Estrogen Deficiency Potentiates Thioacetamide-Induced Hepatic Fibrosis in Sprague-Dawley Rats

 ,
, 

Abstract
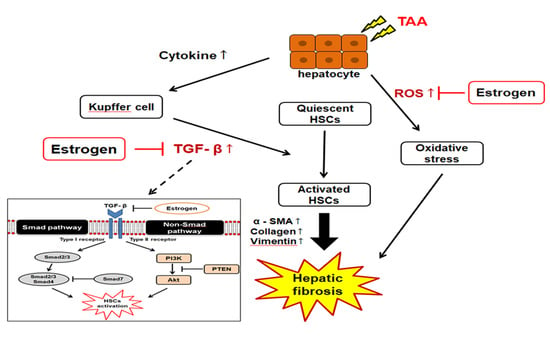
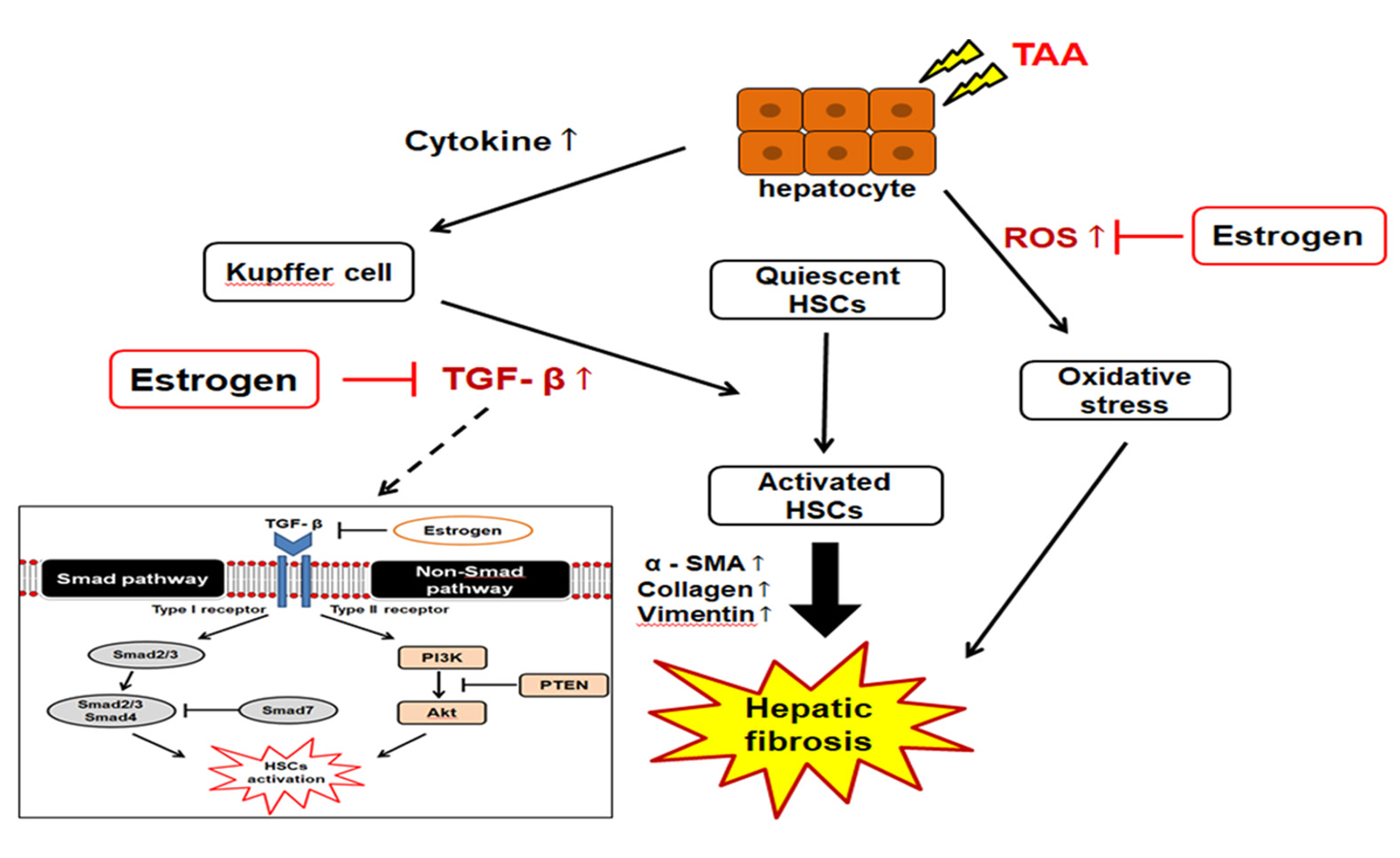
1. Introduction
2. Results
2.1. Effects of ED on TAA-Mediated Body and Liver Weight Changes
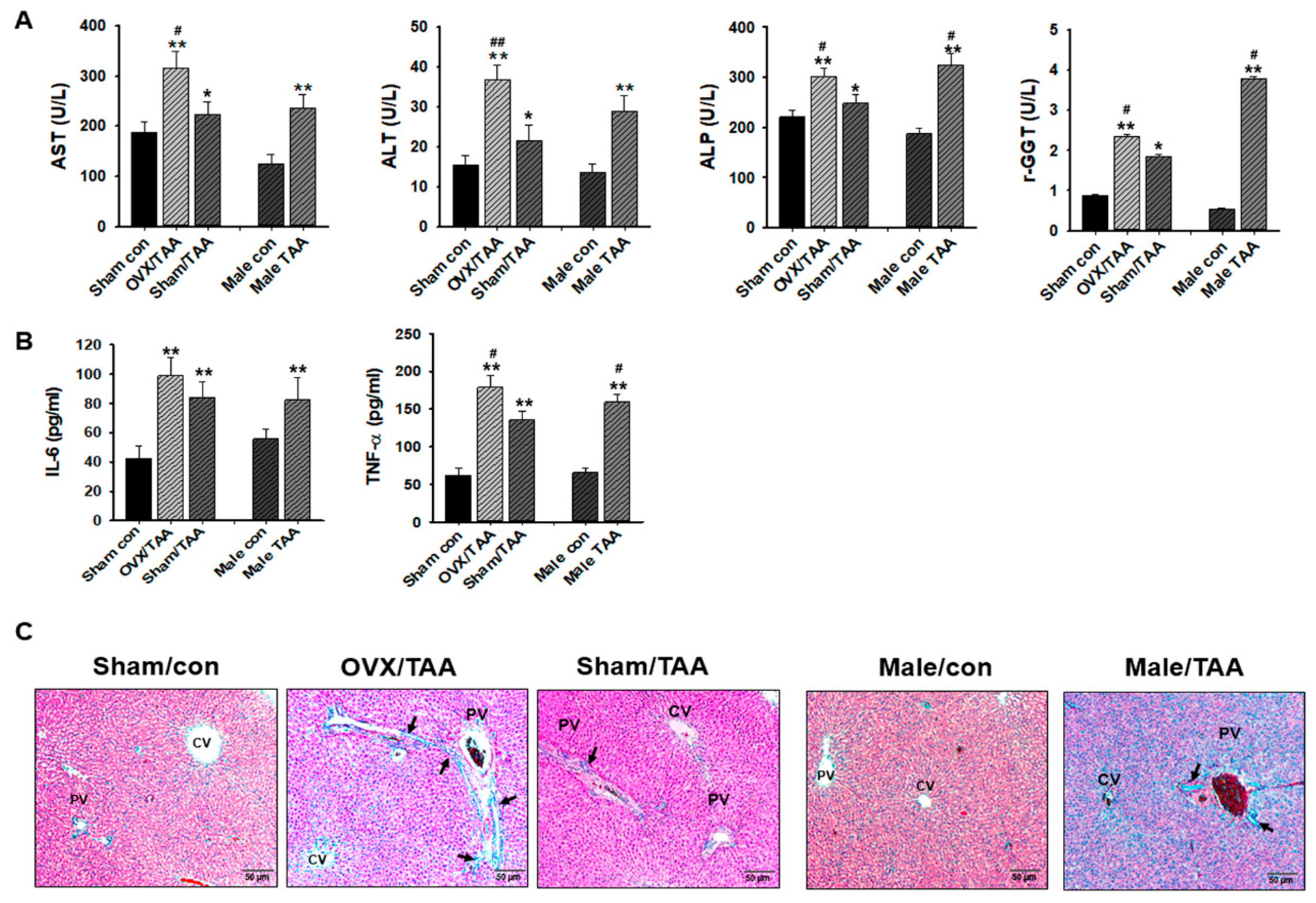
2.2. Effect of ED on TAA-Induced Liver Damage
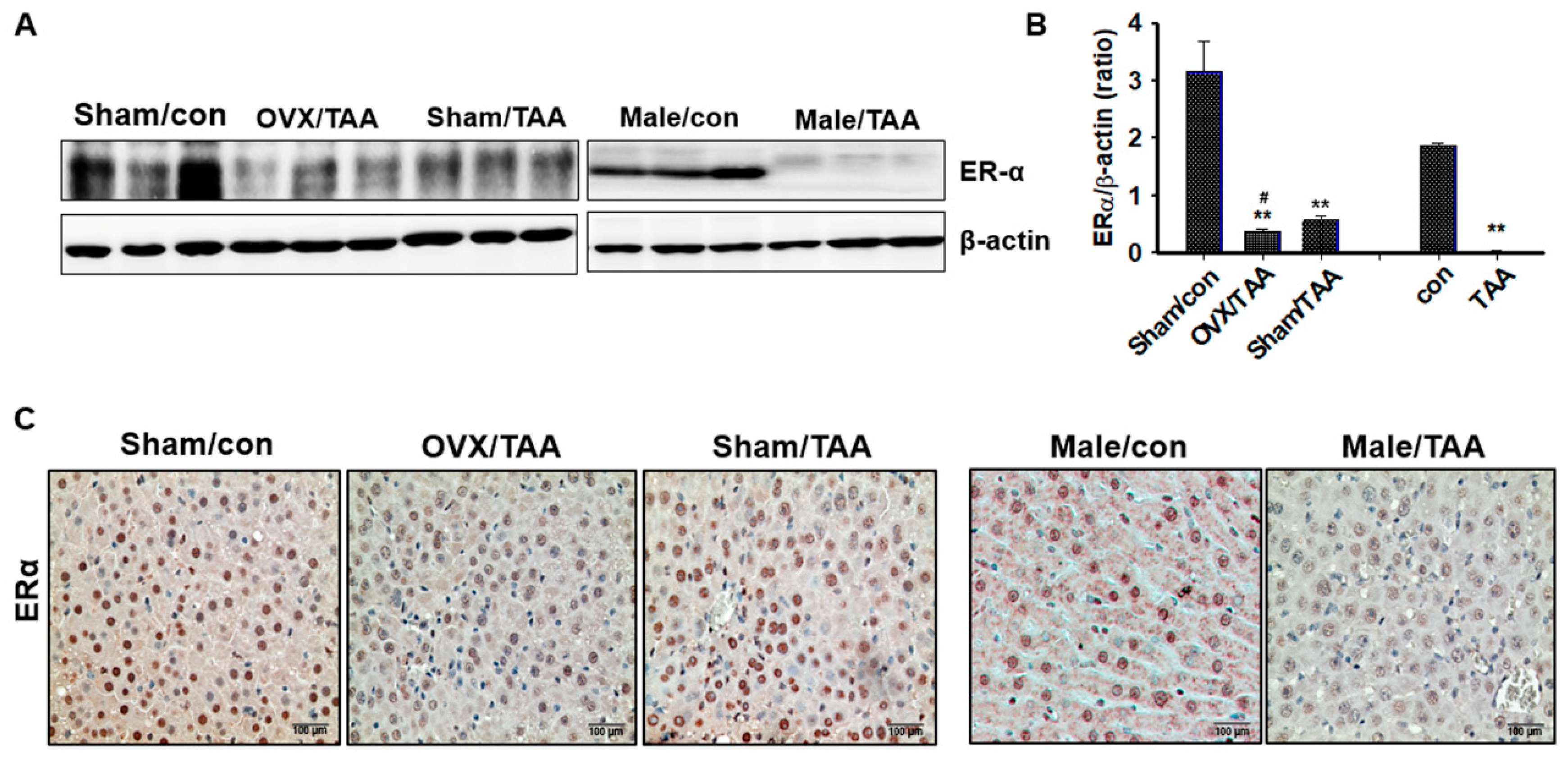
2.3. Effect of ED on ER Expression in the Livers of TAA-Treated Rats
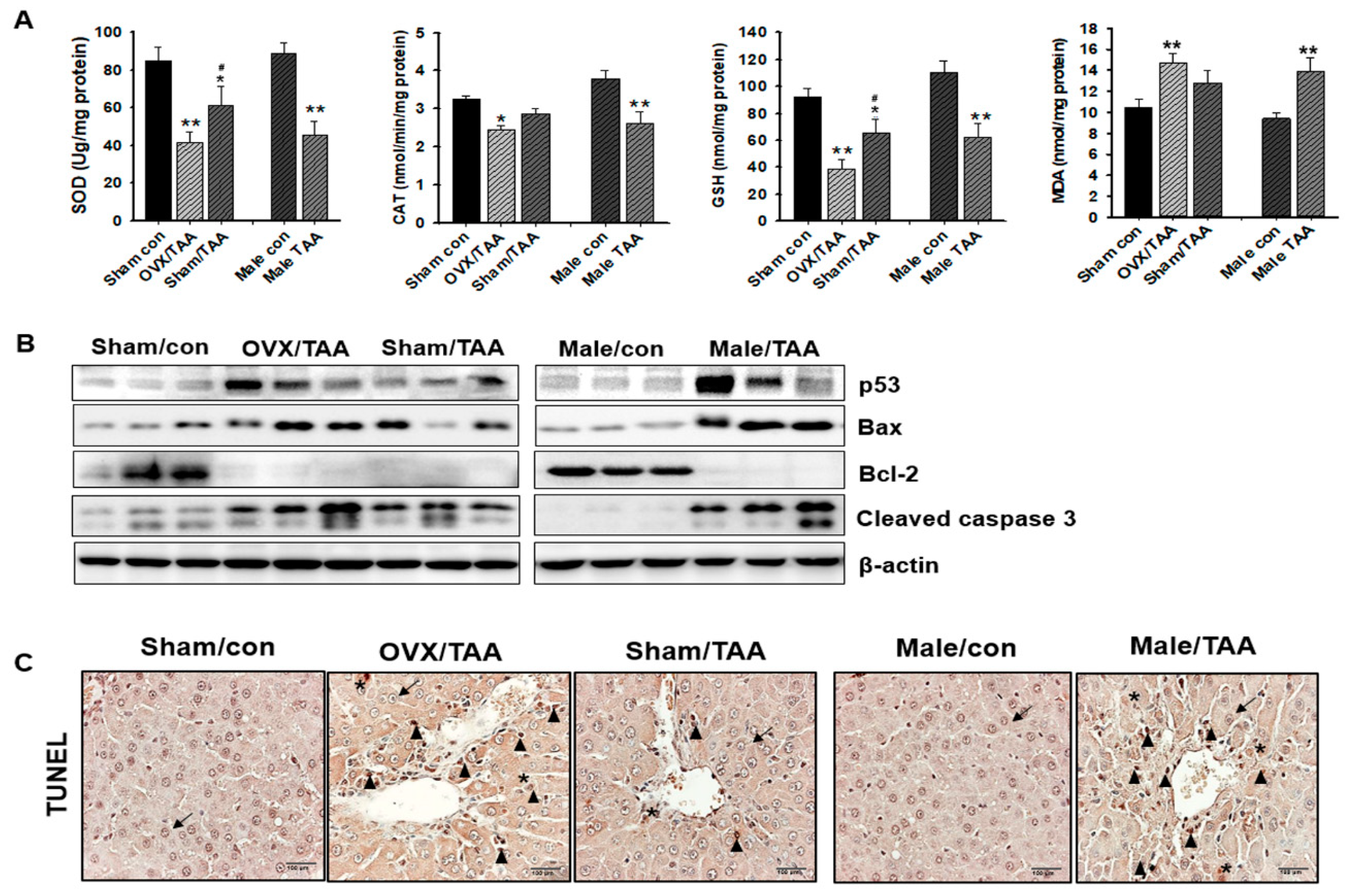
2.4. Influence of ED on Hepatic Antioxidant Enzyme Activities
2.5. Effect of ED on Hepatocyte Apoptosis
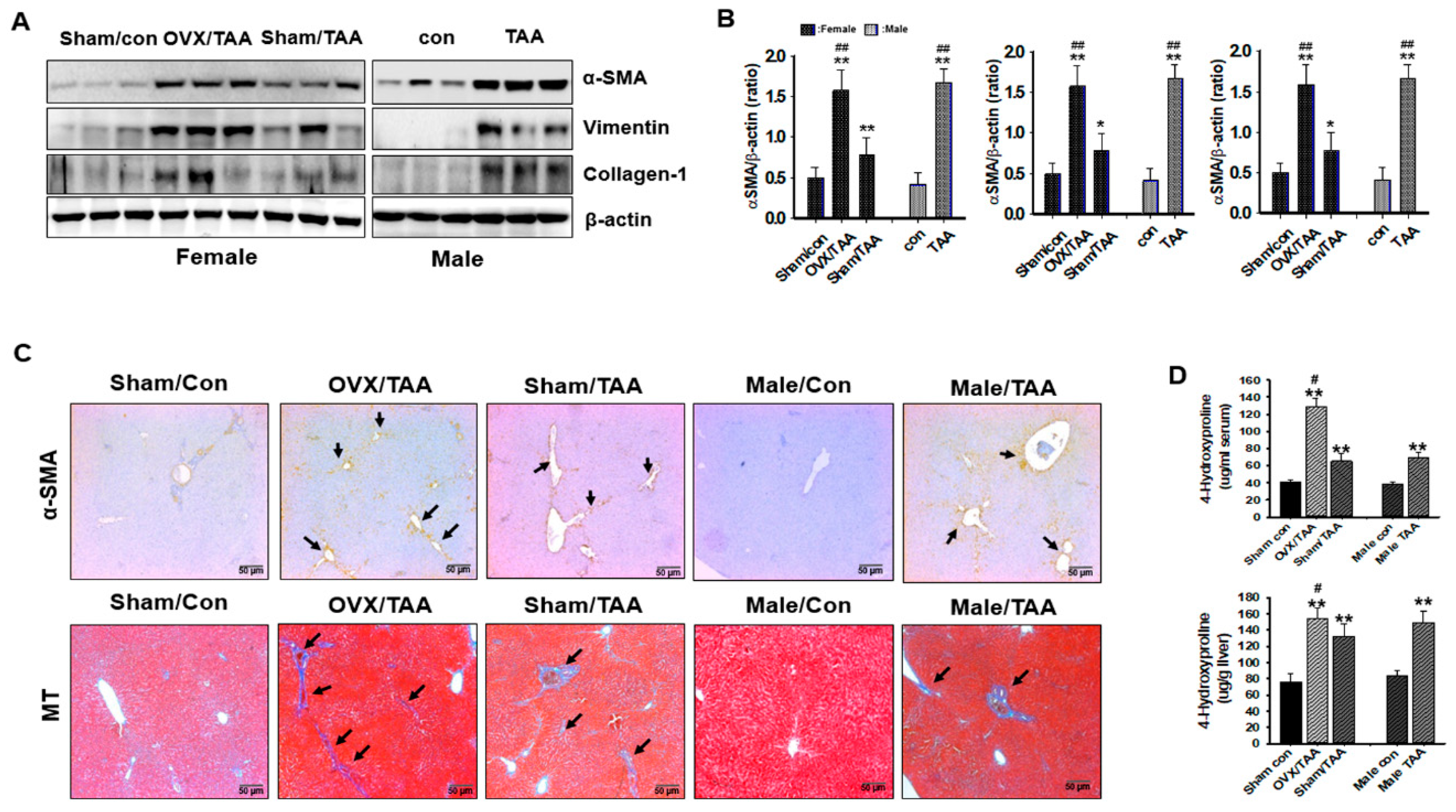
2.6. Influence of ED on Liver Fibrosis
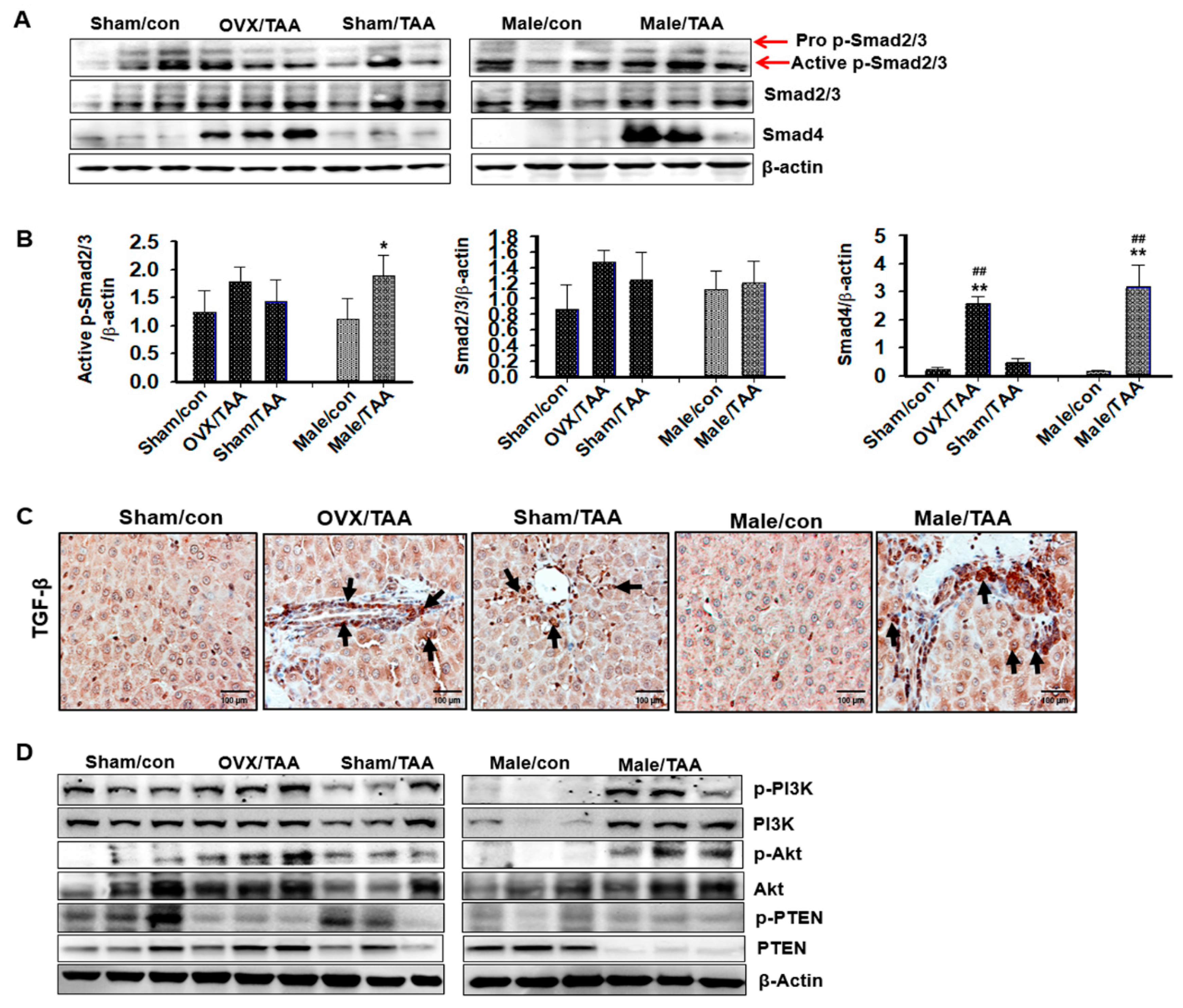
2.7. TGF-β1/Smad Signaling-Mediated Effects of ED on Liver Fibrosis
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Experimental Design
4.3. Serum Biochemical Analysis
4.4. Histopathological Examination and Masson’s Trichrome (MT) Staining
4.5. Glutathione (GSH) Content Determination
4.6. Assay for Antioxidant Enzyme Activities
4.7. Malondialdehyde Assay
4.8. Western Blotting Analysis
4.9. Hydroxyproline Assay
4.10. Immunohistochemical Examination
4.11. TUNEL Assay
4.12. Statistical Analysis
Author Contributions
Funding
Ethical Approval
Conflicts of Interest
Abbreviations
| ALT | alanine aminotransferase |
| ALP | alkaline phosphatase |
| AST | aspartate aminotransferase |
| CAT | catalase |
| ECM | extracellular matrix |
| ED | estrogen deficiency |
| EMT | epithelial- mesenchymal transition |
| OVX | ovariectomized |
| GGT | γ-glutamyl transferase |
| GSH | glutathione |
| H&E | hematoxylin and eosin |
| IL-6 | interleukin-6 |
| MDA | malondialdehyde |
| NAFLD | non-alcoholic fatty liver disease |
| NPC | non-parenchymal cells |
| α-SMA | α-smooth muscle actin |
| SOD | superoxide dismutase |
| TAA | thioacetamide |
| TGF-β | transforming growth factor-β |
| TNF-α | tumor necrosis factor-α |
| ROS | reactive oxygen species |
References
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Invest. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Brady, C.W. Liver disease in menopause. World J. Gastroenterol. 2015, 21, 7613–7620. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.; Peters, M.G. Liver disease in women: the influence of gender on epidemiology, natural history, and patient outcomes. Gastroenterol. Hepatol. 2013, 9, 633–639. [Google Scholar]
- Yang, J.D.; Abdelmalek, M.F.; Pang, H.; Guy, C.D.; Smith, A.D.; Diehl, A.M.; Suzuki, A. Gender and menopause impact severity of fibrosis among patients with nonalcoholic steatohepatitis. Hepatology 2014, 59, 1406–1414. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, M.; Shimizu, I.; Shiba, M.; Ito, S. Suppressive effects of estradiol on dimethylnitrosamine-induced fibrosis of the liver in rats. Hepatology 1999, 29, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.W.; Gong, J.; Chang, X.M.; Luo, J.Y.; Dong, L.; Jia, A.; Xu, G.P. Effects of estradiol on liver estrogen receptor-alpha and its mRNA expression in hepatic fibrosis in rats. World J. Gastroenterol. 2004, 10, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Mokdad, A.A.; Lopez, A.D.; Shahraz, S.; Lozano, R.; Mokdad, A.H.; Stanaway, J.; Murray, C.J.; Naghavi, M. Liver cirrhosis mortality in 187 countries between 1980 and 2010: a systematic analysis. BMC Med. 2014, 12, 145. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.C.; Zhang, Q.B.; Qiao, L. Pathogenesis of liver cirrhosis. World J. Gastroenterol. 2014, 20, 7312–7324. [Google Scholar] [CrossRef]
- Tsukada, S.; Parsons, C.J.; Rippe, R.A. Mechanisms of liver fibrosis. Clin. Chim. Acta 2006, 364, 33–60. [Google Scholar] [CrossRef]
- Gressner, O.A.; Weiskirchen, R.; Gressner, A.M. Evolving concepts of liver fibrogenesis provide new diagnostic and therapeutic options. Comp. Hepatol. 2007, 30, 6–7. [Google Scholar] [CrossRef]
- Schuppan, D. Liver fibrosis: Common mechanisms and antifibrotic therapies. Clin. Res. Hepatol. Gastroenterol. 2015, 39, S51–S59. [Google Scholar] [CrossRef]
- Inagaki, Y.; Okazaki, I. Emerging insights into Transforming growth factor beta Smad signal in hepatic fibrogenesis. Gut 2007, 56, 284–292. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef]
- Li, H.Y.; Zhang, Q.G.; Chen, J.W.; Chen, S.Q.; Chen, S.Y. The fibrotic role of phosphatidylinositol-3-kinase/Akt pathway in injured skeletal muscle after acute contusion. Int. J. Sports Med. 2013, 34, 789–794. [Google Scholar] [CrossRef]
- Wallace, M.C.; Hamesch, K.; Lunova, M.; Kim, Y.; Weiskirchen, R.; Strnad, P.; Friedman, S.L. Standard operating procedures in experimental liver research: thioacetamide model in mice and rats. Lab. Anim. 2015, 49 (Suppl. S1), 21–29. [Google Scholar] [CrossRef]
- Yanguas, S.C.; Cogliati, B.; Willebrords, J.; Maes, M.; Colle, I.; van den Bossche, B.; de Oliveira, C.P.M.S.; Andraus, W.; Alves, V.A.F.; Leclercq, I.; et al. Experimental models of liver fibrosis. Arch. Toxicol. 2016, 90, 1025–1048. [Google Scholar] [CrossRef]
- Amali, A.A.; Rekha, R.D.; Lin, C.J.; Wang, W.L.; Gong, H.Y.; Her, G.M.; Wu, J.L. Thioacetamide induced liver damage in zebrafish embryo as a disease model for steatohepatitis. J. Biomed. Sci. 2006, 13, 225–232. [Google Scholar] [CrossRef]
- Chilakapati, J.; Shankar, K.; Korrapati, M.C.; Hill, R.A.; Mehendale, H.M. Saturation toxicokinetics of thioacetamide: role in initiation of liver injury. Drug Metab. Dispos. 2005, 33, 1877–1885. [Google Scholar] [CrossRef]
- Akhtar, T.; Sheikh, N. An overview of thioacetamide-induced hepatotoxicity. Toxin Rev. 2013, 32, 43–46. [Google Scholar] [CrossRef]
- Gieling, R.G.; Wallace, K.; Han, Y.P. Interleukin-1 participates in the progression from liver injury to fibrosis. Am. J. Physiol. Gastroint. Liver Physiol. 2009, 296, 1324–1331. [Google Scholar] [CrossRef]
- Olsen, N.J.; Kovacs, W.J. Gonadal steroids and immunity. Endocr. Rev. 1996, 17, 369–384. [Google Scholar]
- Cutolo, M.; Wilder, R.L. Different roles for androgens and estrogens in the susceptibility to autoimmune rheumatic diseases. Rheum. Dis. Clin. N. Am. 2000, 26, 825–839. [Google Scholar] [CrossRef]
- Nilsson, S.; Mäkelä, S.; Treuter, E.; Tujague, M.; Thomsen, J.; Andersson, G.; Enmark, E.; Pettersson, K.; Warner, M.; Gustafsson, J.A. Mechanisms of estrogen action. Physiol. Rev. 2001, 81, 1535–1565. [Google Scholar] [CrossRef]
- Villa, E. Role of estrogen in liver cancer. Women’s Health (Lond.) 2008, 4, 41–50. [Google Scholar] [CrossRef]
- Kolios, G.; Valatas, V.; Kouroumalis, E. Role of Kupffer cells in the pathogenesis of liver disease. World J. Gastroenterol. 2006, 12, 7413–7420. [Google Scholar] [CrossRef]
- Osz, J.; Brélivet, Y.; Peluso-Iltis, C.; Cura, V.; Eiler, S.; Ruff, M.; Bourguet, W.; Rochel, N.; Moras, D. Structural basis for a molecular allosteric control mechanism of cofactor binding to nuclear receptors. Proc. Natl. Acad. Sci. USA 2012, 109, E588–E594. [Google Scholar] [CrossRef]
- Li, X.; Huang, J.; Yi, P.; Bambara, R.A.; Hilf, R.; Muyan, M. Single-chain estrogen receptors (ERs) reveal that the ERalpha/beta heterodimer emulates functions of the ER alpha dimer in genomic estrogen signaling pathways. Mol. Cell Biol. 2004, 24, 7681–7694. [Google Scholar] [CrossRef]
- Lane, P.H. Estrogen receptors in the kidney: lessons from genetically altered mice. Gend. Med. 2008, 5, S11–S18. [Google Scholar] [CrossRef]
- Ciocca, D.R.; Jorge, A.D.; Jorge, O.; Milutín, C.; Hosokawa, R.; Díaz Lestren, M.; Muzzio, E.; Schulkin, S.; Schirbu, R. Estrogen receptors, progesterone receptors and heat-shock 27-kD protein in liver biopsy specimens from patients with hepatitis B virus infection. Hepatology 1991, 13, 838–844. [Google Scholar] [CrossRef]
- Weiser, M.J.; Foradori, C.D.; Handa, R.J. Estrogen receptor beta in the brain: from form to function. Brain Res. Rev. 2008, 57, 309–320. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Carlsson, B.; Grandien, K.; Enmark, E.; Häggblad, J.; Nilsson, S.; Gustafsson, J.A. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology 1997, 138, 863–870. [Google Scholar] [CrossRef]
- Ciana, P.; Di Luccio, G.; Belcredito, S.; Pollio, G.; Vegeto, E.; Tatangelo, L.; Tiveron, C.; Maggi, A. Engineering of a mouse for the in vivo profiling of estrogen receptor activity. Mol. Endocrinol. 2001, 15, 1104–1113. [Google Scholar] [CrossRef]
- Fisher, B.; Gunduz, N.; Saffer, E.A.; Zheng, S. Relation of estrogen and its receptor to rat liver growth and regeneration. Cancer Res. 1984, 44, 2410–2415. [Google Scholar]
- Kahn, D.; Eagon, P.K.; Porter, L.E.; Elm, M.S.; Makowka, L.; Podesta, L.; Starzl, T.E.; Van Thiel, D.H. Effect of tamoxifen on hepatic regeneration in male rats. Dig. Dis. Sci. 1989, 34, 27–32. [Google Scholar] [CrossRef]
- Handan, O.B.; Gonca, O.; Aydincan, A.; Gokhan, M.; Mehmet, H. Hepatic estrogen receptor expression prevents liver fibrosis through decreasing the risk of early activation of hepatic stellate cells. Transplantation 2018, 102, S385. [Google Scholar]
- Cengiz, M.; Ozenirler, S.; Yılmaz, G. Estrogen receptor alpha expression and liver Fibrosis in chronic hepatitis C virus genotype 1b: A clinicopathological study. Hepat. Mon. 2014, 14, e21885. [Google Scholar] [CrossRef]
- Parola, M.; Robino, G. Oxidative stress-related molecules and liver fibrosis. J. Hepatol. 2001, 35, 297–306. [Google Scholar] [CrossRef]
- Kitajima, Y.; Doi, H.; Ono, Y.; Urata, Y.; Goto, S.; Kitajima, M.; Miura, K.; Li, T.S.; Masuzaki, H. Estrogen deficiency heterogeneously affects tissue specific stem cells in mice. Sci. Rep. 2015, 5, 12861. [Google Scholar] [CrossRef]
- Li, S.; Tan, H.Y.; Wang, N.; Zhang, Z.J.; Lao, L.; Wong, C.W.; Feng, Y. The role of oxidative stress and antioxidants in liver diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124. [Google Scholar] [CrossRef]
- Shimizu, I.; Ito, S. Protection of estrogens against the progression of chronic liver disease. Hepatol. Res. 2007, 37, 239–247. [Google Scholar] [CrossRef]
- Jurczuk, M.; Brzoska, M.M.; Moniuszko-Jakoniuk, J.; Gałazyn-Sidorczuk, M.; Kulikowska- Karpińska, E. Antioxidant enzymes activity and lipid peroxidation in liver and kidney of rats exposed to cadmium and ethanol. Food Chem. Toxicol. 2004, 42, 429–438. [Google Scholar] [CrossRef]
- Low, T.Y.; Leow, C.K.; Salto-Tellez, M.; Chung, M.C. A proteomic analysis of thioacetamide-induced hepatotoxicity and cirrhosis in rat livers. Proteomics 2004, 4, 3960–3974. [Google Scholar] [CrossRef]
- Wang, K.; Lin, B. Pathophysiological significance of hepatic apoptosis. ISRN Hepatol. 2012, 2013, 740149. [Google Scholar] [CrossRef]
- Canbay, A.; Feldstein, A.E.; Higuchi, H.; Werneburg, N.; Grambihler, A.; Bronk, S.F.; Gores, G.J. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology 2003, 38, 1188–1198. [Google Scholar] [CrossRef]
- Jiang, H.S.; Zhu, L.L.; Zhang, Z.; Chen, H.; Chen, Y.; Dai, Y.T. Estradiol attenuates the TGF-β1-induced conversion of primary TAFs into myofibroblasts and inhibits collagen production and myofibroblast contraction by modulating the Smad and Rho/ROCK signaling pathways. Int. J. Mol. Med. 2015, 36, 801–807. [Google Scholar] [CrossRef]
- Shimizu, I.; Mizobuchi, Y.; Yasuda, M.; Shiba, M.; Ma, Y.R.; Horie, T.; Liu, F.; Ito, S. Inhibitory effect of oestradiol on activation of rat hepatic stellate cells in vivo and in vitro. Gut 1999, 44, 127–136. [Google Scholar] [CrossRef]
- Lu, G.; Shimizu, I.; Cui, X.; Itonaga, M.; Tamaki, K.; Fukuno, H.; Inoue, H.; Honda, H.; Ito, S. Antioxidant and antiapoptotic activities of idoxifene and estradiol in hepatic fibrosis in rats. Life Sci. 2004, 74, 897–907. [Google Scholar] [CrossRef]
- Weber, A.; Boger, R.; Vick, B.; Urbanik, T.; Haybaeck, J.; Zoller, S.; Teufel, A.; Krammer, P.H.; Opferman, J.T.; Galle, P.R.; et al. Hepatocyte-specific deletion of the antiapoptotic protein myeloid cell leukemia-1 triggers proliferation and hepatocarcinogenesis in mice. Hepatology 2010, 51, 1226–1236. [Google Scholar] [CrossRef]
- Chakraborty, J.B.; Oakley, F.; Walsh, M.J. Mechanisms and biomarkers of apoptosis in liver disease and fibrosis. Int. J. Hepatol. 2012, 2012, 648915. [Google Scholar] [CrossRef]
- Blaner, W.S.; O’Byrne, S.M.; Wongsiriroj, N.; Kluwe, J.; D’Ambrosio, D.M.; Jiang, H.; Schwabe, R.F.; Hillman, E.M.; Piantedosi, R.; Libien, J. Hepatic stellate cell lipid droplets: a specialized lipid droplet for retinoid storage. Biochim. Biophys. Acta 2009, 1791, 467–473. [Google Scholar] [CrossRef]
- Yu, L.; Hebert, M.C.; Zhang, Y.E. TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. EMBO J. 2002, 21, 3749–3759. [Google Scholar] [CrossRef]
- Biswas, S.; Ghose, S. Divergent impact of gender in advancement of liver injuries, diseases, and carcinogenesis. Front. Biosci. 2018, 10, 65–100. [Google Scholar] [CrossRef][Green Version]
- Toyoki, Y.; Sasaki, M.; Narumi, S.; Yoshihara, S.; Morita, T.; Konn, M. Semiquantitative evaluation of hepatic fibrosis by measuring tissue hydroxyproline. Hepatogastroenterology 1998, 45, 2261–2264. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Body Weight (g) | Liver Weight (g) | Uterus Weight (g) | |||
|---|---|---|---|---|---|---|
| Initial (g) | Final (g) | Absolute (g) | Relative (%) a | Absolute (g) | Relative (%) b | |
| Sham control | 182.5 ± 2.5 | 245.6 ± 6.3 | 7.91 ± 1.05 | 3.21 ± 0.15 | 1.17 ± 0.05 | 0.48 ± 0.01 |
| OVX TAA | 181.9 ± 3.1 | 194.6 ± 5.7 | 8.69 ± 1.12 ** | 4.58 ± 0.21 ** | 0.11 ± 0.02 ** | 0.07 ± 0.02 ** |
| Sham-operated TAA | 184.1 ± 2.8 | 199.2 ± 4.7 | 7.92 ± 1.37 | 3.98 ± 0.27 ## | 0.73 ± 0.04 ## | 0.39 ± 0.05 ## |
| Male control | 234.2 ± 3.8 | 373.8 ± 9.2 | 9.78 ± 1.52 | 2.74 ± 0.31 | - | - |
| Male TAA | 235.6 ± 4.1 | 245.4 ± 8.6 | 11.63 ± 1.48 ** | 4.73 ± 0.38 ** | - | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.H.; Son, J.Y.; Kim, K.S.; Park, Y.J.; Kim, H.R.; Park, J.H.; Kim, K.-B.; Lee, K.Y.; Kang, K.W.; Kim, I.S.; et al. Estrogen Deficiency Potentiates Thioacetamide-Induced Hepatic Fibrosis in Sprague-Dawley Rats. Int. J. Mol. Sci. 2019, 20, 3709. https://doi.org/10.3390/ijms20153709
Lee YH, Son JY, Kim KS, Park YJ, Kim HR, Park JH, Kim K-B, Lee KY, Kang KW, Kim IS, et al. Estrogen Deficiency Potentiates Thioacetamide-Induced Hepatic Fibrosis in Sprague-Dawley Rats. International Journal of Molecular Sciences. 2019; 20(15):3709. https://doi.org/10.3390/ijms20153709
Chicago/Turabian StyleLee, Yong Hee, Ji Yeon Son, Kyeong Seok Kim, Yoo Jung Park, Hae Ri Kim, Jae Hyeon Park, Kyu-Bong Kim, Kwang Youl Lee, Keon Wook Kang, In Su Kim, and et al. 2019. "Estrogen Deficiency Potentiates Thioacetamide-Induced Hepatic Fibrosis in Sprague-Dawley Rats" International Journal of Molecular Sciences 20, no. 15: 3709. https://doi.org/10.3390/ijms20153709
APA StyleLee, Y. H., Son, J. Y., Kim, K. S., Park, Y. J., Kim, H. R., Park, J. H., Kim, K.-B., Lee, K. Y., Kang, K. W., Kim, I. S., Kacew, S., Lee, B. M., & Kim, H. S. (2019). Estrogen Deficiency Potentiates Thioacetamide-Induced Hepatic Fibrosis in Sprague-Dawley Rats. International Journal of Molecular Sciences, 20(15), 3709. https://doi.org/10.3390/ijms20153709