Endothelium-Dependent Hyperpolarization (EDH) in Diabetes: Mechanistic Insights and Therapeutic Implications


Abstract
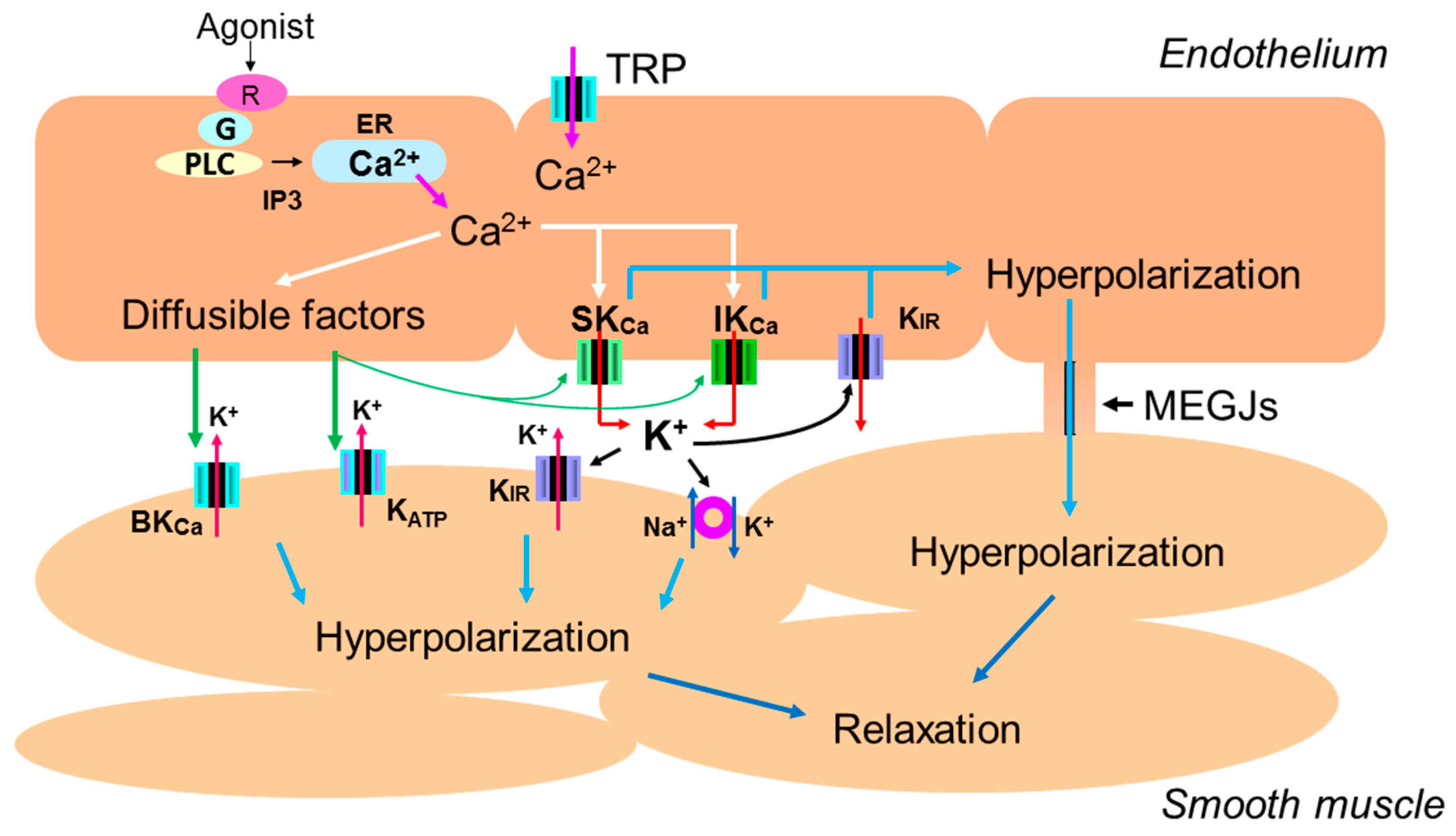
1. Introduction
2. EDH in Animal Models of Diabetes
3. Mechanisms of Impaired EDH in Diabetes
3.1. The Role of Intracellular Ca2+ Mobilization
3.2. The Role of Endothelial Potassium Channels
3.3. The Role of Gap Junctions
3.4. The Role of ROS
3.5. The Role of Inflammatory Cytokines
3.6. The Roles of Diffusible Factors
3.7. Other Factors
4. Therapeutic Implications
4.1. Insulin
4.2. Biguanide (Metformin)
4.3. Dpp-4 Inhibitors And Glp-1r Agonists
4.4. SGLT2 Inhibitors
4.5. Renin Angiotensin System Inhibitors
4.6. Statins
4.7. Protein Kinase C Inhibitors
4.8. Aldose Reductase Inhibitors
4.9. Exercise
5. EDH in Human Diabetes
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
| ACE | angiotensin converting enzyme |
| ACh | Acetylcholine |
| AGE | advanced glycation end product |
| AMPK | AMP-activated protein kinase |
| ARB | angiotensin type 1 receptor blocker |
| ARI | aldose reductase inhibitor |
| BKCa | large conductance Ca2+-activated K+ |
| CNP | C-type natriuretic peptide |
| CYP | cytochrome P450 |
| Cx | connexin |
| DPP-4 | dipeptidyl peptidase-4 |
| EDH | endothelium-dependent hyperpolarization |
| EDHF | endothelium-derived hyperpolarizing factor |
| EETs | epoxyeicosatrienoic acids |
| eNOS | endothelial nitric oxide synthase |
| ER | endoplasmic reticulum |
| GK | Goto-Kakizaki |
| GLP-1R | glucagon-like peptide-1 receptor |
| H2O2 | hydrogen peroxide |
| H2S | hydrogen sulfide |
| HUVEC | human umbilical vein endothelial cell |
| IKCa | intermediate–conductance Ca2+-activated K+ |
| IL-1β | interleukin-1beta |
| IL-6 | interleukin-6 |
| KATP | ATP–sensitive K+ |
| Kir | inward rectifier K+ |
| L-NAME | Nω-nitro-l-arginine |
| LPC | lysophosphatidylcholine |
| MEGJs | myoendothelial gap junctions |
| NEP | neural endopeptidase |
| NADPH | nicotinamide adenine dinucleotide phosphate oxidase |
| NO | nitric oxide |
| Nrf2 | nuclear factor erythroid-2-related factor-2 |
| OLETF | Otsuka long-evans tokushima fatty |
| Ox-LDL | oxidized low-density lipoprotein |
| OZ | obese Zucker |
| PDE | phosphodiesterase |
| PKA | protein kinase A |
| PKC | protein kinase C |
| RAS | renin-angiotensin system |
| ROS | reactive oxygen species |
| sEH | soluble epoxide hydrolase |
| SGLT2 | sodium glucose co-transporter2 |
| SHR | spontaneously hypertensive rats |
| SHRSP | stroke-prone spontaneously hypertensive rats |
| SKCa | small-conductance Ca2+-activated K+ |
| STZ | streptozotocin |
| TNF-α | tumor necrosis factor-alpha |
| TRP | transient receptor potential |
| TRPV1 | TRP vanilloid-type-1 |
| TRPV4 | TRP vanilloid-type-4 thromboxane A2 (TXA2) |
| TXA2 | thromboxane A2 |
| WKY | Wistar–Kyoto |
| ZDF | Zucker diabetic fatty |
References
- Vanhoutte, P.M.; Shimokawa, H.; Feletou, M.; Tang, E.H.C. Endothelial dysfunction and vascular disease–A 30th anniversary update. Acta Physiol. 2017, 219, 22–96. [Google Scholar] [CrossRef] [PubMed]
- Félétou, M. Endothelium-Dependent Hyperpolarization and Endothelial Dysfunction. J. Cardiovasc. Pharm. 2016, 67, 373–387. [Google Scholar] [CrossRef]
- Garland, C.J.; Dora, K.A. EDH: Endothelium-dependent hyperpolarization and microvascular signalling. Acta Physiol. 2017, 219, 152–161. [Google Scholar] [CrossRef]
- Goto, K.; Ohtsubo, T.; Kitazono, T. Endothelium-Dependent Hyperpolarization (EDH) in Hypertension: The Role of Endothelial Ion Channels. Int. J. Mol. Sci. 2018, 19, 315. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.V.; Sandow, S.L. Agonist-evoked endothelial Ca2+ signalling microdomains. Curr. Opin. Pharm. 2019, 45, 8–15. [Google Scholar] [CrossRef]
- Fleming, I. The factor in EDHF: Cytochrome P450 derived lipid mediators and vascular signaling. Vasc. Pharm. 2016, 86, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Coleman, H.A.; Tare, M.; Parkington, H.C. Nonlinear effects of potassium channel blockers on endothelium-dependent hyperpolarization. Acta Physiol. 2017, 219, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Sandow, S.L.; Hill, C.E. Incidence of Myoendothelial Gap Junctions in the Proximal and Distal Mesenteric Arteries of the Rat Is Suggestive of a Role in Endothelium-Derived Hyperpolarizing Factor–Mediated Responses. Circ. Res. 2000, 86, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.H.; Ruddiman, C.A.; Keller, T.S.; Keller, A.S.; Yang, Y.; Good, M.E.; Best, A.K.; Columbus, L.; Isakson, B.E. Heterocellular Contact Can Dictate Arterial Function. Circ. Res. 2019, 124, 1473–1481. [Google Scholar] [CrossRef]
- Earley, S.; Brayden, J.E. Transient Receptor Potential Channels in the Vasculature. Physiol. Rev. 2015, 95, 645–690. [Google Scholar] [CrossRef] [PubMed]
- Behringer, E.; Hakim, M. Functional Interaction among KCa and TRP Channels for Cardiovascular Physiology: Modern Perspectives on Aging and Chronic Disease. Int. J. Mol. Sci. 2019, 20, 1380. [Google Scholar] [CrossRef] [PubMed]
- Grayson, T.H.; Murphy, T.V.; Sandow, S.L. Transient receptor potential canonical type 3 channels: Interactions, role and relevance - A vascular focus. Pharm. Ther. 2017, 174, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Sonkusare, S.K.; Dalsgaard, T.; Bonev, A.D.; Nelson, M.T. Inward rectifier potassium (Kir2.1) channels as end-stage boosters of endothelium-dependent vasodilators. J. Physiol. 2016, 594, 3271–3285. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.; López-Oliva, M.E.; Pinilla, E.; Martínez, M.P.; Sánchez, A.; Rodríguez, C.; García-Sacristán, A.; Hernández, M.; Rivera, L.; Prieto, D. CYP epoxygenase-derived H2O2 is involved in the endothelium-derived hyperpolarization (EDH) and relaxation of intrarenal arteries. Free. Radic. Boil. Med. 2017, 106, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Domingueti, C.P.; Dusse, L.M.S.; Carvalho, M.D.G.; de Sousa, L.P.; Gomes, K.B.; Fernandes, A.P. Diabetes mellitus: The linkage between oxidative stress, inflammation, hypercoagulability and vascular complications. J. Diabetes Complicat. 2016, 30, 738–745. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.-I.; Kao, Y.-H.; Chen, Y.-C.; Huang, J.-H.; Hsiao, F.-C.; Chen, Y.-J. Peroxisome proliferator-activated receptors modulate cardiac dysfunction in diabetic cardiomyopathy. Diabetes Res. Clin. Pract. 2013, 100, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Worldwide trends in diabetes since 1980: A pooled analysis of 751 population-based studies with 4∙4 million participants. Lancet 2016, 387, 1513–1530. [CrossRef]
- Al-Awar, A.; Kupai, K.; Veszelka, M.; Szűcs, G.; Attieh, Z.; Murlasits, Z.; Török, S.; Pósa, A.; Varga, C. Experimental Diabetes Mellitus in Different Animal Models. J. Diabetes Res. 2016, 2016, 1–12. [Google Scholar] [CrossRef]
- Fukao, M.; Hattori, Y.; Kanno, M.; Sakuma, I.; Kitabatake, A. Alterations in endothelium-dependent hyperpolarization and relaxation in mesenteric arteries from streptozotocin-induced diabetic rats. Br. J. Pharm. 1997, 121, 1383–1391. [Google Scholar] [CrossRef]
- Makino, A.; Ohuchi, K.; Kamata, K. Mechanisms underlying the attenuation of endothelium-dependent vasodilatation in the mesenteric arterial bed of the streptozotocin-induced diabetic rat. Br. J. Pharm. 2000, 130, 549–556. [Google Scholar] [CrossRef]
- Wigg, S.J.; Tare, M.; Tonta, M.A.; O’Brien, R.C.; Meredith, I.T.; Parkington, H.C. Comparison of effects of diabetes mellitus on an EDHF-dependent and an EDHF-independent artery. Am. J. Physiol. Circ. Physiol. 2001, 281, H232–H240. [Google Scholar] [CrossRef] [PubMed]
- Leo, C.H.; Hart, J.L.; Woodman, O.L. Impairment of both nitric oxide-mediated and EDHF-type relaxation in small mesenteric arteries from rats with streptozotocin-induced diabetes. Br. J. Pharm. 2011, 162, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Absi, M.; Oso, H.; Khattab, M. The effect of streptozotocin-induced diabetes on the EDHF-type relaxation and cardiac function in rats. J. Adv. Res. 2013, 4, 375–383. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ma, X.; Du, J.; Zhang, P.; Deng, J.X.; Liu, J.; Fu-Yuen Lam, F.; Li, R.A.; Huang, Y.; Jin, J.; Yao, X.Q. Functional role of TRPV4-KCa2.3 signaling in vascular endothelial cells in Normal and streptozotocin-induced diabetic rats. Hypertension 2013, 62, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, K.; Matoba, T.; Kubota, H.; Makoto, H.; Takako, F.; Shosuke, T.; Akira, T.; Hiroaki, S. Influence of diabetes mellitus, hypercholesterolemia, and their combination on EDHF-mediated responses in mice. J. Cardiovasc. Pharm. 2005, 45, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Hashem, M.; Wiehler, W.B.; Lau, W.; Martín, J.; Reid, J.; Triggle, C. Endothelial dysfunction in the streptozotocin-induced diabetic apoE-deficient mouse. Br. J. Pharm. 2005, 146, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Miyamori, K.; Kobayashi, T.; Kamata, K. Specific impairment of endothelium-derived hyperpolarizing factor-type relaxation in mesenteric arteries from streptozotocin-induced diabetic mice. Vascul. Pharm. 2006, 44, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Makino, A.; Platoshyn, O.; Suarez, J.; Yuan, J.X.-J.; Dillmann, W.H. Downregulation of connexin40 is associated with coronary endothelial cell dysfunction in streptozotocin-induced diabetic mice. Am. J. Physiol. 2008, 295, C221–C230. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, M.J.; Edgley, A.J.; Sonobe, T.; Umetani, K.; Schwenke, D.O.; Fujii, Y.; Brown, R.D.; Kelly, D.J.; Shirai, M.; Pearson, J.T. Dynamic synchrotron imaging of diabetic rat coronary microcirculation in vivo. Vasc. Biol. 2012, 32, 370–377. [Google Scholar] [CrossRef]
- Nakazawa, T.; Kaneko, Y.; Mori, A.; Saito, M.; Sakamoto, K.; Nakahara, T.; Ishii, K. Attenuation of nitric oxide- and prostaglandin-independent vasodilation of retinal arterioles induced by acetylcholine in streptozotocin-treated rats. Vascul. Pharm. 2007, 46, 153–159. [Google Scholar] [CrossRef]
- De Vriese, A.S.; Van De Voorde, J.; Blom, H.J.; Vanhoutte, P.M.; Verbeke, M.; Lameire, N.H. The impaired renal vasodilator response attributed to endothelium-derived hyperpolarizing factor in streptozotocin - Induced diabetic rats is restored by 5-methyltetrahydrofolate. Diabetologia 2000, 43, 1116–1125. [Google Scholar] [CrossRef]
- Jack, A.M.; Keegan, A.; Cotter, M.A.; Cameron, N.E. Effects of diabetes and evening primrose oil treatment on responses of aorta, corpus cavernosum and mesenteric vasculature in rats. Life Sci. 2002, 71, 1863–1877. [Google Scholar] [CrossRef]
- Terata, K.; Coppey, L.J.; Davidson, E.P.; Dunlap, J.A.; Gutterman, D.D.; Yorek, M.A. Acetylcholine-induced arteriolar dilation is reduced in streptozotocin-induced diabetic rats with motor nerve dysfunction. Br. J. Pharm. 1999, 128, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Kitada, M.; Ogura, Y.; Koya, D. Rodent models of diabetic nephropathy: Their utility and limitations. Int. J. Nephrol. Renovasc. Dis. 2016, 9, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Burnham, M.P.; Johnson, I.T.; Weston, A.H. Impaired small-conductance Ca2+-activated K+ channel-dependent EDHF responses in Type II diabetic ZDF rats. Br. J. Pharm. 2006, 148, 434–441. [Google Scholar] [CrossRef]
- Young, E.J.; Hill, M.A.; Wiehler, W.B.; Triggle, C.R.; Reid, J.J. Reduced EDHF responses and connexin activity in mesenteric arteries from the insulin-resistant obese Zucker rat. Diabetologia 2008, 51, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Oniki, H.; Fujii, K.; Kansui, Y.; Goto, K.; Iida, M. Effects of angiotensin II receptor antagonist on impaired endothelium-dependent and endothelium-independent relaxations in type II diabetic rats. J. Hypertens. 2006, 24, 331–338. [Google Scholar] [CrossRef]
- Brøndum, E.; Kold-Petersen, H.; Simonsen, U.; Aalkjaer, C. NS309 restores EDHF-type relaxation in mesenteric small arteries from type 2 diabetic ZDF rats. Br. J. Pharm. 2010, 159, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Kobayashi, T.; Kamata, K. Mechanisms underlying the impaired EDHF-type relaxation response in mesenteric arteries from Otsuka Long-Evans Tokushima Fatty (OLETF) rats. Eur. J. Pharm. 2006, 538, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Schach, C.; Resch, M.; Schmid, P.M.; Riegger, G.A.; Endemann, D.H. Type 2 diabetes: Increased expression and contribution of IK Ca channels to vasodilation in small mesenteric arteries of ZDF rats. Am. J. Physiol. Circ. Physiol. 2014, 307, H1093–H1102. [Google Scholar] [CrossRef]
- Matsumoto, T.; Kobayashi, S.; Ando, M.; Watanabe, S.; Iguchi, M.; Taguchi, K.; Kobayashi, T. Impaired endothelium-derived hyperpolarization-type relaxation in superior mesenteric arteries isolated from female Otsuka Long-Evans Tokushima Fatty rats. Eur. J. Pharm. 2017, 807, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Kold-Petersen, H.; Laher, I.; Simonsen, U.; Aalkjaer, C. Impaired endothelial calcium signaling is responsible for the defective dilation of mesenteric resistance arteries from db/db mice to acetylcholine. Eur. J. Pharm. 2015, 767, 17–23. [Google Scholar] [CrossRef]
- Park, Y.; Capobianco, S.; Gao, X.; Falck, J.R.; Dellsperger, K.C.; Zhang, C. Role of EDHF in type 2 diabetes-induced endothelial dysfunction. Am. J. Physiol. Circ. Physiol. 2008, 295, H1982–H1988. [Google Scholar] [CrossRef] [PubMed]
- Yin, D.; Wang, Q.; Zhou, X.; Li, Y. Endothelial dysfunction in renal arcuate arteries of obese Zucker rats: The roles of nitric oxide, endothelium-derived hyperpolarizing factors, and calcium-activated K.+ channels. PLoS ONE 2017, 12, e0183124. [Google Scholar] [CrossRef] [PubMed]
- Tajbakhsh, N.; Sokoya, E.M. Compromised Endothelium-Dependent Hyperpolarization-Mediated Dilations can be Rescued by NS309 in Obese Zucker Rats. Microcirculation 2014, 21, 747–753. [Google Scholar] [CrossRef]
- Schjørring, O.; Kun, A.; Flyvbjerg, A.; Kirkeby, H.J.; Jensen, J.B.; Simonsen, U. Flow-Evoked Vasodilation Is Blunted in Penile Arteries from Zucker Diabetic Fatty Rats. J. Sex. Med. 2012, 9, 1789–1800. [Google Scholar] [CrossRef] [PubMed]
- Coppey, L.J.; Gellett, J.S.; Davidson, E.P.; Dunlap, J.A.; Yorek, M.A. Changes in endoneurial blood flow, motor nerve conduction velocity and vascular relaxation of epineurial arterioles of the sciatic nerve in ZDF-obese diabetic rats. Diabetes Metab. Res. Rev. 2002, 18, 49–56. [Google Scholar] [CrossRef]
- Edgley, A.J.; Tare, M.; Evans, R.G.; Skordilis, C.; Parkington, H.C. In vivo regulation of endothelium-dependent vasodilation in the rat renal circulation and the effect of streptozotocin-induced diabetes. Am. J. Physiol. Integr. Comp. Physiol. 2008, 295, R829–R839. [Google Scholar] [CrossRef]
- Kajikuri, J.; Watanabe, Y.; Ito, Y.; Ito, R.; Yamamoto, T.; Itoh, T. Characteristic changes in coronary artery at the early hyperglycaemic stage in a rat type 2 diabetes model and the effects of pravastatin. Br. J. Pharm. 2009, 158, 621–632. [Google Scholar] [CrossRef]
- Shi, Y. Augmented Endothelium-Derived Hyperpolarizing Factor-Mediated Relaxations Attenuate Endothelial Dysfunction in Femoral and Mesenteric, but Not in Carotid Arteries from Type I Diabetic Rats. J. Pharm. Exp. Ther. 2006, 318, 276–281. [Google Scholar] [CrossRef]
- Pieper, G.M. Enhanced, unaltered and impaired nitric oxide-mediated endothelium- dependent relaxation in experimental diabetes mellitus: Importance of disease duration. Diabetologia 1999, 42, 204–213. [Google Scholar] [CrossRef]
- van den Heuvel, M.; Sorop, O.; van Ditzhuijzen, N.S.; de Vries, R.; van Duin, R.W.B.; Peters, I.; van Loon, J.E.; de Maat, M.P.; van Beusekom, H.M.; van der Giessen, W.J. The effect of bioresorbable vascular scaffold implantation on distal coronary endothelial function in dyslipidemic swine with and without diabetes. Int. J. Cardiol. 2018, 252, 44–51. [Google Scholar] [CrossRef]
- Climent, B.; Moreno, L.; Martinez, P.; Contreras, C.; Sánchez, A.; Perez-Vizcaino, F.; García-Sacristán, A.; Rivera, L.; Prieto, D. Upregulation of SK3 and IK1 Channels Contributes to the Enhanced Endothelial Calcium Signaling and the Preserved Coronary Relaxation in Obese Zucker Rats. PLoS ONE 2014, 9, 109432. [Google Scholar] [CrossRef]
- Yada, T.; Shimokawa, H.; Tachibana, H. Endothelium-dependent hyperpolarization-mediated vasodilatation compensates nitric oxide-mediated endothelial dysfunction during ischemia in diabetes-induced canine coronary collateral microcirculation in vivo. Microcirculation 2018, 25, e12456. [Google Scholar] [CrossRef]
- Chadderdon, S.M.; Belcik, J.T.; Bader, L.; Peters, D.M.; Kievit, P.; Alkayed, N.J.; Kaul, S.; Grove, K.L.; Lindner, J.R. Temporal Changes in Skeletal Muscle Capillary Responses and Endothelial-Derived Vasodilators in Obesity-Related Insulin Resistance. Diabetes 2016, 65, 2249–2257. [Google Scholar] [CrossRef]
- Guerra, G.; Lucariello, A.; Perna, A.; Botta, L.; De Luca, A.; Moccia, F. The Role of Endothelial Ca2+ Signaling in Neurovascular Coupling: A View from the Lumen. Int. J. Mol. Sci. 2018, 19, 938. [Google Scholar] [CrossRef]
- Ottolini, M.; Hong, K.; Sonkusare, S.K. Calcium signals that determine vascular resistance. Wiley Interdiscip. Rev. Syst. Biol. Med. 2019, e1448. [Google Scholar] [CrossRef]
- Graier, W.F.; Wascher, T.C.; Lackner, L.; Toplak, H.; Krejs, G.J.; Kukovetz, W.R. Exposure to elevated D-glucose concentrations modulates vascular endothelial cell vasodilatatory response. Diabetes 1993, 42, 1497–1505. [Google Scholar] [CrossRef]
- Pieper, G.M.; Dondlinger, L. Glucose elevations alter bradykinin-stimulated intracellular calcium accumulation in cultured endothelial cells. Cardiovasc. Res. 1997, 34, 169–178. [Google Scholar] [CrossRef][Green Version]
- Kimura, C.; Oike, M.; Ito, Y. Acute Glucose Overload Abolishes Ca2+ Oscillation in Cultured Endothelial Cells From Bovine Aorta. Circ. Res. 1998, 82, 677–685. [Google Scholar] [CrossRef]
- Kimura, C.; Oike, M.; Kashiwagi, S.; Ito, Y. Effects of Acute Glucose Overload on Histamine H2 Receptor-Mediated Ca2+ Mobilization in Bovine Cerebral Endothelial Cells. Diabetes 1998, 47, 104–112. [Google Scholar] [CrossRef]
- Tang, Y.; Li, G.D. Chronic exposure to high glucose impairs bradykinin-stimulated nitric oxide production by interfering with the phospholipase-C-implicated signalling pathway in endothelial cells: Evidence for the involvement of protein kinase C. Diabetologia 2004, 47, 2093–2104. [Google Scholar] [CrossRef][Green Version]
- Bishara, N.B.; Dunlop, M.E.; Murphy, T.V.; Darby, I.A.; Sharmini Rajanayagam, M.A.; Hill, M.A. Matrix protein glycation impairs agonist-induced intracellular Ca2+ signaling in endothelial cells. J. Cell Physiol. 2002, 193, 80–92. [Google Scholar] [CrossRef]
- Özkan, M.H.; Uma, S. Inhibition of acetylcholine-induced EDHF response by elevated glucose in rat mesenteric artery. Life Sci. 2005, 78, 14–21. [Google Scholar] [CrossRef]
- Gomes, M.B.; Cailleaux, S.; Tibiriçá, E. Metformin prevents the impairment of endothelium-dependent vascular relaxation induced by high glucose challenge in rabbit isolated perfused kidneys. Naunyn. Schmiedebergs Arch. Pharm. 2005, 372, 24–30. [Google Scholar] [CrossRef]
- Tsai, S.H.; Hein, T.W.; Kuo, L.; Yang, V.C. High glucose impairs EDHF-mediated dilation of coronary arterioles via reduced cytochrome P450 activity. Microvasc. Res. 2011. [Google Scholar] [CrossRef]
- Kamata, K.; Nakajima, M. Ca2+ mobilization in the aortic endothelium in streptozotocin-induced diabetic and cholesterol-fed mice. Br. J. Pharm. 1998, 123, 1509–1516. [Google Scholar] [CrossRef][Green Version]
- Estrada, I.A.; Donthamsetty, R.; Debski, P.; Zhou, M.-H.; Zhang, S.L.; Yuan, J.X.-J.; Han, W.; Makino, A. STIM1 Restores Coronary Endothelial Function in Type 1 Diabetic Mice. Circ. Res. 2012, 111, 1166–1175. [Google Scholar] [CrossRef]
- Gokina, N.I.; Bonev, A.D.; Gokin, A.P.; Goloman, G. Role of impaired endothelial cell Ca2+ signaling in uteroplacental vascular dysfunction during diabetic rat pregnancy. Am. J. Physiol. Circ. Physiol. 2013, 304, H935–H945. [Google Scholar] [CrossRef][Green Version]
- Shamsaldeen, Y.A.; Ugur, R.; Benham, C.D.; Lione, L.A. Diabetic dyslipidaemia is associated with alterations in eNOS, caveolin-1, and endothelial dysfunction in streptozotocin treated rats. Diabetes Metab. Res. Rev. 2018, 34, e2995. [Google Scholar] [CrossRef]
- Fukao, M.; Hattori, Y.; Kanno, M.; Sakuma, I.; Kitabatake, A. Evidence for selective inhibition by lysophosphatidylcholine of acetylcholine-induced endothelium-dependent hyperpolarization and relaxation in rat mesenteric artery. Br. J. Pharm. 1995, 116, 1541–1543. [Google Scholar] [CrossRef][Green Version]
- Eizawa, H.; Yui, Y.; Inoue, R.; Kosuga, K.; Hattori, R.; Aoyama, T.; Sasayama, S. Lysophosphatidylcholine Inhibits Endothelium-Dependent Hyperpolarization and N ω -Nitro- l -Arginine/IndomethacinResistant Endothelium-Dependent Relaxation in the Porcine Coronary Artery. Circulation 1995, 92, 3520–3526. [Google Scholar] [CrossRef]
- Seki, T.; Goto, K.; Kiyohara, K.; Kansui, Y.; Murakami, N.; Haga, Y.; Ohtsubo, T.; Matsumura, K.; Kitazono, T. Downregulation of Endothelial Transient Receptor Potential Vanilloid Type 4 Channel and Small-Conductance of Ca2+ -Activated K+ Channels Underpins Impaired Endothelium-Dependent Hyperpolarization in Hypertension. Hypertension 2017, 69, 143–153. [Google Scholar] [CrossRef]
- Boudaka, A.; Al-Suleimani, M.; Al-Lawati, I.; BaOmar, H.; Siyabi, S.A.; Zadjali, F. Downregulation of Endothelial Transient Receptor Potential Vanilloid Type 4 Channel Underlines Impaired Endothelial Nitric Oxide-Mediated Relaxation in the Mesenteric Arteries of Hypertensive Rats Ammar. Physiol. Res. 2019, 219–231. [Google Scholar] [CrossRef]
- Monaghan, K.; Mcnaughten, J.; McGahon, M.K.; Kelly, C.; Kyle, D.; Yong, P.H.; McGeown, J.G.; Curtis, T.M. Hyperglycemia and Diabetes Downregulate the Functional Expression of TRPV4 Channels in Retinal Microvascular Endothelium. PLoS ONE 2015, 10, e0128359. [Google Scholar] [CrossRef]
- Cassuto, J.; Dou, H.; Czikora, I.; Szabo, A.; Patel, V.S.; Kamath, V.; De Chantemele, E.B.; Feher, A.; Romero, M.J.; Bagi, Z. Peroxynitrite Disrupts Endothelial Caveolae Leading to eNOS Uncoupling and Diminished Flow-Mediated Dilation in Coronary Arterioles of Diabetic Patients. Diabetes 2014, 63, 1381–1393. [Google Scholar] [CrossRef]
- Cheng, J.P.X.; Nichols, B.J. Caveolae: One Function or Many? Trends Cell Biol. 2016, 26, 177–189. [Google Scholar] [CrossRef]
- Goedicke-Fritz, S.; Kaistha, A.; Kacik, M.; Markert, S.; Hofmeister, A.; Busch, C.; Bänfer, S.; Jacob, R.; Grgic, I.; Hoyer, J. Evidence for functional and dynamic microcompartmentation of Cav-1/TRPV4/KCa in caveolae of endothelial cells. Eur. J. Cell Boil. 2015, 94, 391–400. [Google Scholar] [CrossRef]
- Goto, K.; Rummery, N.M.; Grayson, T.H.; Hill, C.E. Attenuation of conducted vasodilatation in rat mesenteric arteries during hypertension: Role of inwardly rectifying potassium channels. J. Physiol. 2004, 561, 215–231. [Google Scholar] [CrossRef]
- Sancho, M.; Samson, N.C.; Hald, B.O.; Hashad, A.M.; Marrelli, S.P.; Brett, S.E.; Welsh, D.G. KIR channels tune electrical communication in cerebral arteries. J. Cereb. Blood Flow. Metab. 2017, 37, 2171–2184. [Google Scholar] [CrossRef]
- Hearon, C.M.; Richards, J.C.; Racine, M.L.; Luckasen, G.J.; Larson, D.G.; Dinenno, F.A. Amplification of endothelium-dependent vasodilatation in contracting human skeletal muscle: Role of K IR channels. J. Physiol. 2019, 597, 1321–1335. [Google Scholar] [CrossRef]
- Gokina, N.I.; Bonev, A.D.; Phillips, J.; Gokin, A.P.; Veilleux, K.; Oppenheimer, K.; Goloman, G. Impairment of IK Ca channels contributes to uteroplacental endothelial dysfunction in rat diabetic pregnancy. Am. J. Physiol. Circ. Physiol. 2015, 309, H592–H604. [Google Scholar] [CrossRef]
- Zhu, J.-H.; Jia, R.-P.; Xu, L.-W.; Wu, J.-P.; Wang, Z.-Z.; Wang, S.-K.; Bo, C.-J. Reduced expression of SK3 and IK1 channel proteins in the cavernous tissue of diabetic rats. Asian J. Androl. 2010, 12, 599–604. [Google Scholar] [CrossRef]
- Chadha, P.S.; Haddock, R.E.; Howitt, L.; Morris, M.J.; Murphy, T.V.; Grayson, T.H.; Sandow, S.L. Obesity up-regulates intermediate conductance calcium-activated potassium channels and myoendothelial gap junctions to maintain endothelial vasodilator function. J. Pharm. Exp. Ther. 2010, 335, 284–293. [Google Scholar] [CrossRef]
- Weston, A.H.; Absi, M.; Harno, E.; Geraghty, A.R.; Ward, D.T.; Ruat, M.; Dodd, R.H.; Dauban, P.; Edwards, G. The expression and function of Ca2+-sensing receptors in rat mesenteric artery; comparative studies using a model of type II diabetes. Br. J. Pharm. 2008, 154, 652–662. [Google Scholar] [CrossRef]
- Haddock, R.E.; Grayson, T.H.; Morris, M.J.; Howitt, L.; Chadha, P.S.; Sandow, S.L. Diet-Induced Obesity Impairs Endothelium-Derived Hyperpolarization via Altered Potassium Channel Signaling Mechanisms. PLoS ONE 2011, 6, e16423. [Google Scholar] [CrossRef]
- Dogné, S.; Flamion, B.; Caron, N. Endothelial Glycocalyx as a Shield Against Diabetic Vascular Complications. Arterioscler Thromb. Vasc. Biol. 2018, 38, 1427–1439. [Google Scholar]
- Dogné, S.; Rath, G.; Jouret, F.; Caron, N.; Dessy, C.; Flamion, B. Hyaluronidase 1 deficiency preserves endothelial function and glycocalyx integrity in early streptozotocin-induced diabetes. Diabetes 2016, 65, 2742–2753. [Google Scholar] [CrossRef]
- Mishra, R.C.; Wulff, H.; Cole, W.C.; Braun, A.P. A pharmacologic activator of endothelial KCa channels enhances coronary flow in the hearts of type 2 diabetic rats. J. Mol. Cell. Cardiol. 2014, 72, 364–373. [Google Scholar] [CrossRef]
- Newsholme, P.; Cruzat, V.F.; Keane, K.N.; Carlessi, R.; De Bittencourt, P.I.H. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem. J. 2016, 473, 4527–4550. [Google Scholar] [CrossRef]
- Dal, S.; Sigrist, S. The Protective Effect of Antioxidants Consumption on Diabetes and Vascular Complications. Diseases 2016, 4, 24. [Google Scholar] [CrossRef]
- Hermann, A.; Sitdikova, G.F.; Weiger, T.M. Oxidative stress and maxi calcium-activated potassium (BK) channels. Biomolecules 2015, 5, 1870–1911. [Google Scholar] [CrossRef]
- Choi, S.; Na, H.Y.; Kim, J.A.; Cho, S.E.; Suh, S.H. Contradictory effects of superoxide and hydrogen peroxide on K Ca3.1 in human endothelial cells. Korean. J. Physiol. Pharmacol. 2013, 17, 181–187. [Google Scholar] [CrossRef]
- Cai, S.; Sauvé, R. Effects of thiol-modifying agents on a K(Ca2+) channel of intermediate conductance in bovine aortic endothelial cells. J. Membr. Biol. 1997, 158, 147–158. [Google Scholar] [CrossRef]
- Choi, S.; Kim, J.A.; Na, H.Y.; Kim, J.E.; Park, S.; Han, K.H.; Kim, Y.J.; Suh, S.H. NADPH oxidase 2-derived superoxide downregulates endothelial K Ca3.1 in preeclampsia. Free Radic. Biol. Med. 2013, 57, 10–21. [Google Scholar] [CrossRef]
- Nowotny, K.; Jung, T.; Höhn, A.; Weber, D.; Grune, T. Advanced Glycation End Products and Oxidative Stress in Type 2 Diabetes Mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef]
- Naser, N.; Januszewski, A.S.; Brown, B.E.; Jenkins, A.J.; Hill, M.A.; Murphy, T.V. Advanced Glycation End Products Acutely Impair Ca2+ Signaling in Bovine Aortic Endothelial Cells. Front. Physiol. 2013, 4. [Google Scholar] [CrossRef]
- Zhao, L.M.; Wang, Y.; Ma, X.Z.; Wang, N.P.; Deng, X.L. Advanced glycation end products impair KCa3.1- and K Ca2.3-mediated vasodilatation via oxidative stress in rat mesenteric arteries. Pflugers Arch. Eur. J. Physiol. 2014, 466, 307–317. [Google Scholar] [CrossRef]
- Yi, F.; Ling, T.-Y.; Lu, T.; Wang, X.-L.; Li, J.; Claycomb, W.C.; Shen, W.-K.; Lee, H.-C. Down-regulation of the Small Conductance Calcium-activated Potassium Channels in Diabetic Mouse Atria*. J. Boil. Chem. 2015, 290, 7016–7026. [Google Scholar] [CrossRef]
- Alaaeddine, R.A.; Mroueh, A.; Gust, S.; Eid, A.H.; Plane, F.; El-Yazbi, A.F. Impaired cross-talk between NO and hyperpolarization in myoendothelial feedback: A novel therapeutic target in early endothelial dysfunction of metabolic disease. Curr. Opin. Pharm. 2019, 45, 33–41. [Google Scholar] [CrossRef]
- Fancher, I.S.; Ahn, S.J.; Adamos, C.; Osborn, C.; Oh, M.J.; Fang, Y.; Reardon, C.A.; Getz, G.S.; Phillips, S.A.; Levitan, I. Hypercholesterolemia-Induced Loss of Flow-Induced Vasodilation and Lesion Formation in Apolipoprotein E–Deficient Mice Critically Depend on Inwardly Rectifying K+ Channels. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef]
- Sancho, M.; Fabris, S.; Hald, B.O.; Brett, S.E.; Sandow, S.L.; Poepping, T.L.; Welsh, D.G. Membrane Lipid-K IR 2.x Channel Interactions Enable Hemodynamic Sensing in Cerebral Arteries. Arterioscler Thromb. Vasc. Biol. 2019, 39, 1072–1087. [Google Scholar] [CrossRef]
- Barbera, N.; Levitan, I. Chiral Specificity of Cholesterol Orientation Within Cholesterol Binding Sites in Inwardly Rectifying K+ Channels. Adv. Exp. Med. Biol. 2019, 77–95. [Google Scholar]
- Pogoda, K.; Kameritsch, P.; Mannell, H.; Pohl, U. Connexins in the control of vasomotor function. Acta Physiol. 2019, 225, e13108. [Google Scholar] [CrossRef]
- Molica, F.; Figueroa, X.; Kwak, B.; Isakson, B.; Gibbins, J. Connexins and Pannexins in Vascular Function and Disease. Int. J. Mol. Sci. 2018, 19, 1663. [Google Scholar] [CrossRef]
- Inoguchi, T.; Ueda, F.; Umeda, F.; Yamashita, T.; Nawata, H. Inhibition of intercellular communication via gap junction in cultured aortic endothelial cells by elevated glucose and phorbol ester. Biochem. Biophys. Res. Commun. 1995, 208, 492–497. [Google Scholar] [CrossRef]
- Kuroki, T.; Inoguchi, T.; Umeda, F.; Ueda, F.; Nawata, H. High glucose induces alteration of gap junction permeability and phosphorylation of connexin-43 in cultured aortic smooth muscle cells. Diabetes 1998, 47, 931–936. [Google Scholar] [CrossRef]
- Sato, T.; Haimovici, R.; Kao, R.; Li, A.F.; Roy, S. Downregulation of connexin 43 expression by high glucose reduces gap junction activity in microvascular endothelial cells. Diabetes 2002, 51, 1565–1571. [Google Scholar] [CrossRef]
- Fernandes, R.; Girão, H.; Pereira, P. High glucose down-regulates intercellular communication in retinal endothelial cells by enhancing degradation of connexin 43 by a proteasome-dependent mechanism. J. Biol. Chem. 2004, 279, 27219–27224. [Google Scholar] [CrossRef]
- Stewart, W.W. Lucifer dyes - Highly fluorescent dyes for biological tracing. Nature 1981, 292, 17–21. [Google Scholar] [CrossRef]
- Oku, H.; Kodama, T.; Sakagami, K.; Puro, D.G. Diabetes-induced disruption of gap junction pathways within the retinal microvasculature. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1915–1920. [Google Scholar]
- Lemmey, H.A.L.; Ye, X.; Ding, H.C.; Triggle, C.R.; Garland, C.J.; Dora, K.A. Hyperglycaemia disrupts conducted vasodilation in the resistance vasculature of db/db mice. Vascul. Pharmacol. 2018, 103–105, 29–35. [Google Scholar] [CrossRef]
- Shibata, M.; Nakaizumi, A.; Puro, D.G. Electrotonic transmission in the retinal vasculature: Inhibitory role of the diabetes/ VEGF/aPKC/ pathway. Physiol. Rep. 2019, 7, e14095. [Google Scholar] [CrossRef]
- Makino, A.; Dai, A.; Han, Y.; Youssef, K.D.; Wang, W.; Donthamsetty, R.; Scott, B.T.; Wang, H.; Dillmann, W.H. O-GlcNAcase overexpression reverses coronary endothelial cell dysfunction in type 1 diabetic mice. Am. J. Physiol. Physiol. 2015, 309, C593–C599. [Google Scholar] [CrossRef]
- Ellis, A.; Goto, K.; Chaston, D.J.; Brackenbury, T.D.; Meaney, K.R.; Falck, J.R.; Wojcikiewicz, R.J.H.; Hill, C.E. Enalapril Treatment Alters the Contribution of Epoxyeicosatrienoic Acids but Not Gap Junctions to Endothelium-Derived Hyperpolarizing Factor Activity in Mesenteric Arteries of Spontaneously Hypertensive Rats. J. Pharm. Exp. Ther. 2009, 330, 413–422. [Google Scholar] [CrossRef]
- Pogoda, K.; Kameritsch, P.; Retamal, M.A.; Vega, J.L. Regulation of gap junction channels and hemichannels by phosphorylation and redox changes: A revision. BMC Cell Biol. 2016, 17, 11. [Google Scholar] [CrossRef]
- Griffith, T.M.; Chaytor, A.T.; Taylor, H.J.; Giddings, B.D.; Edwards, D.H. cAMP facilitates EDHF-type relaxations in conduit arteries by enhancing electrotonic conduction via gap junctions. Proc. Natl. Acad. Sci. USA 2002, 99, 6392–6397. [Google Scholar] [CrossRef]
- Kansui, Y.; Goto, K.; Fujii, K.; Oniki, H.; Matsumura, K.; Iida, M. Cilostamide produces hyperpolarization associated with katp channel activation, but does not augment endothelium-derived hyperpolarization in rat mesenteric arteries. Clin. Exp. Pharm. Physiol. 2009, 36, 729–733. [Google Scholar] [CrossRef]
- Sokoya, E.M.; You, J. Impaired cAMP signaling does not account for the attenuated EDHF-mediated dilations in female rat middle cerebral artery. Brain Res. 2007, 1139, 29–33. [Google Scholar] [CrossRef]
- Matsumoto, T.; Wakabayashi, K.; Kobayashi, T.; Kamata, K. Diabetes-related changes in cAMP-dependent protein kinase activity and decrease in relaxation response in rat mesenteric artery. Am. J. Physiol. Circ. Physiol. 2004, 287, H1064–H1071. [Google Scholar] [CrossRef]
- Moreira, H.S.; Lima-Leal, G.A.; Santos-Rocha, J.; Gomes-Pereira, L.; Duarte, G.P.; Xavier, F.E. Phosphodiesterase-3 inhibitor cilostazol reverses endothelial dysfunction with ageing in rat mesenteric resistance arteries. Eur. J. Pharml. 2018, 822, 59–68. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, Q.; Zhu, J.; Xiao, Q.; Zhang, L. Reactive oxygen species: Key regulators in vascular health and diseases. Br. J. Pharm. 2018, 175, 1279–1292. [Google Scholar] [CrossRef]
- Kayama, Y.; Raaz, U.; Jagger, A.; Adam, M.; Schellinger, I.N.; Sakamoto, M.; Suzuki, H.; Toyama, K.; Spin, J.M.; Tsao, P.S. Diabetic Cardiovascular Disease Induced by Oxidative Stress. Int. J. Mol. Sci. 2015, 16, 25234–25263. [Google Scholar] [CrossRef]
- Cameron, N.E.; Jack, A.M.; Cotter, M.A. Effect of α-lipoic acid on vascular responses and nociception in diabetic rats. Free Radic. Biol. Med. 2001, 31, 125–135. [Google Scholar] [CrossRef]
- Agouni, A.; Lagrue-Lak-Hal, A.-H.; Mostefai, H.A.; Tesse, A.; Mulder, P.; Rouet, P.; Desmoulin, F.; Heymes, C.; Martínez, M.C.; Andriantsitohaina, R. Red Wine Polyphenols Prevent Metabolic and Cardiovascular Alterations Associated with Obesity in Zucker Fatty Rats (Fa/Fa). PLoS ONE 2009, 4, e5557. [Google Scholar] [CrossRef]
- Liang, C.F.; Liu, J.T.; Wang, Y.; Xu, A.; Vanhoutte, P.M. Toll-like receptor 4 mutation protects obese mice against endothelial dysfunction by decreasing NADPH oxidase isoforms 1 and 4. Arter. Thromb Vasc. Biol. 2013, 33, 777–784. [Google Scholar] [CrossRef]
- Dunn, S.M.; Das, K.C. Decreased EDHF-mediated relaxation is a major mechanism in endothelial dysfunction in resistance arteries in aged mice on prolonged high-fat sucrose diet. Physiol. Rep. 2017, 5. [Google Scholar] [CrossRef]
- Inkster, M.E.; Cotter, M.A.; Cameron, N.E. Treatment with the xanthine oxidase inhibitor, allopurinol, improves nerve and vascular function in diabetic rats. Eur. J. Pharmacol. 2007, 561, 63–71. [Google Scholar] [CrossRef]
- Boydens, C.; Pauwels, B.; Vanden Daele, L.; Van de Voorde, J. Protective effect of resveratrol and quercetin on in vitro-induced diabetic mouse corpus cavernosum. Cardiovasc. Diabetol. 2016, 15, 46. [Google Scholar] [CrossRef]
- Nangle, M.; Gibson, T.; Cotter, M.; Cameron, N. Effects of Eugenol on Nerve and Vascular Dysfunction in Streptozotocin-Diabetic Rats. Planta Med. 2006, 72, 494–500. [Google Scholar] [CrossRef]
- Oniki, H.; Goto, K.; Fujii, K.; Kansui, Y.; Murakami, N.; Ohtsubo, T.; Matsumura, K.; Kitazono, T. Effects of the Superoxide Dismutase Mimetic Tempol on Impaired Endothelium-Dependent and Endothelium-Independent Relaxations in Type II Diabetic Rats. Clin. Exp. Hypertens 2013, 35, 112–119. [Google Scholar] [CrossRef]
- Wigg, S.J.; Tare, M.; Forbes, J.; Cooper, M.E.; Thomas, M.C.; Coleman, H.A.; Parkington, H.C.; O’Brien, R.C. Early vitamin E supplementation attenuates diabetes-associated vascular dysfunction and the rise in protein kinase C-β in mesenteric artery and ameliorates wall stiffness in femoral artery of Wistar rats. Diabetologia 2004, 47, 1038–1046. [Google Scholar] [CrossRef]
- Leo, C.-H.; Hart, J.L.; Woodman, O.L. 3.′,4′-Dihydroxyflavonol Reduces Superoxide and Improves Nitric Oxide Function in Diabetic Rat Mesenteric Arteries. PLoS ONE 2011, 6, e20813. [Google Scholar] [CrossRef]
- Ma, X.; Li, Y.-F.; Gao, Q.; Ye, Z.-G.; Lu, X.-J.; Wang, H.-P.; Jiang, H.-D.; Bruce, I.C.; Xia, Q. Inhibition of superoxide anion-mediated impairment of endothelium by treatment with luteolin and apigenin in rat mesenteric artery. Life Sci. 2008, 83, 110–117. [Google Scholar] [CrossRef]
- Cooke, C. Endothelial-dependent vasodilation is reduced in mesenteric arteries from superoxide dismutase knockout mice. Cardiovasc. Res. 2003, 60, 635–642. [Google Scholar] [CrossRef][Green Version]
- Ali, S.F.; Woodman, O.L. Tocomin Restores Endothelium-Dependent Relaxation in the Diabetic Rat Aorta by Increasing NO Bioavailability and Improving the Expression of eNOS. Front. Physiol. 2019, 10, 10. [Google Scholar] [CrossRef]
- Onetti, Y.; Dantas, A.P.; Pérez, B.; McNeish, A.J.; Vila, E.; Jiménez-Altayó, F. Peroxynitrite formed during a transient episode of brain ischaemia increases endothelium-derived hyperpolarization-type dilations in thromboxane/prostaglandin receptor-stimulated rat cerebral arteries. Acta Physiol. 2017, 220, 150–166. [Google Scholar] [CrossRef]
- Chidgey, J.; Fraser, P.A.; Aaronson, P.I. Reactive oxygen species facilitate the EDH response in arterioles by potentiating intracellular endothelial Ca2+ release. Free Radic. Biol. Med. 2016, 97, 274–284. [Google Scholar] [CrossRef]
- Ellinsworth, D.C.; Sandow, S.L.; Shukla, N.; Liu, Y.; Jeremy, J.Y.; Gutterman, D.D. Endothelium-Derived Hyperpolarization and Coronary Vasodilation: Diverse and Integrated Roles of Epoxyeicosatrienoic Acids, Hydrogen Peroxide, and Gap Junctions. Microcirculation 2016, 23, 15–32. [Google Scholar] [CrossRef]
- Gutterman, D.D.; Miura, H.; Liu, Y. Redox modulation of vascular tone: Focus of potassium channel mechanisms of dilation. Arter. Thromb Vasc. Biol. 2005. [Google Scholar] [CrossRef]
- Sena, C.M.; Carrilho, F.; Seiça, R.M. Endothelial Dysfunction in Type 2 Diabetes: Targeting Inflammation. In Endothelial Dysfunction—Old Concepts and New Challenges; IntechOpen: London, UK, 2018. [Google Scholar]
- Kessler, P.; Popp, R.; Busse, R.; Schini-Kerth, V.B. Proinflammatory Mediators Chronically Downregulate the Formation of the Endothelium-Derived Hyperpolarizing Factor in Arteries Via a Nitric Oxide/Cyclic GMP–Dependent Mechanism. Circulation 1999, 99, 1878–1884. [Google Scholar] [CrossRef]
- Gillham, J.C.; Myers, J.E.; Baker, P.N.; Taggart, M.J. TNF-α Alters Nitric Oxide- and Endothelium-Derived Hyperpolarizing Factor-Mediated Vasodilatation in Human Omental Arteries. Hypertens Pregnancy 2008, 27, 29–38. [Google Scholar] [CrossRef]
- Wimalasundera, R.; Fexby, S.; Regan, L.; Thom, S.A.M.; Hughes, A.D. Effect of tumour necrosis factor- α and interleukin 1 β on endothelium-dependent relaxation in rat mesenteric resistance arteries in vitro. Br. J. Pharmacol. 2003, 138, 1285–1294. [Google Scholar] [CrossRef]
- Cotter, M.A.; Gibson, T.M.; Nangle, M.R.; Cameron, N.E. Effects of interleukin-6 treatment on neurovascular function, nerve perfusion and vascular endothelium in diabetic rats. Diabetes Obes. Metab. 2010, 12, 689–699. [Google Scholar] [CrossRef]
- Pedersen, B.K. Anti-inflammatory effects of exercise: Role in diabetes and cardiovascular disease. Eur. J. Clin. Investig. 2017, 47, 600–611. [Google Scholar] [CrossRef]
- Huang, H.; Weng, J.; Wang, M.-H. EETs/sEH in diabetes and obesity-induced cardiovascular diseases. Prostaglandins Other Lipid Mediat. 2016, 125, 80–89. [Google Scholar] [CrossRef]
- Zhang, L.-N.; Vincelette, J.; Chen, D.; Gless, R.D.; Anandan, S.-K.; Rubanyi, G.M.; Webb, H.K.; MacIntyre, D.E.; Wang, Y.-X. (Jim) Inhibition of soluble epoxide hydrolase attenuates endothelial dysfunction in animal models of diabetes, obesity and hypertension. Eur. J. Pharm. 2011, 654, 68–74. [Google Scholar] [CrossRef]
- Roche, C.; Besnier, M.; Cassel, R.; Harouki, N.; Coquerel, D.; Guerrot, D.; Nicol, L.; Loizon, E.; Remy-Jouet, I.; Morisseau, C.; et al. Soluble epoxide hydrolase inhibition improves coronary endothelial function and prevents the development of cardiac alterations in obese insulin-resistant mice. Am. J. Physiol. Circ. Physiol. 2015, 308, H1020–H1029. [Google Scholar] [CrossRef]
- Cheng, Z.; Shen, X.; Jiang, X.; Shan, H.; Cimini, M.; Fang, P.; Ji, Y.; Park, J.Y.; Drosatos, K.; Yang, X.; et al. Hyperhomocysteinemia potentiates diabetes-impaired EDHF-induced vascular relaxation: Role of insufficient hydrogen sulfide. Redox Boil. 2018, 16, 215–225. [Google Scholar] [CrossRef]
- Wang, X.-C.; Sun, W.-T.; Yu, C.-M.; Pun, S.-H.; Underwood, M.J.; He, G.-W.; Yang, Q. ER stress mediates homocysteine-induced endothelial dysfunction: Modulation of IKCa and SKCa channels. Atherosclerosis 2015, 242, 191–198. [Google Scholar] [CrossRef]
- Emoto, M.; Kanda, H.; Shoji, T.; Kawagishi, T.; Komatsu, M.; Mori, K.; Tahara, H.; Ishimura, E.; Inaba, M.; Okuno, Y.; et al. Impact of Insulin Resistance and Nephropathy on Homocysteine in Type 2 Diabetes. Diabetes Care 2001, 24, 533–538. [Google Scholar] [CrossRef]
- Centeno, J.M.; López-Morales, M.A.; Aliena-Valero, A.; Jover-Mengual, T.; Burguete, M.C.; Castelló-Ruiz, M.; Miranda, F.J. Potassium channels contribute to the increased sensitivity of the rabbit carotid artery to hydrogen sulfide in diabetes. Eur. J. Pharm. 2019, 853, 33–40. [Google Scholar] [CrossRef]
- Bełtowski, J.; Wojcicka, G.; Jamroz-Wiśniewska, A. Hydrogen sulfide in the regulation of insulin secretion and insulin sensitivity: Implications for the pathogenesis and treatment of diabetes mellitus. Biochem. Pharm. 2018, 149, 60–76. [Google Scholar] [CrossRef]
- Liu, M.; Li, Y.; Liang, B.; Li, Z.; Jiang, Z.; Chu, C.; Yang, J. Hydrogen sulfide attenuates myocardial fibrosis in diabetic rats through the JAK/STAT signaling pathway. Int. J. Mol. Med. 2018, 41, 1867–1876. [Google Scholar] [CrossRef]
- Lembo, G.; Vecchione, C.; Fratta, L.; Marino, G.; Trimarco, V.; D’Amati, G.; Trimarco, B. Leptin induces direct vasodilation through distinct endothelial mechanisms. Diabetes 2000, 49, 293–297. [Google Scholar] [CrossRef]
- Jamroz-Wiśniewska, A.; Gertler, A.; Solomon, G.; Wood, M.E.; Whiteman, M.; Bełtowski, J. Leptin-Induced Endothelium-Dependent Vasorelaxation of Peripheral Arteries in Lean and Obese Rats: Role of Nitric Oxide and Hydrogen Sulfide. PLoS ONE 2014, 9, e86744. [Google Scholar] [CrossRef]
- Bełtowski, J.; Jamroz-Wiśniewska, A. Hydrogen Sulfide in the Adipose Tissue—Physiology, Pathology and a Target for Pharmacotherapy. Molecules 2016, 22, 63. [Google Scholar] [CrossRef]
- Bełtowski, J.; Wójcicka, G.; Jamroz-Wiśniewska, A. Role of nitric oxide and endothelium-derived hyperpolarizing factor (EDHF) in the regulation of blood pressure by leptin in lean and obese rats. Life Sci. 2006, 79, 63–71. [Google Scholar] [CrossRef]
- Godo, S.; Shimokawa, H. Divergent roles of endothelial nitric oxide synthases system in maintaining cardiovascular homeostasis. Free Radic. Biol. Med. 2017, 109, 4–10. [Google Scholar] [CrossRef]
- Dellostritto, D.J.; Connell, P.J.; Dick, G.M.; Fancher, I.S.; Klarich, B.; Fahmy, J.N.; Kang, P.T.; Chen, Y.-R.; Damron, D.S.; Thodeti, C.K.; et al. Differential regulation of TRPV1 channels by H2O2: Implications for diabetic microvascular dysfunction. Basic Res. Cardiol. 2016, 111, 21. [Google Scholar] [CrossRef]
- Muangman, P.; Spenny, M.L.; Tamura, R.N.; Gibran, N.S. Fatty Acids and Glucose Increase Neutral Endopeptidase Activity in Human Microvascular Endothelial Cells. Shock 2003, 19, 508–512. [Google Scholar] [CrossRef]
- Davidson, E.P.; Kleinschmidt, T.L.; Oltman, C.L.; Lund, D.D.; Yorek, M.A. Treatment of Streptozotocin-Induced Diabetic Rats With AVE7688, a Vasopeptidase Inhibitor: Effect on Vascular and Neural Disease. Diabetes 2007, 56, 355–362. [Google Scholar] [CrossRef][Green Version]
- Seki, T.; Goto, K.; Kansui, Y.; Ohtsubo, T.; Matsumura, K.; Kitazono, T. Angiotensin II Receptor–Neprilysin Inhibitor Sacubitril/Valsartan Improves Endothelial Dysfunction in Spontaneously Hypertensive Rats. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef]
- Lu, T.; Sun, X.; Li, Y.; Chai, Q.; Wang, X.-L.; Lee, H.-C. Role of Nrf2 Signaling in the Regulation of Vascular BK Channel β1 Subunit Expression and BK Channel Function in High-Fat Diet–Induced Diabetic Mice. Diabetes 2017, 66, 2681–2690. [Google Scholar] [CrossRef]
- Sun, X.; Chai, Q.; Shen, W.-K.; Li, Y.; Wang, X.-L.; Li, J.; Thompson, B.; Lu, T.; Lee, H.-C. Regulation of vascular large-conductance calcium-activated potassium channels by Nrf2 signalling. Diabetes Vasc. Dis. Res. 2017, 14, 353–362. [Google Scholar]
- Rumble, J.R.; Cooper, M.E.; Soulis, T.; Cox, A.; Wu, L.; Youssef, S.; Jasik, M.; Jerums, G.; Gilbert, R.E. Vascular hypertrophy in experimental diabetes. Role of advanced glycation end products. J. Clin. Investig. 1997, 99, 1016–1027. [Google Scholar] [CrossRef]
- Haddock, R.E.; Hill, C.E. Sympathetic overdrive in obesity involves purinergic hyperactivity in the resistance vasculature. J. Physiol. 2011, 589, 3289–3307. [Google Scholar] [CrossRef]
- Zimmermann, P.A.; Knot, H.J.; Stevenson, A.S.; Nelson, M.T. Increased Myogenic Tone and Diminished Responsiveness to ATP-Sensitive K+ Channel Openers in Cerebral Arteries From Diabetic Rats. Circ. Res. 1997, 81, 996–1004. [Google Scholar] [CrossRef]
- Pereira, C.A.; Carneiro, F.S.; Matsumoto, T.; Tostes, R.C. Bonus Effects of Antidiabetic Drugs: Possible Beneficial Effects on Endothelial Dysfunction, Vascular Inflammation and Atherosclerosis. Basic. Clin. Pharm. Toxicol. 2018, 123, 523–538. [Google Scholar] [CrossRef]
- Mayhan, W.G.; Trauernicht, A.K.; Irvine, S.D. Insulin reverses impaired acetylcholine-induced dilatation of the rat basilar artery during diabetes mellitus. Brain Res. 2001, 893, 195–201. [Google Scholar] [CrossRef]
- Misurski, D.A.; Wu, S.-Q.; McNeill, J.R.; Wilson, T.W.; Gopalakrishnan, V. Insulin-Induced Biphasic Responses in Rat Mesenteric Vascular Bed. Hypertension 2001, 37, 1298–1302. [Google Scholar] [CrossRef]
- Iida, S.; Taguchi, H.; Watanabe, N.; Kushiro, T.; Kanmatsuse, K. Insulin-induced relaxation of rat mesenteric artery is mediated by Ca2+-activated K+ channels. Eur. J. Pharmacol. 2001, 411, 155–160. [Google Scholar] [CrossRef]
- Imaeda, K.; Okayama, N.; Okouchi, M.; Omi, H.; Kato, T.; Akao, M.; Imai, S.; Uranishi, H.; Takeuchi, Y.; Ohara, H.; et al. Effects of insulin on the acetylcholine-induced hyperpolarization in the guinea pig mesenteric arterioles. J. Diabetes its Complicat. 2004, 18, 356–362. [Google Scholar] [CrossRef]
- Duflot, T.; Moreau-Grangé, L.; Roche, C.; Iacob, M.; Wils, J.; Rémy-Jouet, I.; Cailleux, A.-F.; Leuillier, M.; Renet, S.; Li, D.; et al. Altered bioavailability of epoxyeicosatrienoic acids is associated with conduit artery endothelial dysfunction in type 2 diabetic patients. Cardiovasc. Diabetol. 2019, 18, 35. [Google Scholar] [CrossRef]
- Kimura, M.; Jefferis, A.-M.; Watanabe, H.; Chin-Dusting, J. Insulin Inhibits Acetylcholine Responses in Rat Isolated Mesenteric Arteries via a Non-Nitric Oxide Nonprostanoid Pathway. Hypertension 2002, 39, 35–40. [Google Scholar] [CrossRef]
- Katakam, P.V.G.; Ujhelyi, M.R.; Miller, A.W. EDHF-Mediated Relaxation is Impaired in Fructose-Fed Rats. J. Cardiovasc. Pharm. 1999, 34, 461–467. [Google Scholar] [CrossRef]
- Triggle, C.R.; Ding, H. Metformin is not just an antihyperglycaemic drug but also has protective effects on the vascular endothelium. Acta Physiol. 2017, 219, 138–151. [Google Scholar] [CrossRef]
- Matsumoto, T.; Noguchi, E.; Ishida, K.; Kobayashi, T.; Yamada, N.; Kamata, K. Metformin normalizes endothelial function by suppressing vasoconstrictor prostanoids in mesenteric arteries from OLETF rats, a model of type 2 diabetes. Am. J. Physiol. Circ. Physiol. 2008, 295, H1165–H1176. [Google Scholar] [CrossRef]
- Hamidi Shishavan, M.; Henning, R.H.; van Buiten, A.; Goris, M.; Deelman, L.E.; Buikema, H. Metformin Improves Endothelial Function and Reduces Blood Pressure in Diabetic Spontaneously Hypertensive Rats Independent from Glycemia Control: Comparison to Vildagliptin. Sci. Rep. 2017, 7, 10975. [Google Scholar] [CrossRef]
- Zhao, L.-M.; Wang, Y.; Yang, Y.; Guo, R.; Wang, N.-P.; Deng, X.-L. Metformin Restores Intermediate-Conductance Calcium-Activated K+ channel- and Small-Conductance Calcium-Activated K+ channel-Mediated Vasodilatation Impaired by Advanced Glycation End Products in Rat Mesenteric Artery. Mol. Pharm. 2014, 86, 580–591. [Google Scholar] [CrossRef]
- Chen, H.; Vanhoutte, P.M.; Leung, S.W.S. Acute activation of endothelial AMPK surprisingly inhibits EDH-like relaxations in rat mesenteric arteries. Br. J. Pharm. 2019, 14716. [Google Scholar] [CrossRef]
- Avogaro, A.; Fadini, G.P. The Effects of Dipeptidyl Peptidase-4 Inhibition on Microvascular Diabetes Complications. Diabetes Care. 2014, 37, 2884–2894. [Google Scholar] [CrossRef]
- Almutairi, M.; Al Batran, R.; Ussher, J.R. Glucagon-like peptide-1 receptor action in the vasculature. Peptides 2019, 111, 26–32. [Google Scholar] [CrossRef]
- Shah, Z.; Pineda, C.; Kampfrath, T.; Maiseyeu, A.; Ying, Z.; Racoma, I.; Deiuliis, J.; Xu, X.; Sun, Q.; Moffatt-Bruce, S.; et al. Acute DPP-4 inhibition modulates vascular tone through GLP-1 independent pathways. Vasc. Pharm. 2011, 55, 2–9. [Google Scholar] [CrossRef]
- Salheen, S.M.; Panchapakesan, U.; Pollock, C.A.; Woodman, O.L. The DPP-4 inhibitor linagliptin and the GLP-1 receptor agonist exendin-4 improve endothelium-dependent relaxation of rat mesenteric arteries in the presence of high glucose. Pharm. Res. 2015, 94, 26–33. [Google Scholar] [CrossRef]
- Salheen, S.M.; Panchapakesan, U.; Pollock, C.A.; Woodman, O.L. The Dipeptidyl Peptidase-4 Inhibitor Linagliptin Preserves Endothelial Function in Mesenteric Arteries from Type 1 Diabetic Rats without Decreasing Plasma Glucose. PLoS ONE 2015, 10, e0143941. [Google Scholar] [CrossRef]
- Bayram, Z.; Nacitarhan, C.; Ozdem, S.S. Effects of Glucagon-like Peptide-1 in Diabetic Rat Small Resistance Arteries. J. Cardiovasc. Pharm. 2014, 64, 277–284. [Google Scholar] [CrossRef]
- Sukumaran, V.; Tsuchimochi, H.; Sonobe, T.; Shirai, M.; Pearson, J.T. Liraglutide Improves Renal Endothelial Function in Obese Zucker Rats on a High-Salt Diet. J. Pharm. Exp. Ther. 2019, 369, 375–388. [Google Scholar] [CrossRef]
- Verma, S.; McMurray, J.J.V. SGLT2 inhibitors and mechanisms of cardiovascular benefit: A state-of-the-art review. Diabetologia 2018, 61, 2108–2117. [Google Scholar] [CrossRef]
- Sayour, A.A.; Korkmaz-Icöz, S.; Loganathan, S.; Ruppert, M.; Sayour, V.N.; Oláh, A.; Benke, K.; Brune, M.; Benkő, R.; Horváth, E.M.; et al. Acute canagliflozin treatment protects against in vivo myocardial ischemia–reperfusion injury in non-diabetic male rats and enhances endothelium-dependent vasorelaxation. J. Transl. Med. 2019, 17, 127. [Google Scholar] [CrossRef]
- Hanf, A.; Steven, S.; Oelze, M.; Kroeller-Schoen, S.; Kashani, F.; Roohani, S.; Welschof, P.; Kopp, M.; Goedtel-Armbrust, U.; Xia, N.; et al. The SGLT2 Inhibitor Empagliflozin Improves the Primary Diabetic Complications in ZDF Rats. Free. Radic. Boil. Med. 2017, 112, 112–113. [Google Scholar] [CrossRef][Green Version]
- Cooper, S.; Teoh, H.; Campeau, M.A.; Verma, S.; Leask, R.L. Empagliflozin restores the integrity of the endothelial glycocalyx in vitro. Mol. Cell Biochem. 2019, 1–10. [Google Scholar] [CrossRef]
- Ola, M.S.; Alhomida, A.S.; Ferrario, C.M.; Ahmad, S. Role of Tissue Renin-angiotensin System and the Chymase/angiotensin-( 1-12) Axis in the Pathogenesis of Diabetic Retinopathy. Curr. Med. Chem. 2017, 24. [Google Scholar] [CrossRef]
- Matsumoto, T.; Ishida, K.; Taguchi, K.; Kobayashi, T.; Kamata, K. Losartan Normalizes Endothelium-Derived Hyperpolarizing Factor–Mediated Relaxation by Activating Ca2+-Activated K+ Channels in Mesenteric Artery From Type 2 Diabetic GK Rat. J. Pharm. Sci. 2010, 112, 299–309. [Google Scholar] [CrossRef]
- Hosoya, M.; Ohashi, J.; Sawada, A.; Takaki, A.; Shimokawa, H. Combination Therapy With Olmesartan and Azelnidipine Improves EDHF-Mediated Responses in Diabetic Apolipoprotein E-Deficient Mice. Circ. J. 2010, 74, 798–806. [Google Scholar] [CrossRef]
- Goto, K.; Fujii, K.; Onaka, U.; Abe, I.; Fujishima, M. Renin-Angiotensin System Blockade Improves Endothelial Dysfunction in Hypertension. Hypertension 2000, 36, 575–580. [Google Scholar] [CrossRef]
- Kansui, Y.; Fujii, K.; Goto, K.; Abe, I.; Iida, M. Angiotensin II receptor antagonist improves age-related endothelial dysfunction. J. Hypertens. 2002, 20, 439–446. [Google Scholar] [CrossRef]
- van Thiel, B.S.; van der Pluijm, I.; te Riet, L.; Essers, J.; Danser, A.H.J. The renin–angiotensin system and its involvement in vascular disease. Eur. J. Pharm. 2015, 763, 3–14. [Google Scholar] [CrossRef]
- Oesterle, A.; Laufs, U.; Liao, J.K. Pleiotropic Effects of Statins on the Cardiovascular System. Circ. Res. 2017, 120, 229–243. [Google Scholar] [CrossRef]
- Cameron, N.; Cotter, M.; Inkster, M.; Nangle, M. Looking to the future: Diabetic neuropathy and effects of rosuvastatin on neurovascular function in diabetes models. Diabetes Res. Clin. Pract. 2003, 61, S35–S39. [Google Scholar] [CrossRef]
- Kansui, Y.; Fujii, K.; Goto, K.; Abe, I.; Iida, M. Effects of fluvastatin on endothelium-derived hyperpolarizing factor- and nitric oxide-mediated relaxations in arteries of hypertensive rats. Clin. Exp. Pharm. Physiol. 2004, 31, 354–359. [Google Scholar] [CrossRef]
- Kizub, I.V.; Klymenko, K.I.; Soloviev, A.I. Protein kinase C in enhanced vascular tone in diabetes mellitus. Int. J. Cardiol. 2014, 174, 230–242. [Google Scholar] [CrossRef]
- Cotter, M.; Jack, A.; Cameron, N. Effects of the Protein Kinase C Beta Inhibitor LY333531 on Neural and Vascular Function in Rats with Streptozotocin-induced Diabetes. J. Peripher. Nerv. Syst. 2003, 8, 128–133. [Google Scholar] [CrossRef]
- Matsumoto, T.; Takaoka, E.; Ishida, K.; Nakayama, N.; Noguchi, E.; Kobayashi, T.; Kamata, K. Abnormalities of endothelium-dependent responses in mesenteric arteries from Otsuka Long-Evans Tokushima Fatty (OLETF) rats are improved by chronic treatment with thromboxane A2 synthase inhibitor. Atheroscler 2009, 205, 87–95. [Google Scholar] [CrossRef]
- Fujii, K.; Onaka, U.; Ohya, Y.; Ohmori, S.; Tominaga, M.; Abe, I.; Takata, Y.; Fujishima, M. Role of eicosanoids in alteration of membrane electrical properties in isolated mesenteric arteries of salt-loaded, Dahl salt-sensitive rats. Br. J. Pharm. 1997, 120, 1207–1214. [Google Scholar] [CrossRef][Green Version]
- Goto, K.; Edwards, F.R.; Hill, C.E. Depolarization evoked by acetylcholine in mesenteric arteries of hypertensive rats attenuates endothelium-dependent hyperpolarizing factor. J. Hypertens. 2007, 25, 345–359. [Google Scholar] [CrossRef]
- Maccari, R.; Ottanà, R. Targeting Aldose Reductase for the Treatment of Diabetes Complications and Inflammatory Diseases: New Insights and Future Directions. J. Med. Chem. 2015, 58, 2047–2067. [Google Scholar] [CrossRef]
- Keegan, A.; Jack, A.M.; Cotter, M.A.; Cameron, N.E. Effects of Aldose Reductase Inhibition on Responses of the Corpus Cavernosum and Mesenteric Vascular Bed of Diabetic Rats. J. Cardiovasc. Pharm. 2000, 35, 606–613. [Google Scholar] [CrossRef]
- Akamine, E.H. Minalrestat, an Aldose Reductase Inhibitor, Corrects the Impaired Microvascular Reactivity in Diabetes. J. Pharm. Exp. Ther. 2002, 304, 1236–1242. [Google Scholar] [CrossRef]
- Delbin, M.A.; Trask, A.J. The diabetic vasculature: Physiological mechanisms of dysfunction and influence of aerobic exercise training in animal models. Life Sci. 2014, 102, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Minami, A.; Ishimura, N.; Harada, N.; Sakamoto, S.; Niwa, Y.; Nakaya, Y. Exercise training improves acetylcholine-induced endothelium-dependent hyperpolarization in type 2 diabetic rats, Otsuka Long-Evans Tokushima fatty rats. Atherosclerosis 2002, 162, 85–92. [Google Scholar] [CrossRef]
- Kazemi, F. Myostatin alters with exercise training in diabetic rats; possible interaction with glycosylated hemoglobin and inflammatory cytokines. Cytokine 2019, 120, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Bellien, J.; Thuillez, C.; Joannides, R. Contribution of endothelium-derived hyperpolarizing factors to the regulation of vascular tone in humans. Fundam. Clin. Pharmacol. 2008, 22, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Hearon, C.M.; Dinenno, F.A. Escape, lysis, and feedback: Endothelial modulation of sympathetic vasoconstriction. Curr. Opin. Pharm. 2019, 45, 81–86. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, A.; Cooper, E.J.; Dowell, F.J. Differential effects of glucose on agonist-induced relaxations in human mesenteric and subcutaneous arteries. Br. J. Pharm. 2008, 153, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Angulo, J.; Cuevas, P.; Fernández, A.; Gabancho, S.; Allona, A.; Martín-Morales, A.; Moncada, I.; Videla, S.; De Tejada, I.S. Diabetes impairs endothelium-dependent relaxation of human penile vascular tissues mediated by NO and EDHF. Biochem. Biophys. Res. Commun. 2003, 312, 1202–1208. [Google Scholar] [CrossRef]
- Liu, Y.; Xie, A.; Singh, A.K.; Ehsan, A.; Choudhary, G.; Dudley, S.; Sellke, F.W.; Feng, J. Inactivation of Endothelial Small/Intermediate Conductance of Calcium-Activated Potassium Channels Contributes to Coronary Arteriolar Dysfunction in Diabetic Patients. J. Am. Hear. Assoc. 2015, 4, e002062. [Google Scholar] [CrossRef]
- Liu, Y.; Cole, V.; Lawandy, I.; Ehsan, A.; Sellke, F.W.; Feng, J. Decreased coronary arteriolar response to KCa channel opener after cardioplegic arrest in diabetic patients. Mol. Cell. Biochem. 2018, 445, 187–194. [Google Scholar] [CrossRef]
- Marche, P.; Dubois, S.; Abraham, P.; Parot-Schinkel, E.; Gascoin, L.; Humeau-Heurtier, A.; Ducluzeau, P.; Mahe, G. Neurovascular microcirculatory vasodilation mediated by C-fibers and Transient receptor potential vanilloid-type-1 channels (TRPV 1) is impaired in type 1 diabetes. Sci. Rep. 2017, 7, 44322. [Google Scholar] [CrossRef]
- Mokhtar, S.S.; Vanhoutte, P.M.; Leung, S.W.S.; Yusof, M.I.; Sulaiman, W.A.W.; Saad, A.Z.M.; Suppian, R.; Rasool, A.H.G. Endothelium dependent hyperpolarization-type relaxation compensates for attenuated nitric oxide-mediated responses in subcutaneous arteries of diabetic patients. Nitric Oxide 2016, 53, 35–44. [Google Scholar] [CrossRef]


{kind=link}
| Species | Model | Duration of DM | Glucose (mmol/L) | Vascular Bed | Function EDH | Function KCa | Expression SKCa IKCa | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| Rat | STZ | 8 w | 31 | mesenteric | ↓ | ↓ 1-EBIO | ND | ND | [20] |
| Rat | STZ | 10 w | >33 | mesenteric | ↓ | ND | ↑ | ↑ | [21] |
| Rat | STZ | 4 w | 24 | mesenteric | ↓ | ↓ NS309 | ND | ND | [22] |
| Rat | STZ | 12−15 w | >15 | mesenteric | ↓ | ND | ↓ | ND | [23] |
| Rat | STZ | 12 w | 21 | mesenteric | ↑ | ND | ND | ND | [49] |
| Rat | STZ | 18 day | 21 | uteroplacental | ND | ↓ NS309 | → | → | [81] |
| Rat | STZ | 8 w | 22 | corpus cavernosum | ↓ | ND | ↓ | ↓ | [31,82] |
| Mice | STZ+ApoE−/− | 10 w | 32 | mesenteric | ↓ | ↓ NS1619 | ND | ND | [24] |
| Mice | STZ+ApoE−/− | 12–16 w | >20 | mesenteric | ↓ | ND | ↓ | → | [25] |
| Mice | STZ | 10 w | 44 | mesenteric | ↓ | ND | → | ↑ | [26] |
| Species | Model | Duration of DM | Glucose (mmol/L) | Vascular Bed | Function EDH | Function KCa | Expression SKCa IKCa | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| Rat | ZDF | 17–20 w | 38 | mesenteric | ↓ | → 1-EBIO | ↑ | → | [34] |
| Rat | ZDF | 21 w | 24 | mesenteric | ↓ | ↓ NS309 | → | ND | [37] |
| Rat | ZDF | 18 w | 21 | mesenteric | ↓ | ↓ 1-EBIO | → | ↑ | [39] |
| Rat | ZDF | 12–14 w | ND | mesenteric | ND | → 1-EBIO | ND | ↓ | [84] |
| Rat | OZ | 20 w | 32 | renal | ↓ | ↓ NS1619 | ND | ND | [43] |
| Rat | OZ | 7–10 w | 8.4 | cerebral | ↓ | → NS309 | ND | → | [44] |
| Rat | OZ | 17–18 w | 9.1 | coronary | ND | ↑ NS309 | ↑ | ↑ | [50] |
| Rat | OLETF | 60 w | 19 | mesenteric | ↓ | ↓ 1-EBIO | ND | ND | [38] |
| Rat | OLETF | 50–53 w | 8.4 | mesenteric | ↓ | ↓ NS309 | ND | ND | [40] |
| Rat | Diet | 16–20 w | 9.8 | saphenous | ND | → 1-EBIO | → | ↑ | [83] |
| Rat | Diet | 16–20 w | 9.7 | mesenteric | ↓ | ↑ 1-EBIO | ND | ↑ | [85] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goto, K.; Kitazono, T. Endothelium-Dependent Hyperpolarization (EDH) in Diabetes: Mechanistic Insights and Therapeutic Implications. Int. J. Mol. Sci. 2019, 20, 3737. https://doi.org/10.3390/ijms20153737
Goto K, Kitazono T. Endothelium-Dependent Hyperpolarization (EDH) in Diabetes: Mechanistic Insights and Therapeutic Implications. International Journal of Molecular Sciences. 2019; 20(15):3737. https://doi.org/10.3390/ijms20153737
Chicago/Turabian StyleGoto, Kenichi, and Takanari Kitazono. 2019. "Endothelium-Dependent Hyperpolarization (EDH) in Diabetes: Mechanistic Insights and Therapeutic Implications" International Journal of Molecular Sciences 20, no. 15: 3737. https://doi.org/10.3390/ijms20153737
APA StyleGoto, K., & Kitazono, T. (2019). Endothelium-Dependent Hyperpolarization (EDH) in Diabetes: Mechanistic Insights and Therapeutic Implications. International Journal of Molecular Sciences, 20(15), 3737. https://doi.org/10.3390/ijms20153737