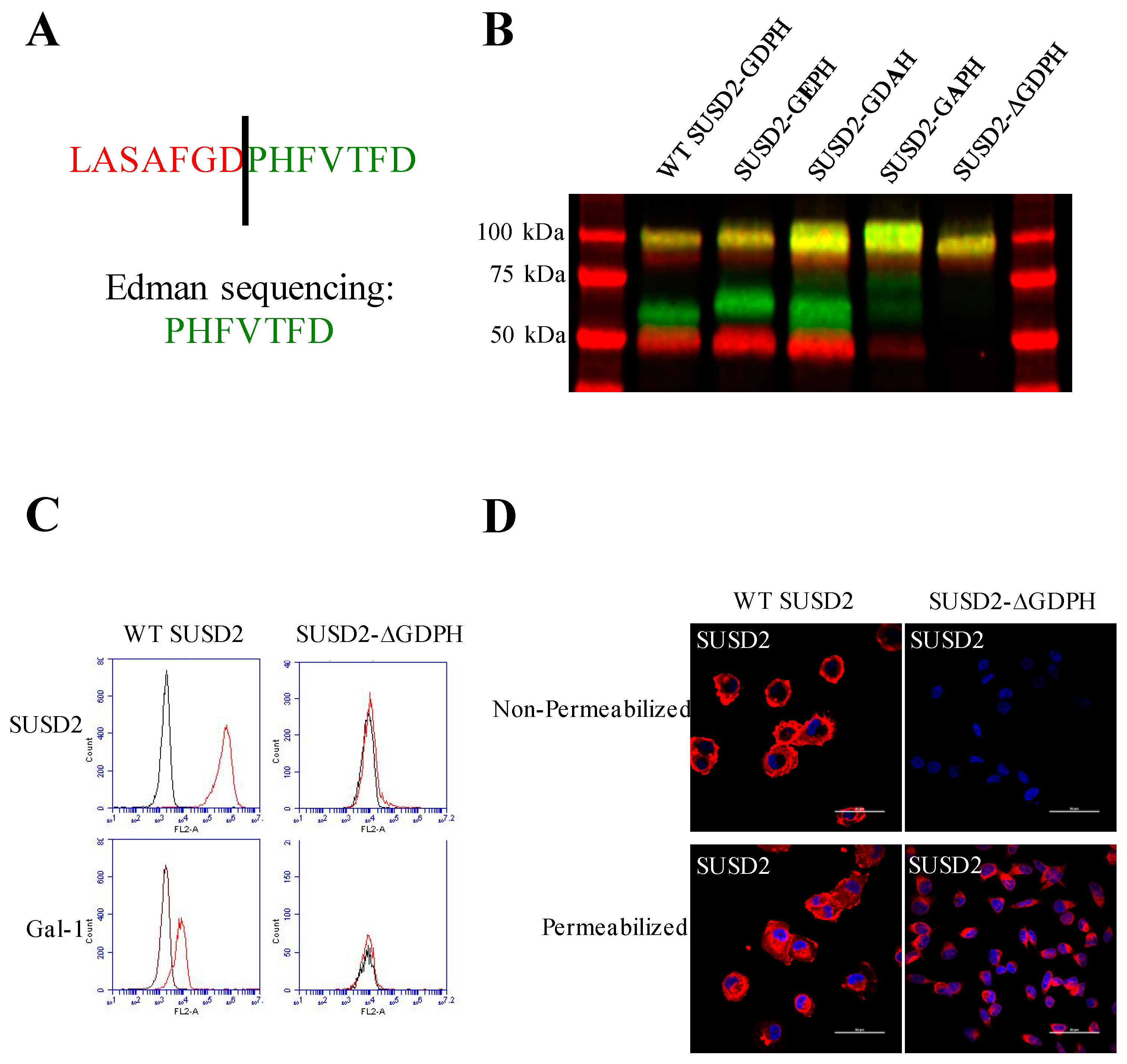
Cleavage in the GDPH sequence has been shown to occur in multiple proteins and by two distinct mechanisms, autocatalytic- and protease-dependent [
13,
14,
15,
21,
22]. The mechanism of MUC5AC cleavage was a pH dependent autolysis at the GDPH sequence. This type of autocatalytic cleavage between aspartic acid and proline is proposed to operate through generation of an unstable five-membered ring intermediate [
22]. This cleavage was abolished by mutagenesis of the GDPH sequence to GEPH in MUC5AC, which would prevent formation of this intermediate [
13]. Interestingly, cleavage of SUSD2-GEPH was not impaired (
Figure 2B). This mutation did not inhibit SUSD2 cleavage, suggesting that SUSD2 cleavage may not be dependent on an autocatalytic mechanism like MUC5AC. To further support a nonautocatalytic cleavage, in vitro transcription/translation was used to produce IVT SUSD2. Unlike the processing of SUSD2 in mammalian cells (
Figure 1B), IVT SUSD2 was not cleaved (
Figure S3B). These data are more consistent with an enzymatic mechanism of cleavage than an autocatalytic mechanism.
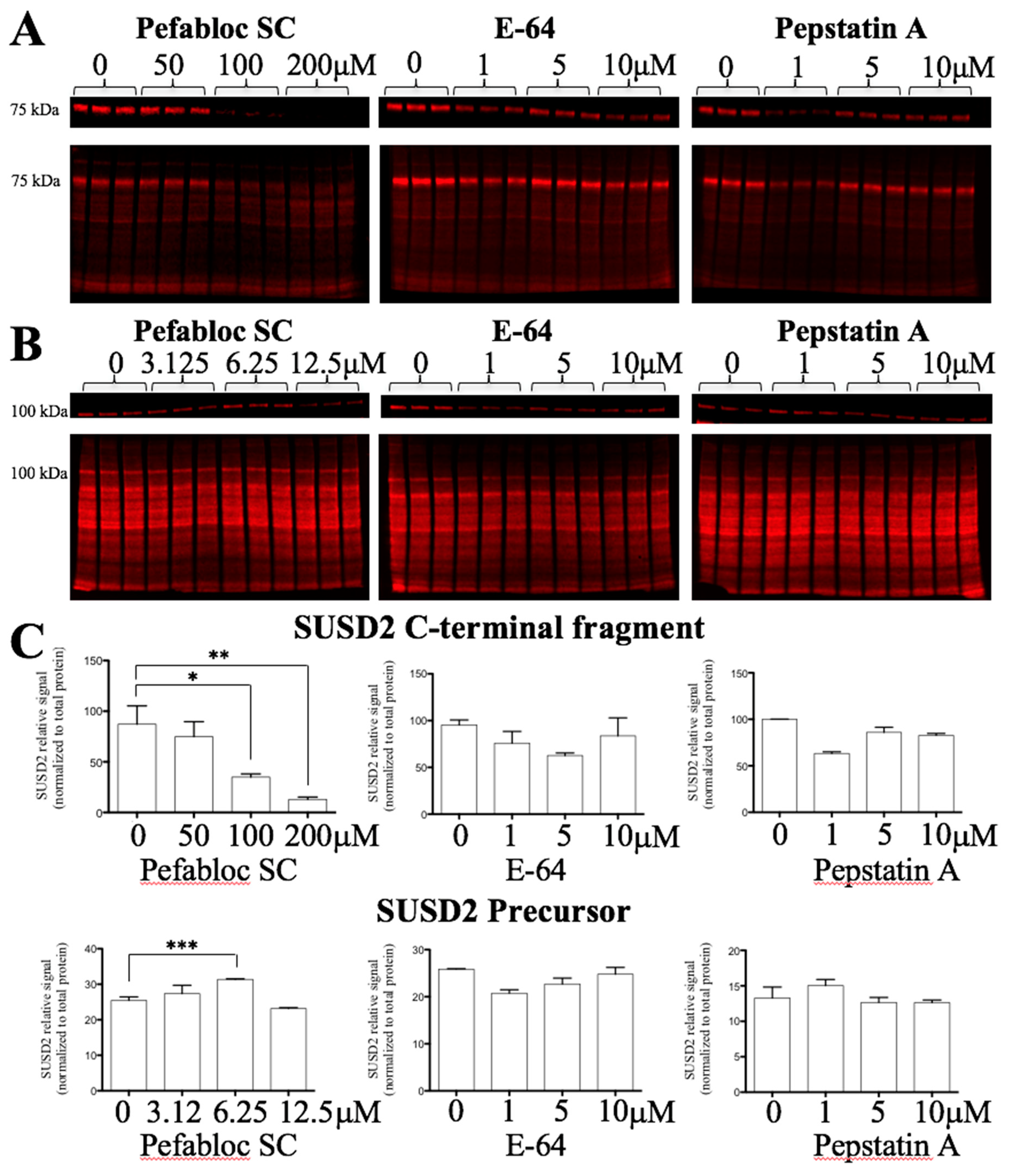
MUC4 is cleaved at its GDPH sequence through a proteolytic mechanism involving an unknown serine protease [
15]. Since MUC4 is a paralog of SUSD2, we hypothesized that SUSD2 may have the same cleavage mechanism as MUC4. Treating stable MDA-MB-231-SUSD2-SNAP cells for 24 h with Pefabloc SC, a pan serine protease inhibitor, demonstrated a significant dose dependent decrease in the SUSD2 C-terminal fragment (
Figure 4A,C). One would predict that inhibition of SUSD2 cleavage would result in the accumulation of the precursor polypeptide as amounts of cleaved fragments decreased. However, we found that at 50–200 µM concentrations of Pefabloc SC, SUSD2 full-length precursor levels decreased. The disappearance of the precursor SUSD2 at these higher doses can likely be attributed to the degradation of unprocessed SUSD2 as observed with MUC4 precursor [
18]. To identify an initial increase in the precursor before its degradation, we analyzed SUSD2 precursor levels at lower doses of Pefabloc SC and at an earlier time point (6 h). We observed a significant increase in SUSD2 precursor over 6 h at 6.25 µM Pefabloc SC, while E-64 and pepstatin A did not affect SUSD2 precursor levels significantly (
Figure 4B,C).
Reciprocal IP of dual tagged FLAG-SUSD2-Myc confirmed that the SUSD2 fragments remained associated after cleavage and that neither fragment was secreted into the supernatant (
Figure 6). Other groups have demonstrated that the secretion of MUC2 and MUC5AC does not require GDPH cleavage [
19,
20]. In contrast, MUC4 must be cleaved for the N-terminal fragment to be secreted into the extracellular environment [
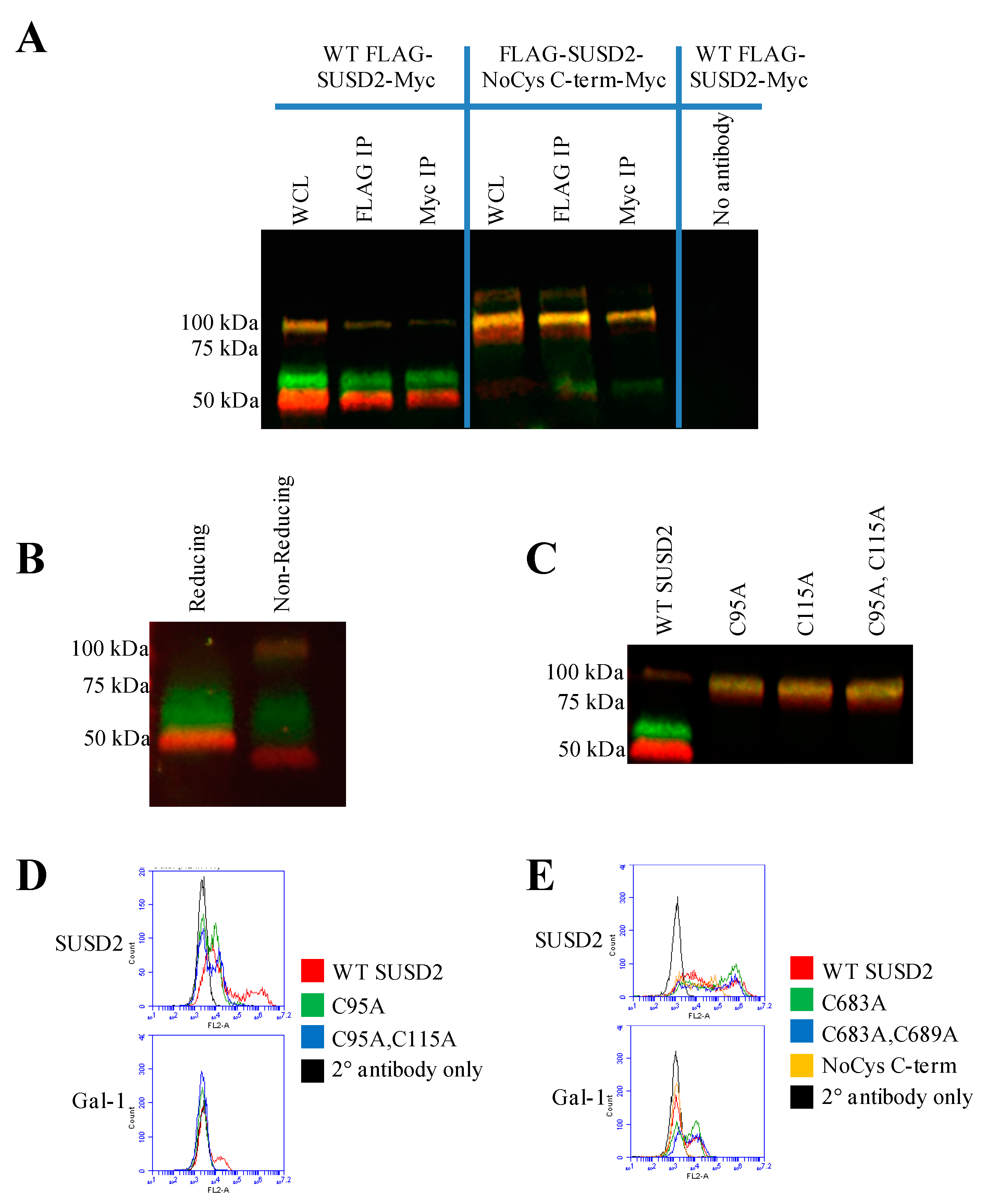
16]. Western immunoblot analysis in nonreducing conditions suggested that the SUSD2 fragments remained associated by at least one inter-fragment disulfide bond (
Figure 5B). Mutagenesis of targeted cysteines on SUSD2 revealed that inter-fragment disulfide bonding is required in order for SUSD2 to be cleaved (
Figure 5E).
SUSD2 contains two N-terminal (C95, C115) and two C-terminal (C683, C689) cysteines that are located outside a defined domain. When both sets of cysteines were substituted with alanine alone or in combination, C95A, C115A, or both combined, a complete inhibition of SUSD2 cleavage was observed (
Figure 5C), suggesting that both amino acids C95 and C115 participate in inter-fragment disulfide bonds and are required for cleavage of SUSD2. Consistent with previous data, C95A, C115A, as well as combined C95A and C115A SUSD2 mutants were unable to present Gal-1 at the cell surface (
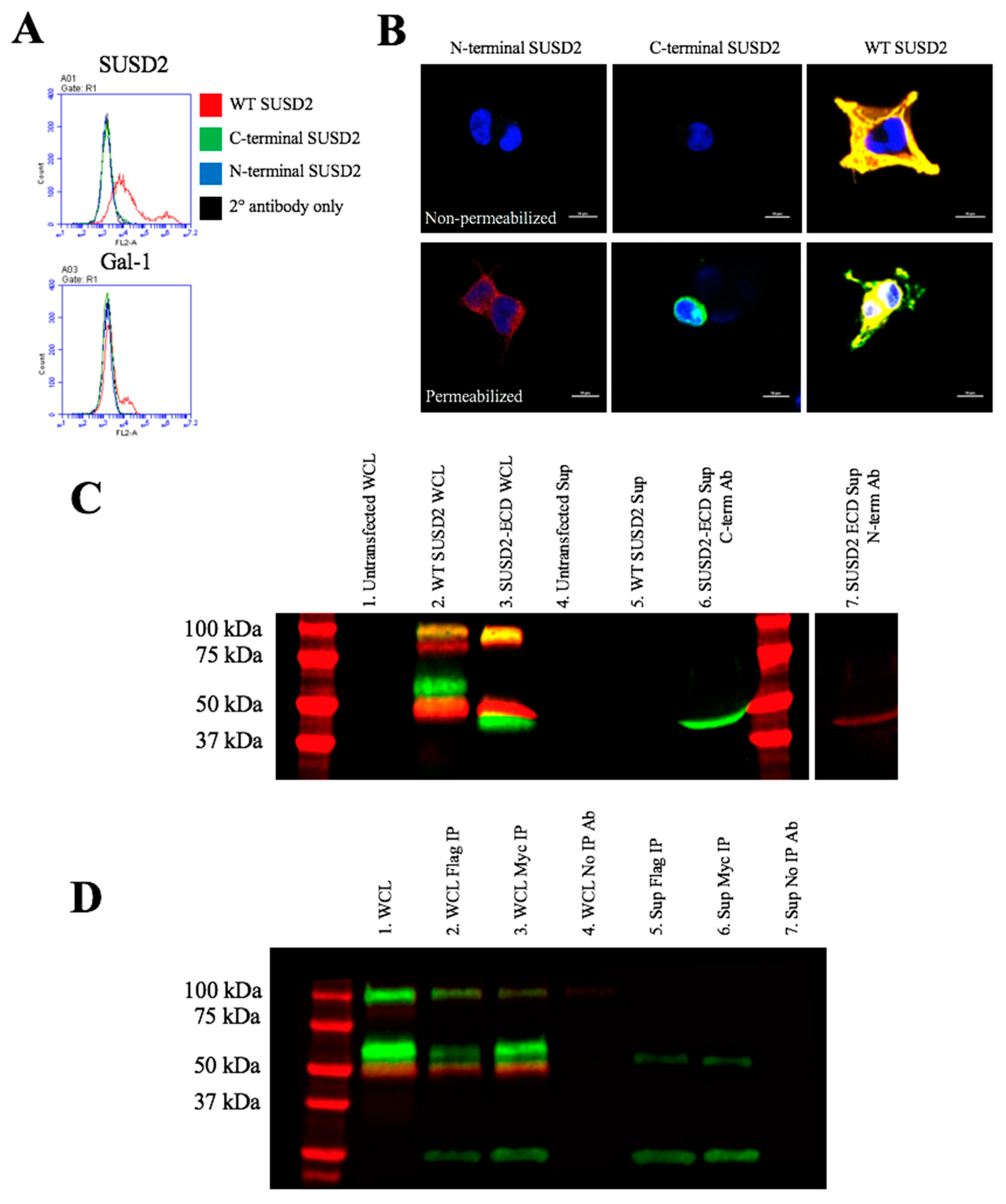
Figure 5D). Since disulfide bonding of SUSD2 fragments was critical for its cleavage, we hypothesized that the mature form of SUSD2 is composed of both fragments. As expected, neither the C- nor N-terminal SUSD2 fragments alone were able to present Gal-1 at the cell surface as analyzed by flow cytometry (
Figure 6A). Confocal microscopy analysis utilizing C-terminal and N-terminal specific antibodies further demonstrated that fragments expressed alone were localized in a perinuclear pattern similar to SUSD2-∆GDPH (
Figure 2D and
Figure 6B). However, WT SUSD2 fragments colocalized strongly at the cell surface in nonpermeabilized cells and throughout the cell in permeabilized conditions (
Figure 6B).
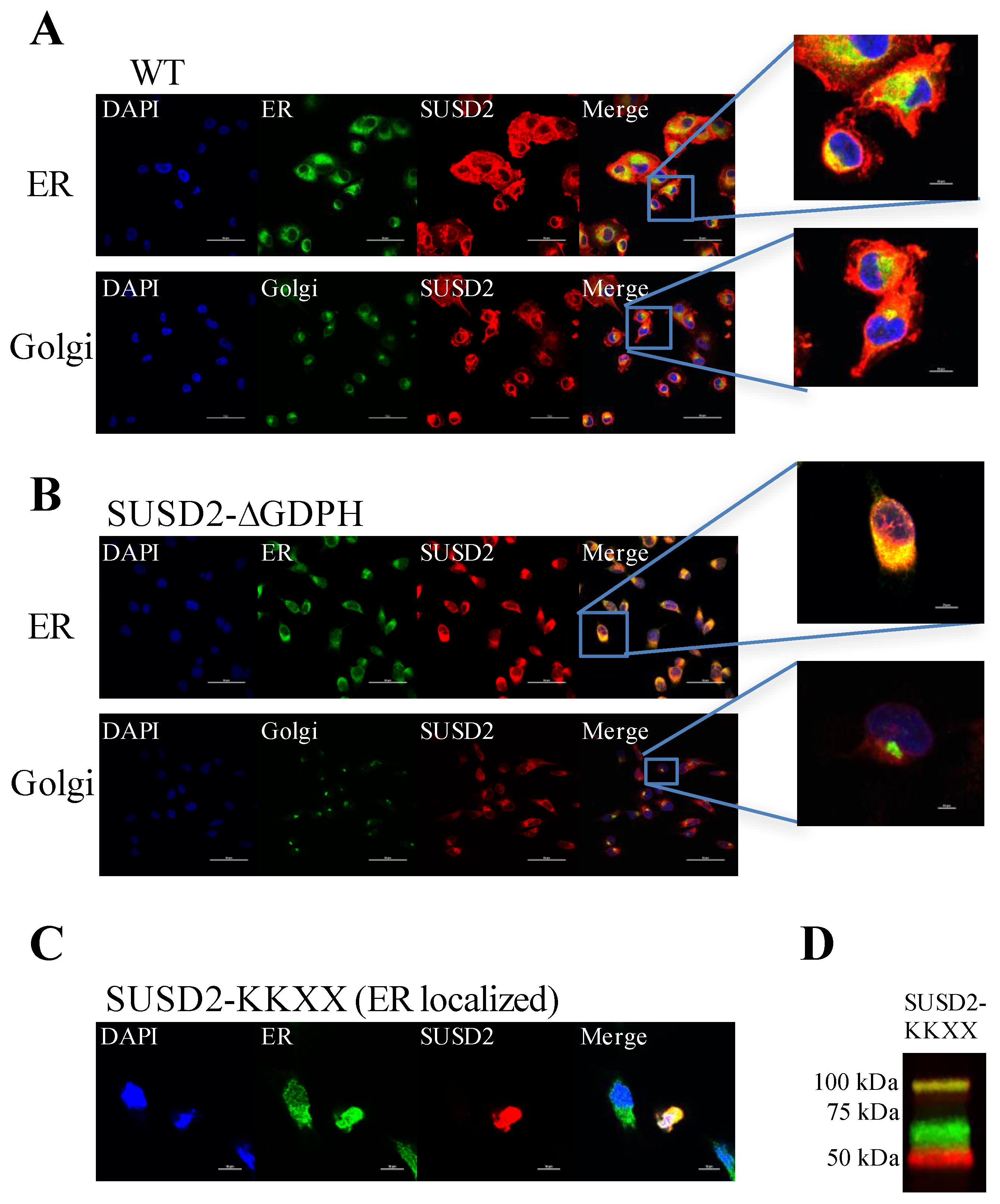
Immunofluorescence staining of WT SUSD2 confirmed surface localization and colocalization with ER and Golgi markers (
Figure 3A). These results indicate that WT SUSD2 is found in the traditional secretory pathway. However, SUSD2-∆GDPH is not found at the cell surface, and it colocalizes with the ER marker but not Golgi marker (
Figure 3B), suggesting that SUSD2 cleavage occurs in the ER and appears to be necessary for SUSD2 to transit out of the ER. In addition, SUSD2-KKXX was cleaved while being retained in the ER (
Figure 3C,D).
Taken together, our results indicate that SUSD2 is cleaved at the GDPH site in the ER by a yet unidentified serine protease. The SUSD2 N- and C-terminal fragments are both critical for surface localization of SUSD2 and Gal-1. SUSD2 must form disulfide bonds before cleavage and is transported to the membrane as a heterodimer with the fragments connected via disulfide bonds. SUSD2 chaperones Gal-1 to the cell surface of breast cancer cells, which has potential implications for immune evasion in breast cancer. Discovery of the identity of this protease could prove useful in targeting SUSD2 production as a therapeutic intervention in breast cancer by preventing Gal-1 surface presentation.
4. Materials and Methods
4.1. Design of SUSD2 Mutant Plasmids
SUSD2 mutants were generated using the Quikchange XL site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA). The pcDNA3.1-myc/his containing wild-type
SUSD2 was generated previously [
1] and used as the template for mutagenesis. The SUSD2 extracellular domain (SUSD2-ECD) was generated by inserting a stop codon directly before the transmembrane domain. Primers were synthesized by IDT, Coralville, IA, USA and shown in
Table S1. The expression construct encoding SUSD2 with the ER retention signal, KKXX, at the C-terminal end, referred to as pSUSD2-KKXX, was generated by inserting two lysine codons at positions immediately 5′ to the final 2 histidine codons of SUSD2 myc/his. This strategy generated a SUSD2 protein with lysines at the -3 and -4 position from the C-terminus.
SUSD2-GDPH mutants included SUSD2-GDPH deletion (SUSD2-∆GDPH), SUSD2-GEPH, SUSD2-GDAH, and SUSD2-GAPH. SUSD2-GDPH deletion primers were also used for mutagenesis with pLXSN-SUSD2 as a template. pLXSN-SUSD2 was generated previously by inserting the open reading frame of SUSD2 into the pLXSN plasmid without any tags [
1]. pLXSN-SUSD2-∆GDPH was used to generate stable cell lines expressing
SUSD2-∆GDPH without the myc/his tag.
SUSD2 cysteine mutants were generated by mutagenesis using pFLAG-SUSD2-Myc as a template. SUSD2 cysteine mutants included single mutations: C683A, C689A, C95A, and C115A; double mutations: C683A and C689A and C95A and C115A; and mutation of all six C-terminal fragment cysteines to alanine: C683A, C689A, C725A, C751A, C765A, and C778A. Mutagenesis primers are shown in
Table S1.
The expression plasmid encoding the SUSD2-SNAP fusion protein was generated by inserting SUSD2 into the pSNAPf plasmid (NEB, Ipswich, MA, USA) using the EcoRV and EcoRI cloning sites. Genscript’s Gene Synthesis service generated pFLAG-SUSD2-Myc in pcDNA3.1 with the FLAG sequence on the 5′ end of SUSD2 and myc sequence 3′ of SUSD2. The plasmid encoding the SUSD2 C-terminal fragment was synthesized by Genscript (Piscataway, NJ, USA), and the plasmid encoding the SUSD2 N-terminal fragment was generated using site-directed mutagenesis to insert a stop codon (TAA) directly 5′ of the nucleotides encoding the PH of the GDPH sequence. All plasmids generated were sequence verified using Simple Seq kit per manufacturer instructions (Eurofins Genomics, Louisville, Kentucky, USA). Sequences were analyzed using Sequencher DNA sequence analysis software (Gene Codes Corporation, Ann Arbor, Michigan, USA).
4.2. Cell Lines
MDA-MB-231, SKBR3, and 293T cells were maintained in DMEM supplemented with 10% fetal bovine serum (FBS) and penicillin and streptomycin at 37 °C in 5% CO
2 in a humidified air incubator. Stable MDA-MB-231-SUSD2 cells have been described previously [
1]. Stable MDA-MB-231-SUSD2 GDPH deletion (MDA-MB-231-SUSD2-∆GDPH) cells were generated using retroviral packaging Phoenix cells and pLXSN SUSD2-∆GDPH. Viral particles were used to infect MDA-MB-231 wild-type cells. Cells expressing SUSD2-∆GDPH were selected using 500 µM G418. Two single clones were isolated and designated MDA-MB-231-SUSD2-∆GDPH 1 and 9. Data shown in this paper are from MDA-MB-231-SUSD2-∆GDPH 1, and data are representative of results from both clones. MDA-MB-231-SUSD2-SNAP stable cells were generated by transfection with pSNAPf-SUSD2 and selection with G418. A clonal population was isolated and used for SUSD2-SNAP labeling assays. All cell lines were authenticated and tested negatively for mycoplasma.
4.3. Western Immunoblot Analysis
Western immunoblot analysis was performed as previously described [
1] with modifications. Following gel electrophoresis, proteins were transferred to Immobilon-FL polyvinylidene fluoride (PVDF) membranes (EMD Millipore, Billerica, MA, USA). Membranes were allowed to dry completely after transfer. Methanol was used to rehydrate membranes prior to sequential washing with Tris buffered saline (TBS) and blocking with 10% blocking buffer in TBS. Primary antibodies against the SUSD2 N-terminus and C-terminus were incubated on the same membrane overnight at 4 °C. The N-terminal specific primary is a rabbit monoclonal antibody (Abcam, Cambridge, UK), and the C-terminal specific primary is a mouse monoclonal antibody clone 944812 (R&D Systems, Minneapolis, MN, USA). Membranes were washed and blocked again with 5% blocking solution in TBS. IRdye 680RD (red) anti-rabbit secondary and IRdye 800 (green) anti-mouse secondary were used to detect the N-terminal and C-terminal specific primaries, respectively. (LI-COR, Lincoln, NE, USA). Membranes were dried and then imaged using an Odyssey Fc imaging system. (LI-COR)
4.4. Edman Sequencing
Expi293 cells have been adapted to higher density culture and optimized for high production of proteins [
23]. To facilitate secretion of SUSD2-ECD into the supernatant, an expression plasmid encoding the SUSD2 extracellular domain (pSUSD2-ECD) was designed by inserting a stop codon immediately prior to the transmembrane domain (diagram in
Figure S1). Supernatant of transfected Expi293 cells were harvested one week after transfection. Duplicate gels were run, and protein from each gel was transferred to a separate PVDF membrane. The first membrane was probed using a mouse monoclonal C-terminal specific SUSD2 antibody clone #944812 (R&D Systems) and detected using an anti-mouse secondary antibody conjugated to alkaline phosphatase (
Figure S1). Bands were visualized using colorimetric detection by addition of NBT/BCIP. This blot served as a reference that allowed the identification of the C-terminal band on the second membrane. The second membrane was stained with Coomassie to visualize the band that would be subjected to Edman degradation (
Figure S2). The C-terminal band was harvested in sample buffer containing 5% β-mercaptoethanol from the Coomassie stained membrane and sequenced by Edman degradation at the UC Davis Molecular Structure Facility, Davis, CA, USA.
4.5. Flow Cytometry
Cells were harvested using trypsin and resuspended in phosphate buffered saline (PBS) containing 1% FBS and 0.1% NaN3. Surface staining of SUSD2 and Gal-1 was performed using primary antibodies against SUSD2 (Abcam) or Gal-1 (R&D Systems) with nonpermeabilized cells. The antibody to SUSD2 was generated using an immunogen composed of amino acids 544–691 of human SUSD2, corresponding to sequence from the N-terminal fragment of SUSD2. Excess primary antibody was washed away, and cells were incubated with PE conjugated secondary antibodies. Cells were analyzed using an Accuri C6 flow cytometer (BD Biosciences, San Jose, CA, USA).
4.6. Immunofluorescence Microscopy
Immunofluorescence microscopy was performed as previously described [
1]. Triton X-100 was omitted in nonpermeabilized conditions. SUSD2 staining was performed using an N-terminal specific rabbit anti-SUSD2 antibody (Abcam) or a C-terminal specific mouse anti-SUSD2 antibody (Abnova, Taoyuan City, Taiwan). Mouse monoclonal anti-KDEL and mouse monoclonal anti-58K Golgi protein antibodies were used to visualize the endoplasmic reticulum (ER) and Golgi, respectively (Abcam). Slides were imaged using a Nikon A1 confocal microscope and analyzed using NIS elements software (Nikon, Melville, NY, USA).
4.7. Immunoprecipitation
Cell lysates were incubated with either M2 anti-FLAG (Sigma-Aldrich Corp., St Louis, MO, USA) or 4A6 anti-Myc (EMD Millipore) antibodies overnight at 4 °C on a rocking platform. Immune complexes were incubated with PureProteome Protein G magnetic beads (EMD Millipore) for 10 min at room temperature with constant inversion. Beads were then washed three times with 0.1% Tween 20 in PBS. Beads were separated from supernatant using a magnetic separator (EMD Millipore). Immune complexes were then boiled off the beads.
4.8. Fluorescent Pulse-chase
Stable MDA-MB-231-SUSD2-SNAP cells were utilized for fluorescent labeling of the SUSD2-SNAP fusion protein [
24]. Cells were treated with 0.5 µM bromothenylpteridine (BTP) for 30 min, which blocks any previously produced SUSD2-SNAP fusion protein. Cells were washed 3 times with complete media and placed in the incubator until harvest at the appropriate time point. Lysates were labeled with 10 µM SNAP-Surface 682 in PBS with 1mM dithiothreitol (DTT), separated by gel electrophoresis, and proteins were transferred to a Immobilon-FL PVDF membrane (EMD Millipore). Membranes were imaged using LI-COR Odyssey Fc imager.
To assay the effect of protease inhibitors and pH on SUSD2 cleavage, fluorescence pulse chase was utilized. MDA-MB-231-SUSD2-SNAP cells were blocked with BTP as described above. After the final wash step to remove BTP, protease inhibitors were added to assess the effects of inhibitors on nascent SUSD2 only. MDA-MB-231-SUSD2-SNAP cells were treated with various protease inhibitors for 6 or 24 h. 1, 5, and 10 µM Pepstatin A (aspartic) (ThermoFisher Scientific), 3.125, 6.25, and 12.5 µM Pefabloc SC (serine) (Sigma-Aldrich Corp.) and 1, 5, and 10 µM E-64 (cysteine) (Sigma-Aldrich Corp.) were used separately to inhibit different classes of proteases. For pH neutralization experiments, complete media with 25 mM ammonium chloride was added to MDA-MB-231-SUSD2-SNAP cells. The pH was neutralized directly following the final wash step to remove BTP in order to visualize the effect of pH on only nascent SUSD2. After 6 h of protease inhibitor treatment or pH neutralization, cell lysates were harvested and labeled with 10 µM SNAP-surface 682 in PBS with 1 mM DTT for one hour at 37 °C. Following labeling, lysates were heated at 95 °C for 3 min and separated by gel electrophoresis before transfer to a PVDF membrane. The membrane was imaged using LI-COR Odyssey Fc imager. Subsequently, membranes were stained for total protein as a loading control using REVERT total protein stain per manufacturer instructions (LI-COR) and imaged again using LI-COR Odyssey Fc imager.
4.9. In Vitro Transcription/Translation of SUSD2
In vitro transcription/translation (IVT) of SUSD2 was produced using the 1-Step Human High-Yield Mini IVT Kit (ThermoFisher). SUSD2 was cloned into the pT7CFE1-NHis-GST vector using EcoRI and EcoRV restriction sites. IVT of SUSD2 was carried out according to manufacturer instructions.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}