bHLH–PAS Proteins: Their Structure and Intrinsic Disorder


Abstract
1. Introduction to bHLH–PAS Proteins
2. bHLH–PAS Protein Conservation between Organisms
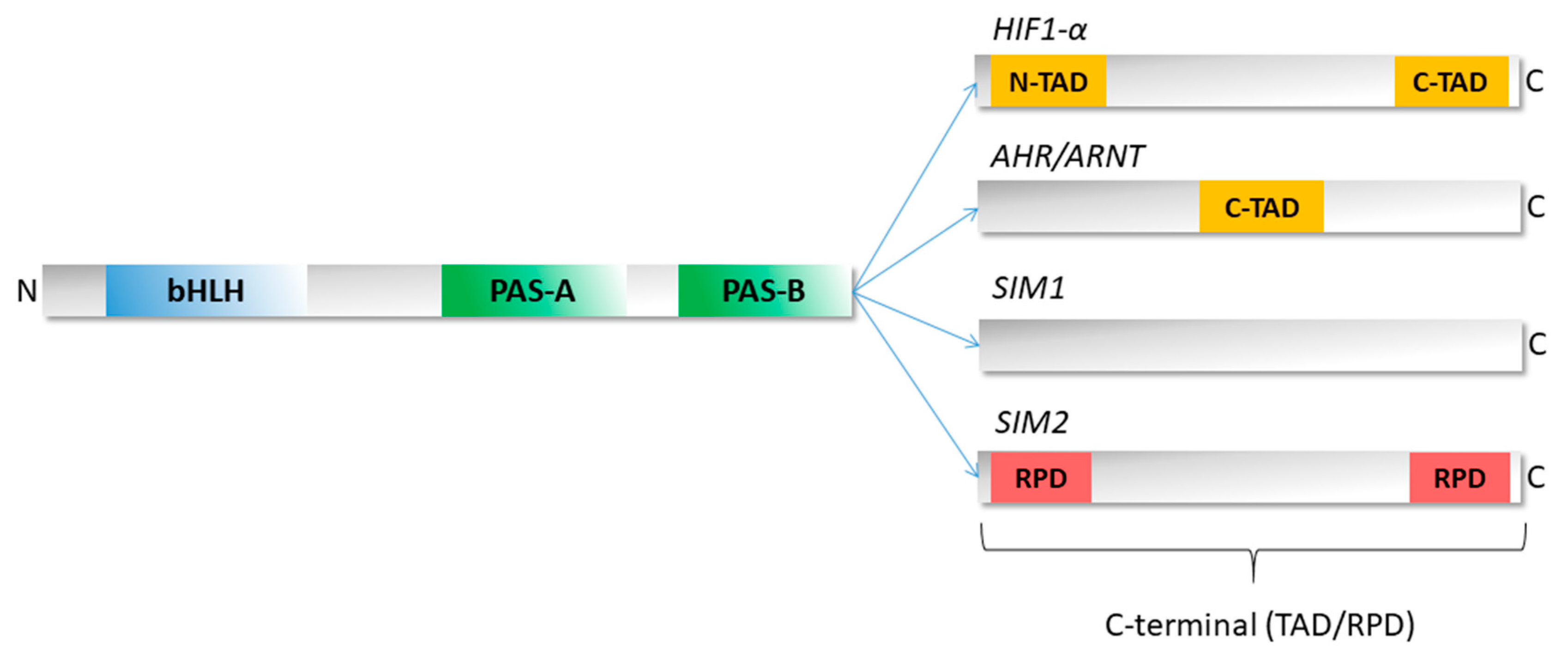
3. Structure of bHLH–PAS Proteins
4. Unique Properties of the C-Terminal Domains of bHLH–PAS Proteins as IDRs
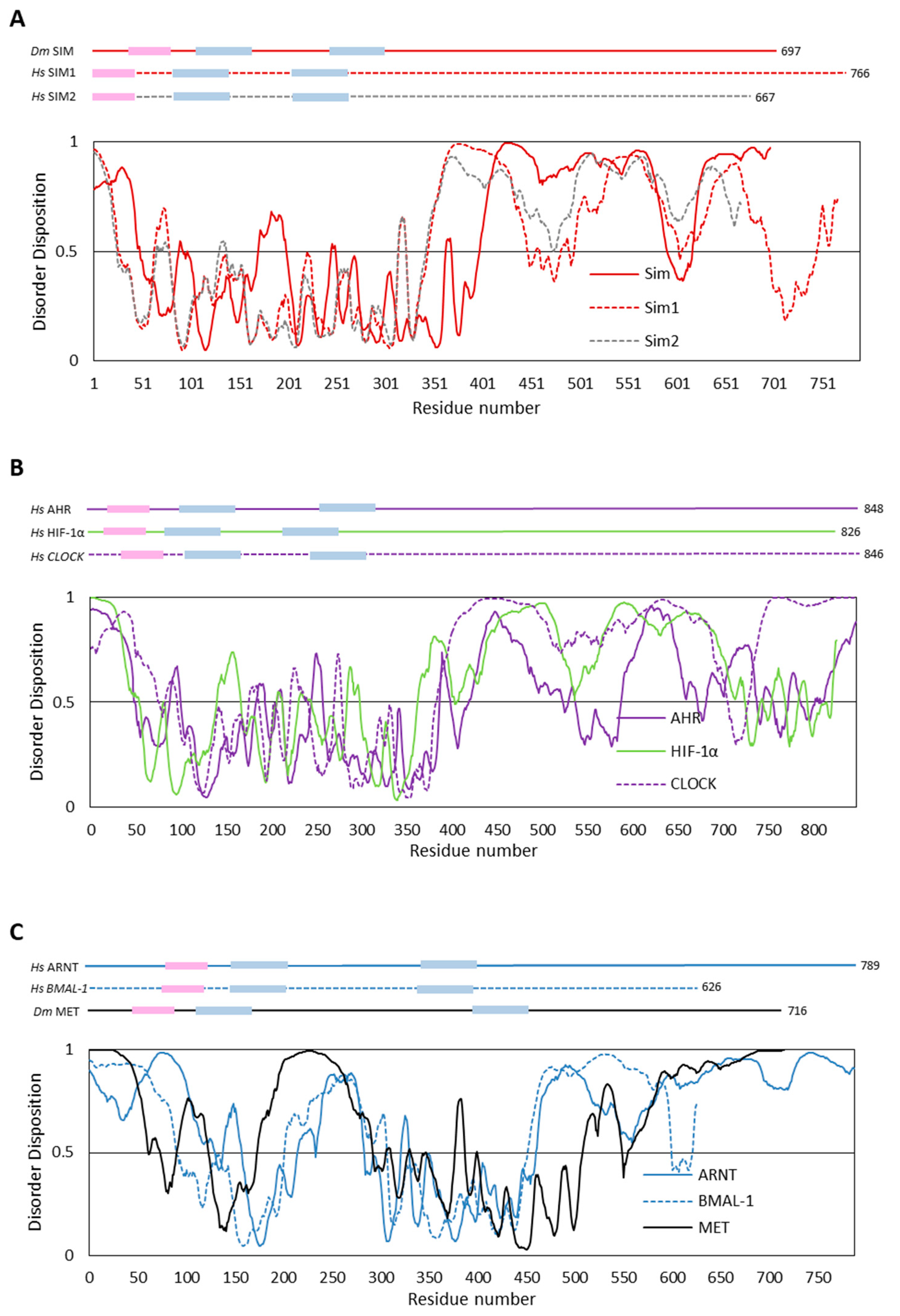
4.1. In Silico Analyses of Selected bHLH–PAS Proteins
4.2. The Impact of Disordered Regions on Protein Function
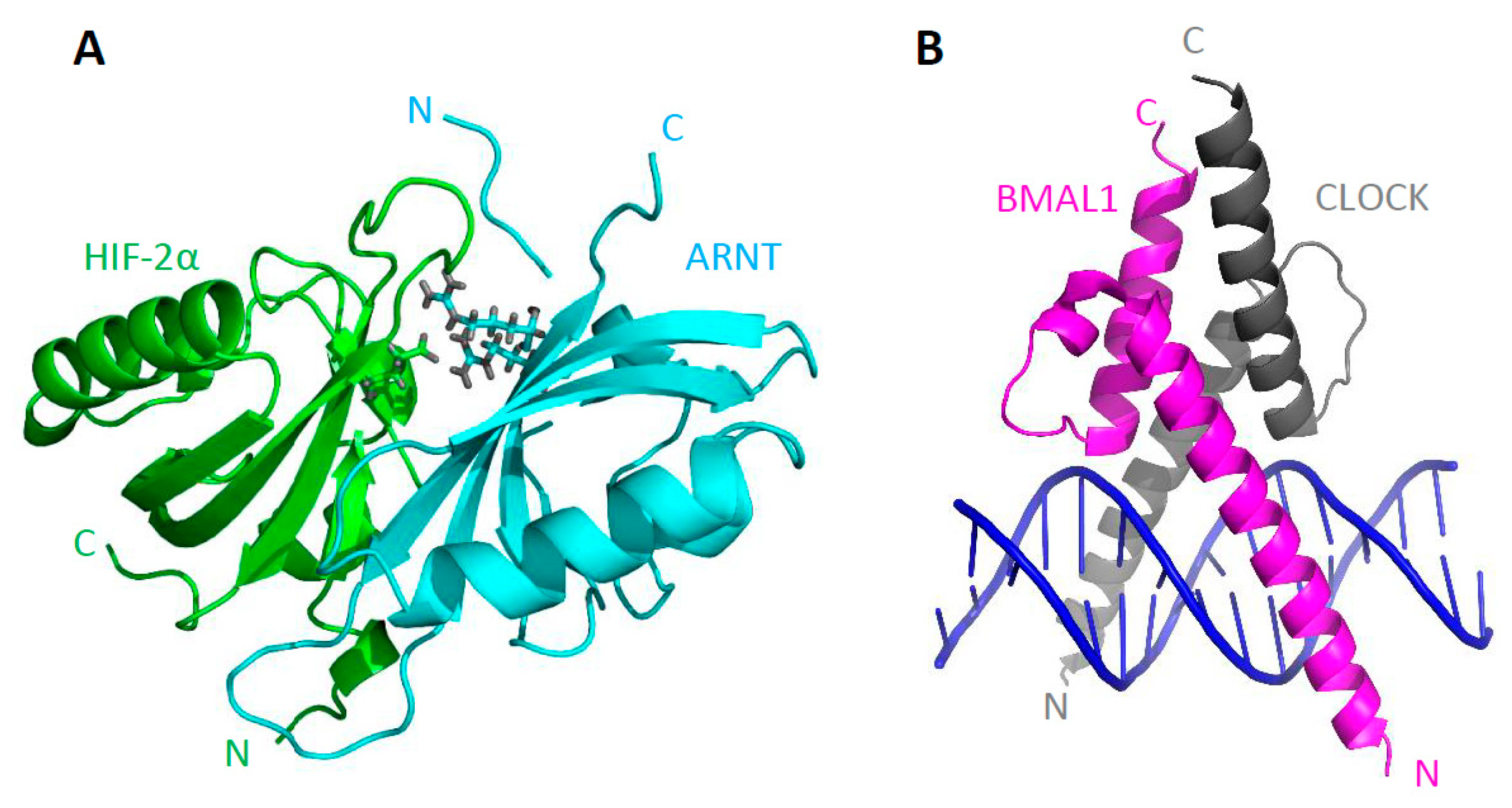
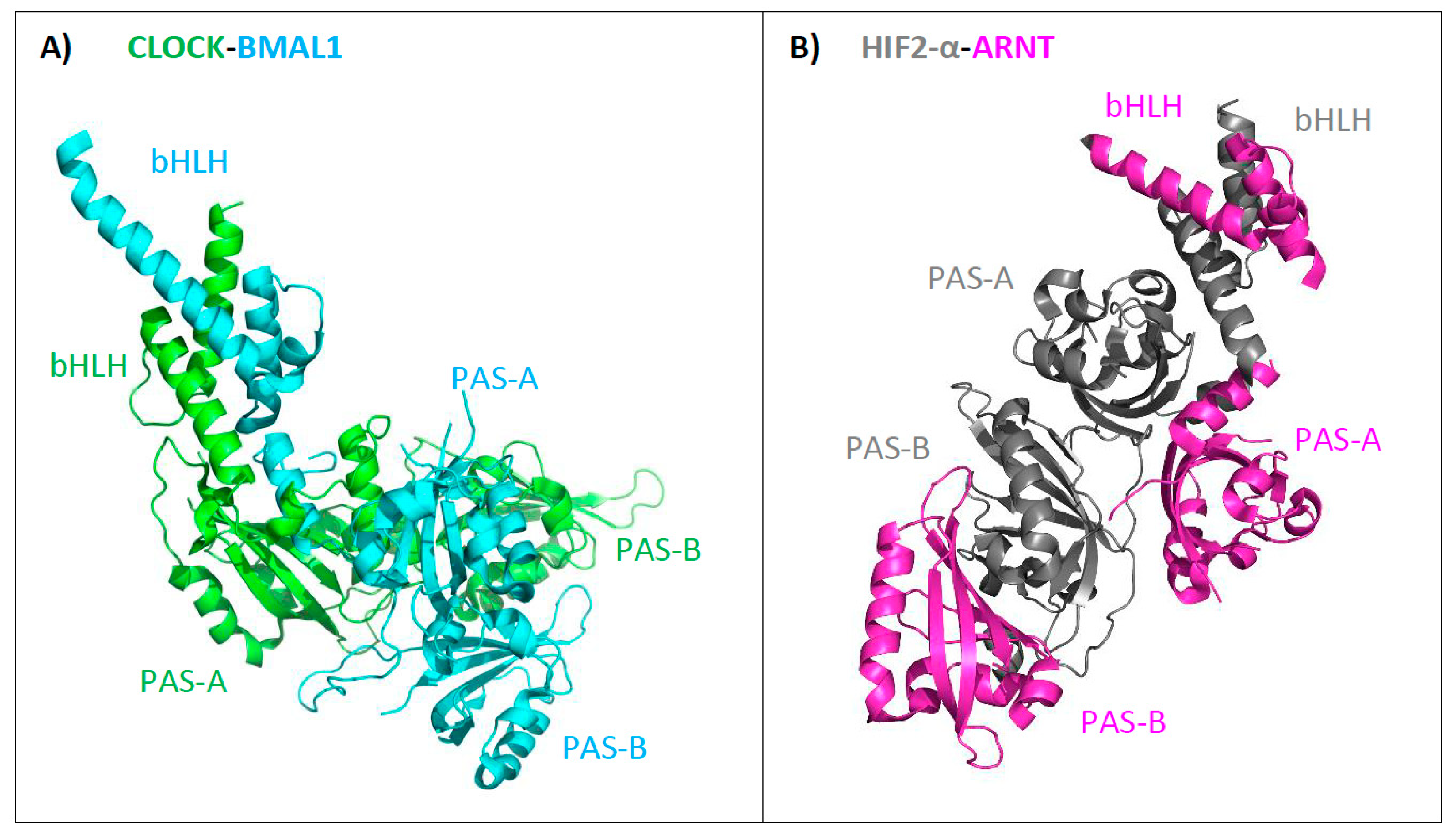
4.3. Structural Analysis of bHLH–PAS C-Terminal Fragments
4.4. Structural Analysis of IDPs
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Aa | amino acid |
| AHR | aryl hydrocarbon receptor |
| AHRR | aryl hydrocarbon receptor repressor |
| ARNT | aryl hydrocarbon receptor nuclear translocator |
| bHLH–PAS | helix–loop–helix/Per-ARNT-SIM |
| CLOCK | circadian locomotor output cycles protein kaput |
| CRY1 | cryptochrome |
| DYS | dysfusion protein |
| ER | estrogen receptor |
| GCE | germ cell-expressed protein |
| HAT | histone transacetylase |
| HIF | hypoxia-inducible factors |
| IDPs | intrinsically disordered proteins |
| IDRs | intrinsically disordered regions |
| JH | juvenile hormone |
| LBD | ligand-binding domain |
| MET | methoprene-tolerant protein |
| MoREs | molecular recognition elements |
| MET/C | C-terminus of the MET protein |
| NES | nuclear export signal |
| NLS | nuclear localization signal |
| NMR | Nuclear Resonance Magnetism |
| NPAS | neuronal PAS domain-containing proteins |
| NR | boxes nuclear receptor boxes |
| PAC | PAS-associated C-terminal motif |
| PDB | Protein Data Bank |
| PREs | paramagnetic relaxation enhancements |
| RDCs | residual dipolar couplings |
| SAXS | Small-angle X-ray Scattering |
| SIM | single-minded proteins |
| SIMA | similar protein |
| SS | spineless protein |
| TAD or TRD | transactivation or repression domain |
| TGO | TANGO protein |
| TRH | trachealess protein |
References
- Crews, S.T. Control of cell lineage-specific development and transcription by bHLH-PAS proteins. Genes Dev. 1998, 12, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-W.; Bae, S.-H.; Jeong, J.-W.; Kim, S.-H.; Kim, K.-W. Hypoxia-inducible factor (HIF-1)α: Its protein stability and biological functions. Exp. Mol. Med. 2004, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ema, M.; Hirota, K.; Mimura, J.; Abe, H.; Yodoi, J.; Sogawa, K.; Poellinger, L.; Fujii-Kuriyama, Y. Molecular mechanisms of transcription activation by HLF and HIF1alpha in response to hypoxia: Their stabilization and redox signal-induced interaction with CBP/p300. EMBO J. 1999, 18, 1905–1914. [Google Scholar] [CrossRef] [PubMed]
- Petrulis, J.R.; Kusnadi, A.; Ramadoss, P.; Hollingshead, B.; Perdew, G.H. The hsp90 Co-chaperone XAP2 Alters Importin β Recognition of the Bipartite Nuclear Localization Signal of the Ah Receptor and Represses Transcriptional Activity. J. Biol. Chem. 2003, 278, 2677–2685. [Google Scholar] [CrossRef] [PubMed]
- Xue, P.; Fu, J.; Zhou, Y. The Aryl Hydrocarbon Receptor and Tumor Immunity. Front. Immunol. 2018, 9, 286. [Google Scholar] [CrossRef] [PubMed]
- Neavin, D.; Liu, D.; Ray, B.; Weinshilboum, R. The Role of the Aryl Hydrocarbon Receptor (AHR) in Immune and Inflammatory Diseases. Int. J. Mol. Sci. 2018, 19, 3851. [Google Scholar] [CrossRef]
- Freer, S.M.; Lau, D.C.; Pearson, J.C.; Talsky, K.B.; Crews, S.T. Molecular and functional analysis of Drosophila single-minded larval central brain expression. Gene Expr. Patterns 2011, 11, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Pielage, J.; Steffes, G.; Lau, D.C.; Parente, B.A.; Crews, S.T.; Strauss, R.; Klämbt, C. Novel behavioral and developmental defects associated with Drosophila single-minded. Dev. Biol. 2002, 249, 283–299. [Google Scholar] [CrossRef]
- Chen, H.; Chrast, R.; Rossier, C.; Gos, A.; Antonarakis, S.E.; Kudoh, J.; Yamaki, A.; Shindoh, N.; Maeda, H.; Minoshima, S.; et al. Single–minded and Down syndrome? Nat. Genet. 1995, 10, 9–10. [Google Scholar] [CrossRef]
- Bersten, D.C.; Sullivan, A.E.; Peet, D.J.; Whitelaw, M.L. bHLH–PAS proteins in cancer. Nat. Rev. Cancer 2013, 13, 827–841. [Google Scholar] [CrossRef]
- Safe, S.; Lee, S.O.; Jin, U.H. Role of the aryl hydrocarbon receptor in carcinogenesis and potential as a drug target. Toxicol. Sci. 2013, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Beamer, C.A.; Shepherd, D.M. Role of the aryl hydrocarbon receptor (AhR) in lung inflammation. Semin. Immunopathol. 2013, 35, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Puccetti, M.; Paolicelli, G.; Oikonomou, V.; De Luca, A.; Renga, G.; Borghi, M.; Pariano, M.; Stincardini, C.; Scaringi, L.; Giovagnoli, S.; et al. Towards Targeting the Aryl Hydrocarbon Receptor in Cystic Fibrosis. Mediat. Inflamm. 2018, 2018, 1601486. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Fei, P.; Mu, J.; Li, W.; Song, J. Hippocampal expression of aryl hydrocarbon receptor nuclear translocator 2 and neuronal PAS domain protein 4 in a rat model of depression. Neurol. Sci. 2014, 35, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Choy, F.C.; Klarić, T.S.; Koblar, S.A.; Lewis, M.D. The Role of the Neuroprotective Factor Npas4 in Cerebral Ischemia. Int. J. Mol. Sci. 2015, 16, 29011–29028. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, P.V.; Lynn, F.C. All-encomPASsing regulation of β-cells: PAS domain proteins in β-cell dysfunction and diabetes. Trends Endocrinol. Metab. 2015, 26, 49–57. [Google Scholar] [CrossRef]
- Speckmann, T.; Sabatini, P.V.; Nian, C.; Smith, R.G.; Lynn, F.C. Npas4 Transcription Factor Expression Is Regulated by Calcium Signaling Pathways and Prevents Tacrolimus-induced Cytotoxicity in Pancreatic Beta Cells. J. Biol. Chem. 2016, 291, 2682–2695. [Google Scholar] [CrossRef]
- Li, X.; Duan, X.; Jiang, H.; Sun, Y.; Tang, Y.; Yuan, Z.; Guo, J.; Liang, W.; Chen, L.; Yin, J.; et al. Genome-Wide Analysis of Basic/Helix-Loop-Helix Transcription Factor Family in Rice and Arabidopsis. Plant Physiol. 2006, 141, 1167–1184. [Google Scholar] [CrossRef]
- Ponting, C.P.; Aravind, L. PAS: A multifunctional domain family comes to light. Curr. Biol. 1997, 7, R674–R677. [Google Scholar] [CrossRef]
- Kewley, R.J.; Whitelaw, M.L.; Chapman-Smith, A. The mammalian basic helix–loop–helix/PAS family of transcriptional regulators. Int. J. Biochem. Cell Biol. 2004, 36, 189–204. [Google Scholar] [CrossRef]
- Wu, D.; Rastinejad, F. Structural characterization of mammalian bHLH-PAS transcription factors. Curr. Opin. Struct. Biol. 2017, 43, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Partch, C.L.; Gardner, K.H. Coactivator recruitment, a new role for PAS domains in transcriptional regulation by the bHLH-PAS family. J. Cell. Physiol. 2010, 223, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Woods, S.L.; Whitelaw, M.L. Differential Activities of Murine Single Minded 1 (SIM1) and SIM2 on a Hypoxic Response Element. J. Biol. Chem. 2002, 277, 10236–10243. [Google Scholar] [CrossRef] [PubMed]
- Moffett, P.; Reece, M.; Pelletier, J. The murine Sim-2 gene product inhibits transcription by active repression and functional interference. Mol. Cell. Biol. 1997, 17, 4933–4947. [Google Scholar] [CrossRef] [PubMed]
- Moffett, P.; Pelletier, J. Different transcriptional properties of mSim-1 and mSim-2. FEBS Lett. 2000, 466, 80–86. [Google Scholar] [CrossRef]
- Vorrink, S.U.; Domann, F.E. Regulatory crosstalk and interference between the xenobiotic and hypoxia sensing pathways at the AhR-ARNT-HIF1α signaling node. Chem. Biol. Interact. 2014, 218, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Tal, R.; Shaish, A.; Bangio, L.; Peled, M.; Breitbart, E.; Harats, D. Activation of C-transactivation domain is essential for optimal HIF-1α-mediated transcriptional and angiogenic effects. Microvasc. Res. 2008, 76, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, B.N.; Probst, M.R.; Reisz-Porszasz, S.; Hankinson, O. Identification of functional domains of the aryl hydrocarbon receptor. J. Biol Chem. 1995, 270, 29270–29278. [Google Scholar] [CrossRef]
- Fribourgh, J.L.; Partch, C.L. Assembly and function of bHLH-PAS complexes. Proc. Natl. Acad. Sci. USA 2017, 114, 5330–5332. [Google Scholar] [CrossRef]
- Hoffman, E.C.; Reyes, H.; Chu, F.F.; Sander, F.; Conley, L.H.; Brooks, B.A.; Hankinson, O. Cloning of a factor required for activity of the Ah (dioxin) receptor. Science 1991, 252, 954–958. [Google Scholar] [CrossRef] [PubMed]
- Andreasen, E.A.; Spitsbergen, J.M.; Tanguay, R.L.; Stegeman, J.J.; Heideman, W.; Peterson, R.E. Tissue-Specific Expression of AHR2, ARNT2, and CYP1A in Zebrafish Embryos and Larvae: Effects of Developmental Stage and 2,3,7,8-Tetrachlorodibenzo-p-dioxin Exposure. Toxicol. Sci. 2002, 68, 403–419. [Google Scholar] [CrossRef] [PubMed]
- Crews, S.T.; Fan, C.-M. Remembrance of things PAS: Regulation of development by bHLH–PAS proteins. Curr. Opin. Genet. Dev. 1999, 9, 580–587. [Google Scholar] [CrossRef]
- Sonnenfeld, M.; Ward, M.; Nystrom, G.; Mosher, J.; Stahl, S.; Crews, S. The Drosophila tango gene encodes a bHLH-PAS protein that is orthologous to mammalian Arnt and controls CNS midline and tracheal development. Development 1997, 124, 4571–4582. [Google Scholar] [PubMed]
- Panda, S.; Hogenesch, J.B.; Kay, S.A. Circadian rhythms from flies to human. Nature 2002, 417, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Paranjpe, D.A.; Sharma, V.K. Evolution of temporal order in living organisms. J. Circadian Rhythm. 2005, 3, 7. [Google Scholar] [CrossRef]
- Li, K.-L.; Lu, T.-M.; Yu, J.-K. Genome-wide survey and expression analysis of the bHLH-PAS genes in the amphioxus Branchiostoma floridae reveal both conserved and diverged expression patterns between cephalochordates and vertebrates. Evodevo 2014, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Meng, M.; Peng, J.; Qian, W.; Kang, L.; Xia, Q. Genome-wide comparison of genes involved in the biosynthesis, metabolism, and signaling of juvenile hormone between silkworm and other insects. Genet. Mol. Biol. 2014, 37, 444–459. [Google Scholar] [CrossRef] [PubMed]
- Abdou, M.; Peng, C.; Huang, J.; Zyaan, O.; Wang, S.; Li, S.; Wang, J. Wnt Signaling Cross-Talks with JH Signaling by Suppressing Met and gce Expression. PLoS ONE 2011, 6, e26772. [Google Scholar] [CrossRef]
- Jindra, M.; Bellés, X.; Shinoda, T. Molecular basis of juvenile hormone signaling. Curr. Opin. Insect. Sci. 2015, 11, 39–46. [Google Scholar] [CrossRef]
- Erbel, P.J.A.; Card, P.B.; Karakuzu, O.; Bruick, R.K.; Gardner, K.H. Structural basis for PAS domain heterodimerization in the basic helix-loop-helix-PAS transcription factor hypoxia-inducible factor. Proc. Natl. Acad. Sci. USA 2003, 100, 15504–15509. [Google Scholar] [CrossRef] [PubMed]
- Card, P.B.; Erbel, P.J.A.; Gardner, K.H. Structural Basis of ARNT PAS-B Dimerization: Use of a Common Beta-sheet Interface for Hetero- and Homodimerization. J. Mol. Biol. 2005, 353, 664–677. [Google Scholar] [CrossRef] [PubMed]
- Henry, J.T.; Crosson, S. Ligand-binding PAS domains in a genomic, cellular, and structural context. Annu. Rev. Microbiol. 2011, 65, 261–286. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, Y.; Li, L.; Su, X.-D. Intermolecular recognition revealed by the complex structure of human CLOCK-BMAL1 basic helix-loop-helix domains with E-box DNA. Cell Res. 2013, 23, 213. [Google Scholar] [CrossRef] [PubMed]
- Jones, S. An overview of the basic helix-loop-helix proteins. Genome Biol. 2004, 5, 226. [Google Scholar] [CrossRef] [PubMed]
- Scheuermann, T.H.; Tomchick, D.R.; Machius, M.; Guo, Y.; Bruick, R.K.; Gardner, K.H. Artificial ligand binding within the HIF2 PAS-B domain of the HIF2 transcription factor. Proc. Natl. Acad. Sci. USA 2009, 106, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Chelliah, Y.; Shan, Y.; Taylor, C.A.; Yoo, S.-H.; Partch, C.; Green, C.B.; Zhang, H.; Takahashi, J.S. Crystal structure of the heterodimeric CLOCK:BMAL1 transcriptional activator complex. Science 2012, 337, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Potluri, N.; Lu, J.; Kim, Y.; Rastinejad, F. Structural integration in hypoxia-inducible factors. Nature 2015, 524, 303–308. [Google Scholar] [CrossRef]
- Bernardo, T.J.; Dubrovsky, E.B. The Drosophila juvenile hormone receptor candidates methoprene-tolerant (MET) and germ cell-expressed (GCE) utilize a conserved LIXXL motif to bind the FTZ-F1 nuclear receptor. J. Biol. Chem. 2012, 287, 7821–7833. [Google Scholar] [CrossRef]
- Furness, S.G.B.; Lees, M.J.; Whitelaw, M.L. The dioxin (aryl hydrocarbon) receptor as a model for adaptive responses of bHLH/PAS transcription factors. FEBS Lett. 2007, 581, 3616–3625. [Google Scholar] [CrossRef]
- Mirsky, A.E.; Pauling, L. On the Structure of Native, Denatured, and Coagulated Proteins. Proc. Natl. Acad. Sci. USA 1936, 22, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2014, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Flexible Nets of Malleable Guardians, Intrinsically Disordered Chaperones in Neurodegenerative Diseases. Chem. Rev. 2011, 111, 1134–1166. [Google Scholar] [CrossRef] [PubMed]
- Ball, K.A.; Wemmer, D.E.; Head-Gordon, T. Comparison of Structure Determination Methods for Intrinsically Disordered Amyloid-β Peptides. J. Phys. Chem. B 2014, 118, 6405–6416. [Google Scholar] [CrossRef] [PubMed]
- Godlewski, J.; Wang, S.; Wilson, T.G. Interaction of bHLH-PAS proteins involved in juvenile hormone reception in Drosophila. Biochem. Biophys. Res. Commun. 2006, 342, 1305–1311. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Romero, P.; Rani, M.; Dunker, A.K.; Obradovic, Z. Predicting Protein Disorder for N-, C-, and Internal Regions. Genome Inform. 1999, 10, 30–40. [Google Scholar]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 2010, 1804, 996–1010. [Google Scholar] [CrossRef]
- Dosztanyi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef]
- Lobanov, M.Y.; Sokolovskiy, I.V.; Galzitskaya, O.V. IsUnstruct: Prediction of the residue status to be ordered or disordered in the protein chain by a method based on the Ising model. J. Biomol. Struct. Dyn. 2013, 31, 1034–1043. [Google Scholar] [CrossRef]
- Dziedzic-Letka, A.; Ozyhar, A. Intrinsically disordered proteins. Postepy Biochem. 2012, 58, 100–109. [Google Scholar] [PubMed]
- Vacic, V.; Oldfield, C.J.; Mohan, A.; Radivojac, P.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Characterization of Molecular Recognition Features, MoRFs, and Their Binding Partners. J. Proteome Res. 2007, 6, 2351–2366. [Google Scholar] [CrossRef] [PubMed]
- Zelzer, E.; Wappner, P.; Shilo, B.-Z. The PAS domain confers target gene specificity of Drosophila bHLH/PAS proteins. Genes Dev. 1997, 11, 2079–2089. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002, 11, 739–756. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.; Klein, C.; Müller, L.; Hansen, S.; Buchner, J. p53 contains large unstructured regions in its native state. J. Mol. Biol. 2002, 322, 917–927. [Google Scholar] [CrossRef]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Dolwick, K.M.; Schmidt, J.V.; Bradfield, C.A. Potent transactivation domains of the Ah receptor and the Ah receptor nuclear translocator map to their carboxyl termini. J. Biol. Chem. 1994, 269, 31518–31524. [Google Scholar] [PubMed]
- Brunnberg, S.; Pettersson, K.; Rydin, E.; Matthews, J.; Hanberg, A.; Pongratz, I. The basic helix-loop-helix-PAS protein ARNT functions as a potent coactivator of estrogen receptor-dependent transcription. Proc. Natl. Acad. Sci. USA 2003, 100, 6517–6522. [Google Scholar] [CrossRef]
- Ren, L.; Thompson, J.D.; Cheung, M.; Ngo, K.; Sung, S.; Leong, S.; Chan, W.K. Selective suppression of the human aryl hydrocarbon receptor function can be mediated through binding interference at the C-terminal half of the receptor. Biochem. Pharmacol. 2016, 107, 91–100. [Google Scholar] [CrossRef]
- Kawajiri, K.; Fujii-Kuriyama, Y. The aryl hydrocarbon receptor: A multifunctional chemical sensor for host defense and homeostatic maintenance. Exp. Anim. 2017, 66, 75–89. [Google Scholar] [CrossRef]
- Kallio, P.J.; Okamoto, K.; Brien, S.O.; Carrero, P.; Makino, Y.; Tanaka, H.; Poellinger, L. Signal transduction in hypoxic cells: Inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-inducible factor-1 α. EMBO J. 1998, 17, 6573–6586. [Google Scholar] [CrossRef] [PubMed]
- Mylonis, I.; Chachami, G.; Paraskeva, E.; Simos, G. Atypical CRM1-dependent nuclear export signal mediates regulation of hypoxia-inducible factor-1alpha by MAPK. J. Biol. Chem. 2008, 283, 27620–27627. [Google Scholar] [CrossRef] [PubMed]
- Doi, M.; Hirayama, J.; Sassone-Corsi, P. Circadian regulator CLOCK is a histone acetyltransferase. Cell 2006, 125, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Gustafson, C.L.; Sammons, P.J.; Khan, S.K.; Parsley, N.C.; Ramanathan, C.; Lee, H.-W.; Liu, A.C.; Partch, C.L. Cryptochrome 1 regulates the circadian clock through dynamic interactions with the BMAL1 C terminus. Nat. Struct. Mol. Biol. 2015, 22, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Kolonko, M.; Ożga, K.; Hołubowicz, R.; Taube, M.; Kozak, M.; Ożyhar, A.; Greb-Markiewicz, B. Intrinsic Disorder of the C-Terminal Domain of Drosophila Methoprene-Tolerant Protein. PLoS ONE 2016, 11, e0162950. [Google Scholar] [CrossRef] [PubMed]
- Broadus, J.; McCabe, J.R.; Endrizzi, B.; Thummel, C.S.; Woodard, C.T. The Drosophila beta FTZ-F1 orphan nuclear receptor provides competence for stage-specific responses to the steroid hormone ecdysone. Mol. Cell 1999, 3, 143–149. [Google Scholar] [CrossRef]
- Fuxreiter, M.; Simon, I.; Bondos, S. Dynamic protein–DNA recognition: Beyond what can be seen. Trends Biochem. Sci. 2011, 36, 415–423. [Google Scholar] [CrossRef]
- Guo, X.; Bulyk, M.L.; Hartemink, A.J. Intrinsic disorder within and flanking the DNA-binding domains of human transcription factors. Biocomputing 2012, 104–115. [Google Scholar] [CrossRef]
- Roschger, C.; Schubert, M.; Regl, C.; Andosch, A.; Marquez, A.; Berger, T.; Huber, C.G.; Lütz-Meindl, U.; Cabrele, C. The Recombinant Inhibitor of DNA Binding Id2 Forms Multimeric Structures via the Helix-Loop-Helix Domain and the Nuclear Export Signal. Int. J. Mol. Sci. 2018, 19, 1105. [Google Scholar] [CrossRef]
- Eliezer, D. Biophysical characterization of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2009, 19, 23–30. [Google Scholar] [CrossRef]
- Jensen, M.R.; Zweckstetter, M.; Huang, J.; Blackledge, M. Exploring Free-Energy Landscapes of Intrinsically Disordered Proteins at Atomic Resolution Using NMR Spectroscopy. Chem. Rev. 2014, 114, 6632–6660. [Google Scholar] [CrossRef] [PubMed]
- Levine, Z.A.; Larini, L.; LaPointe, N.E.; Feinstein, S.C.; Shea, J.-E. Regulation and aggregation of intrinsically disordered peptides. Proc. Natl. Acad. Sci. USA 2015, 112, 2758–2763. [Google Scholar] [CrossRef] [PubMed]
- Tolkatchev, D.; Plamondon, J.; Gingras, R.; Su, Z.; Ni, F. Recombinant Production of Intrinsically Disordered Proteins for Biophysical and Structural Characterization. In Instrumental Analysis of Intrinsically Disordered Proteins; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; pp. 653–670. [Google Scholar]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef]
- Vacic, V.; Uversky, V.N.; Dunker, A.K.; Lonardi, S. Composition Profiler: A tool for discovery and visualization of amino acid composition differences. BMC Bioinform. 2007, 8, 211. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar]
- Cilia, E.; Pancsa, R.; Tompa, P.; Lenaerts, T.; Vranken, W.F. From protein sequence to dynamics and disorder with DynaMine. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Cilia, E.; Pancsa, R.; Tompa, P.; Lenaerts, T.; Vranken, W.F. The DynaMine webserver: Predicting protein dynamics from sequence. Nucleic Acids Res. 2014, 42, W264–W270. [Google Scholar] [CrossRef]
- Uversky, V.N. Size-exclusion chromatography in structural analysis of intrinsically disordered proteins. Methods Mol. Biol. 2012, 896, 179–194. [Google Scholar] [CrossRef]
- Schuck, P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 2000, 78, 1606–1619. [Google Scholar] [CrossRef]
- Greenfield, N.; Fasman, G.D. Computed circular dichroism spectra for the evaluation of protein conformation. Biochemistry 1969, 8, 4108–4116. [Google Scholar] [CrossRef]
- Kikhney, A.G.; Svergun, D.I. A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Lett. 2015, 589, 2570–2577. [Google Scholar] [CrossRef] [PubMed]
- Kachala, M.; Valentini, E.; Svergun, D.I. Application of SAXS for the Structural Characterization of IDPs. In Intrinsically Disordered Proteins Studied by NMR Spectroscopy; Springer: Cham, Switzerland, 2015; pp. 261–289. [Google Scholar]
- Konrat, R. NMR contributions to structural dynamics studies of intrinsically disordered proteins. J. Magn. Reson. 2014, 241, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Solyom, Z.; Schwarten, M.; Geist, L.; Konrat, R.; Willbold, D.; Brutscher, B. BEST-TROSY experiments for time-efficient sequential resonance assignment of large disordered proteins. J. Biomol. NMR 2013, 55, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Kosol, S.; Contreras-Martos, S.; Cedeño, C.; Tompa, P. Structural Characterization of Intrinsically Disordered Proteins by NMR Spectroscopy. Molecules 2013, 18, 10802–10828. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class I | Class II | Type of Signal |
|---|---|---|
| hypoxia-inducible factors (HIF; HIF1-α, HIF2-α, and HIF3-α) | aryl hydrocarbon receptor nuclear translocator (ARNT), also known as HIF1-β and ARNT2 | regulated by hypoxia |
| aryl hydrocarbon receptor (AHR); aryl hydrocarbon receptor repressor (AHRR) | regulated by xenobiotics | |
| single-minded proteins (SIM1 and SIM2) | developmentally regulated | |
| neuronal PAS domain proteins (NPAS) | developmentally regulated | |
| circadian locomotor output cycles protein kaput (CLOCK) | Circadian rhythm proteins (BMAL1 and BMAL2, also known as ARNTL and ARNTL2) | circadian rhythms |
| Form | Protein | Segment | Organism | PDB ID |
|---|---|---|---|---|
| monomers | HIF2-α | PAS-B domain | Homo sapiens | 1P97 |
| ARNT | PAS-B domain | Homo sapiens | 1X0O | |
| dimer | HIF-2a:ARNT | PAS-B domains | Homo sapiens | 2A24 |
| Form | Protein | Segment | Organism | PDB ID |
|---|---|---|---|---|
| monomers | AHR | PAS-A | Mus musculus | 4M4X |
| ARNT | PAS-B | Homo sapiens | 2B02 | |
| HIF1-α | PAS-B | Homo sapiens | 4H6J | |
| dimers | ARNT Homodimer | PAS-B | Homo sapiens | 4EQ1 |
| HIF2-α:ARNT | PAS-B | Homo sapiens | 3F1P | |
| HIF2-α:ARNT with artificial ligand | PAS-B | Homo sapiens | 3F1O | |
| HIF2-α:ARNT | PAS-B | Homo sapiens | 6D0C | |
| ARNT/HIF transcription factor/coactivator complex | PAS-B | Homo sapiens, Mus musculus | 4PKY | |
| ARNT transcription factor/coactivator complex | PAS-B domain | Homo sapiens, Mus musculus | 4LPZ | |
| HIF2-α:ARNT | bHLH; PAS-A; PAS-B | Mus musculus | 4ZP4 | |
| HIF2-α:ARNT with HRE DNA | bHLH; PAS-A; PAS-B | Mus musculus | 4ZPK | |
| HIF1-α:ARNT with HRE DNA | bHLH; PAS-A; PAS-B | Mus musculus | 4ZPR | |
| AHR:ARNT | bHLH; PAS-A | Homo sapiens | 5NJ8 | |
| AHR:ARNT bound to the dioxin response element (DRE) | bHLH; PAS-A | Homo sapiens, Mus musculus | 5V0L | |
| AHRR:ARNT | bHLH; PAS-A; PAS-B | Homo sapiens, Bos taurus | 5Y7Y | |
| NPAS1:ARNT | bHLH; PAS-A; PAS-B | Mus musculus | 5SY5 | |
| NPAS3:ARNT in complex with HRE DNA | bHLH; PAS-A; PAS-B | Mus musculus | 5SY7 | |
| CLOCK-BMAL1 | bHLH | Homo sapiens | 4H10 | |
| CLOCK:BMAL1 | bHLH; PAS-A; PAS-B | Mus musculus | 4F3L |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolonko, M.; Greb-Markiewicz, B. bHLH–PAS Proteins: Their Structure and Intrinsic Disorder. Int. J. Mol. Sci. 2019, 20, 3653. https://doi.org/10.3390/ijms20153653
Kolonko M, Greb-Markiewicz B. bHLH–PAS Proteins: Their Structure and Intrinsic Disorder. International Journal of Molecular Sciences. 2019; 20(15):3653. https://doi.org/10.3390/ijms20153653
Chicago/Turabian StyleKolonko, Marta, and Beata Greb-Markiewicz. 2019. "bHLH–PAS Proteins: Their Structure and Intrinsic Disorder" International Journal of Molecular Sciences 20, no. 15: 3653. https://doi.org/10.3390/ijms20153653
APA StyleKolonko, M., & Greb-Markiewicz, B. (2019). bHLH–PAS Proteins: Their Structure and Intrinsic Disorder. International Journal of Molecular Sciences, 20(15), 3653. https://doi.org/10.3390/ijms20153653