Genetics and genomics of
Populus received attention in many laboratories throughout the world in the past 30–40 years since this genus has become a model system for forest tree species [
28,
29,
39,
40]. Trees play a key role in many terrestrial eco-systems as they are involved in valuable biogeochemical cycles (water, oxygen, and nitrogen). Forests are highly important for carbon sequestration and regulation of CO
2 concentration. In the beginning of the genomics area, basic science was feasible with tree species such as poplar and eucalyptus with small genomes, but challenging with species like beech, oak, spruce, or pine. These species have huge genome sizes with DNA full of retrotransposons, and characterized by large intron sizes and a high number of mini- and microsatellite repeats [
41]. The poplar genome is with about 550 Mbp relatively small in size [
42], and complete genome sequences are available for
P. trichocarpa [
42],
P. euphratica [
43],
P. tremula [
44],
P. deltoides (
https://phytozome.jgi.doe.gov/pz/portal.html #!info?alias=Org_PdeltoidesWV94_er), and partially for
P. pruinosa [
45] and
P. alba [
43].
All different poplar genomes are very similar and collinear to each other, but with particular features like higher number of salt tolerance genes in
P. euphratica [
43]. For
P. trichocarpa, [
42] assumed a quite recent whole-genome duplication event (about 60–65 Mya ago), resulting in about 8000 pairs of duplicated genes. Thus, for many genes, including
SOC1, FUL,
NFP-likes, and
TOZ19, a paralogous copy is available in the
P. trichocarpa genome. This also holds true for the poplar clones used for transformation in this study, INRA 717-1B4 (
P. ×
canescens; [
23,
46,
47,
48]) and W52 (
P. tremula; [
44,
49]). Thus, to obtain clear knockout lines for both functional and physiological analyses, all paralog copies should be targeted by the CRISPR/Cas9 approach.
In poplar, few studies on gene editing by CRISPR/Cas9 have been published in the last four to five years [
22,
23,
30,
50,
51,
52,
53]. All studies have in common that CRISPR/Cas9 has successfully been applied to insert a mutation in one or more genes for knockout (KO) of the poplar target gene. [
53] knocked out all
NST/SND orthologs and confirmed their central role in the secondary cell wall (SCW) formation in wood fibres, xylem ray parenchyma cells and phloem fibers in hybrid aspen. [
50] discovered an epigenetic mechanism that modified anthocyanin biosynthesis in poplar. Following knockout of a histone H3K9 demethylase gene
JMJ25,
MYB182 expression is altered leading to a change of the histone methylation status in the chromatin. Muhr et al. [
51] knocked-out the two transcription factors involved in shaping plant architecture,
BRANCHED1, and
BRANCHED2, and found that in contrast to
Arabidopsis, in poplar
BRANCHED2 plays an even more critical role in bud outgrowth regulation.
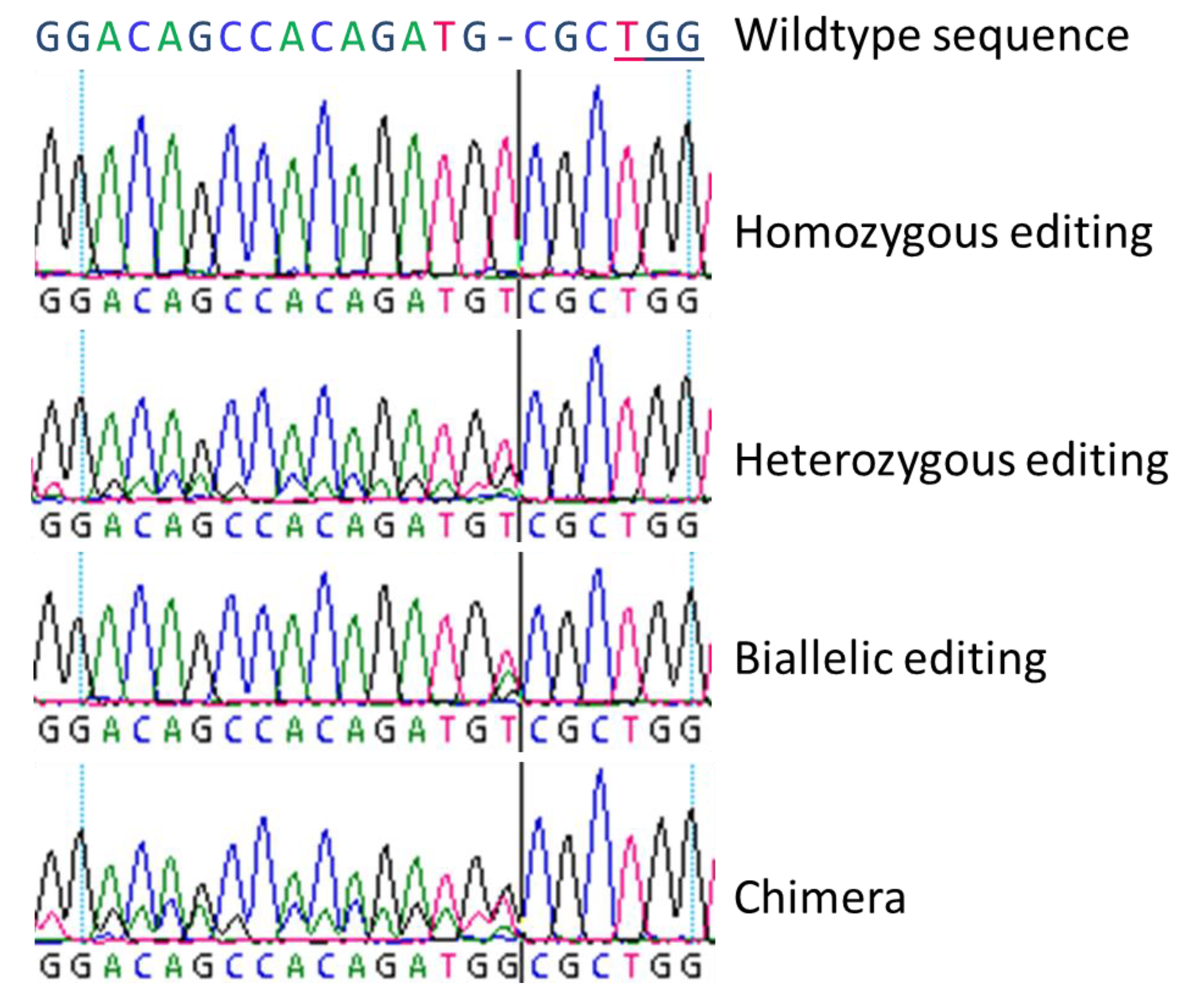
3.1. Biallelic and Homozygous Editings
In general, we observed all three basic types of modifications as described by Fan et al. [
22] by Sanger-sequencing, and we could clearly distinguish homozygous, biallelic, and chimeric lines. Heterozygous mutations could not be revealed by simple Sanger-sequencing since the obtained sequencing results are very similar to chimeric lines with a mix of wildtype and homozygously mutated sequences.
For the nine DNA targets in the twelve different genes included in this study, in total we found 30 biallelic and 44 homozygous editing-based mutations, varying between the different genes. Both, the highest number of biallelic and homozygous editing were found in gene SOC1. However, for three of the twelve DNA targets neither biallelically nor homozygously edited lines were revealed. Especially homozygous genome editing is of special interest in trees, where either long vegetative phases exist or, as in poplar, selfing of parent lines is impossible due to dioecy. As shown for the eight DNA targets under study here, homozygous genome editing occurred and in the majority of cases, by insertion or deletion of just one nucleotide, resulting in a frame shift of the coding region. This causes a more or less precocious stop-codon in the predicted transcript sequence, leading to an incomplete protein.
The sequences revealed the preferable insertion of A/T, that was also reported for plants by Bortesi et al. [
5]. Nucleotide substitutions have not been revealed in all approaches which are published here. This is in accordance with earlier studies [
15] which were summarized by Bortesi et al. [
5], which revealed only rare substitution events except in the case of soybean protoplasts [
55].
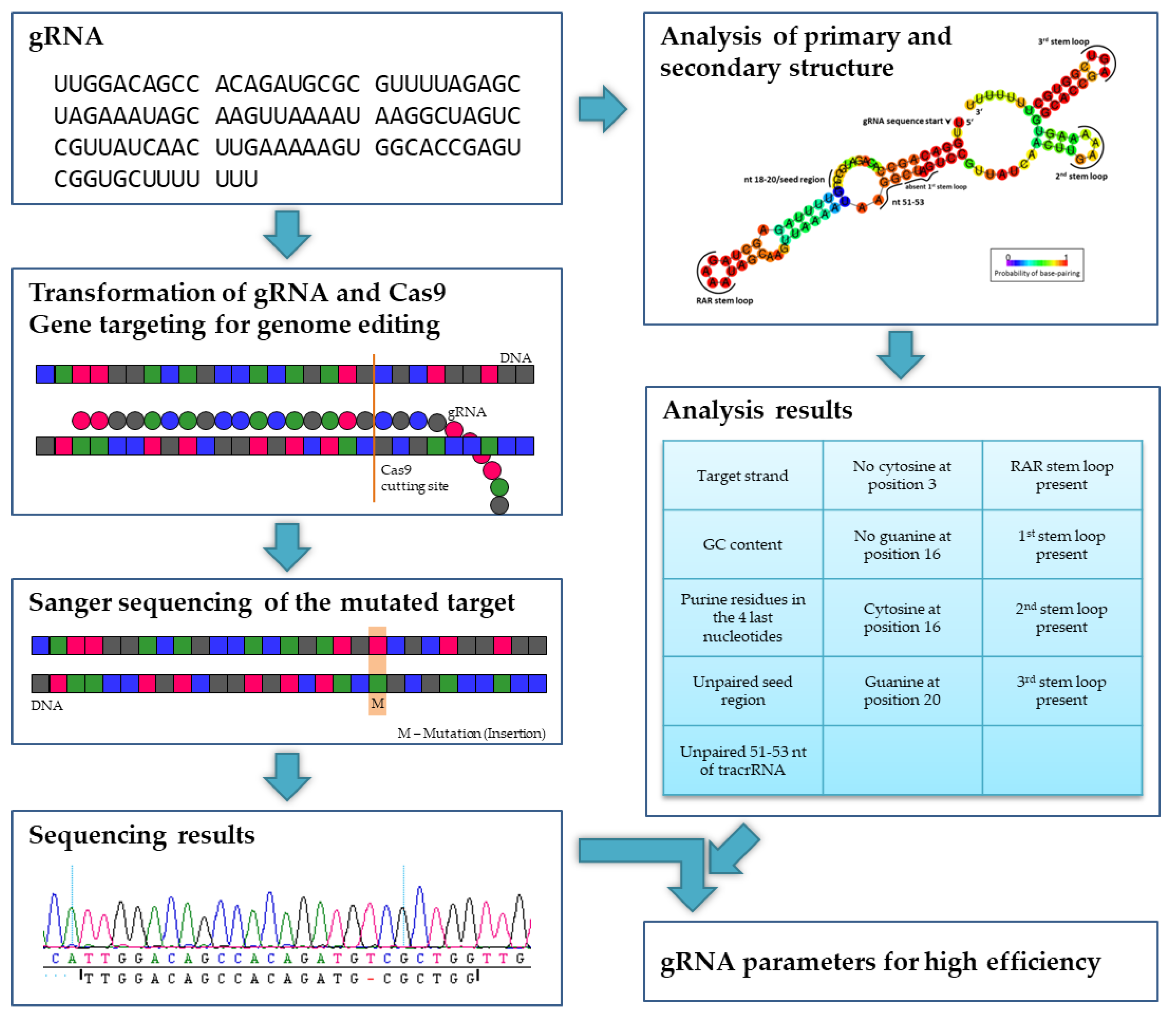
3.2. gRNA Efficiency
Since Doench et al. [
17] found that gRNA structure elements have an impact on cleavage efficiency, a structural analysis was performed with each gRNA to verify the assumption that the tested gRNAs differ in their activity. A GC content between 40% and 60% and purine residues in the four last nucleotides of the gRNA seem to support the editing efficiency argument of [
3]. Also in the perennial
Vitis vinifera, a high GC content improved the CRISPR/Cas9-mediated editing efficiency [
56]. For gRNA secondary structure calculation the bioinformatical tool RNAfold Webserver [
19] was used that is based on the minimum free energy (MFE) algorithm from Zuker and Stiegler [
21] and that utilized the RNA folding parameters from the Andronescu model, 2007 [
20]. Doench et al. [
17] unveiled several determined gRNA locations with an influence on the editing efficiency: Guanine was preferred at nucleotide position 20 of the gRNA and a cytosine was adverse. Furthermore, they reported that no cytosine should be located at position 3, but a cytosine is preferred at position 16, while a guanine at this location has a disadvantageous impact on gRNA efficiency. Wong et al. [
18] further analyzed the dataset of Doench et al. [
17] and reported that nucleotides at position 18–20 are the seed region of the gRNA. This region and the nucleotides located at 51–53 should be unbound and accessible to form efficient gRNAs. If the seed region would bind to position 51–53 of the gRNA, it would be non-functional. The findings from both of these publications can be correlated with the efficiency of the used gRNAs. Comparing between the two DNA single strands, the non-transcribed DNA strand should be targeted by the gRNA for an enhanced efficiency [
3].
For genome editing of the well-known flowering time-determining genes
SOC1 and
FUL, including their paralogous genes, four different gRNAs were designed and integrated by a transgenic approach into poplar plant cells. The obtained transgenic lines were Sanger-sequenced and analyzed in relation to the gRNA efficiency factors mentioned above. For these four gRNAs, it was notable that both gRNA1 and gRNA3 which were targeting the non-transcribed strand of the intended gene worked with higher efficiency than both gRNA2 and gRNA4 which were targeting the transcribed strand. This effect was previously observed in CRISPR/Cas9 approaches in human cells [
16].
The gRNA1 for the editing of SOC1 from poplar clone P1 worked with the highest efficiency. In contrast to the other three gRNAs (gRNA2-4), gRNA1 had the highest GC content (60%) and only partly paired nucleotides 51–53, and gRNA2 revealed the lowest efficiency in this approach. This is comprehensible regarding the target SOC1 Paralog 2, since the gRNA sequence was not completely complementary to the genomic sequence, but contained a heterozygous SNP at position 13 of the gRNA. Regarding target SOC1 Paralog 1, the editing efficiency is indeed better in comparison to Paralog 2 since the gRNA sequence is 100% complementary to the genomic sequence. Compared to gRNA1, the lower efficiency of gRNA2 could be found in the paired nucleotide region 51–53, only one purine residue in the last four nucleotides, and a lower GC content. However, the GC content is still within an acceptable range (40%).
Both gRNA3 and gRNA4 work with a lower efficiency than gRNA1. Between the
AGL8 paralogs targeting gRNAs, gRNA3 had a higher efficiency than gRNA4 although the seed region was paired in gRNA3. In gRNA4, the seed region was only partly paired and all other parameters were identical to gRNA3 (except the target strand). Thus a decreasing editing efficiency by a paired seed region mentioned by Wong et al. [
18] could not be verified here.
NFP-like genes were selected to investigate their impact in poplar to enhance mycorrhization. In
P. trichocarpa, four genes seemed to have an impact on mycorrhizal formation, named as
NFP-like1,
NFP-like2,
NFP-like3, and
NFP-like4. Since the poplar genome was recently duplicated [
42], we had to target two genes simultaneously to obtain a CRISPR/Cas9 induced knockout of the genes. Therefore, we searched for target sequences that were similar in both paralog genes (Pair 1:
NFP-like1 and
NFP-like2, Pair 2:
NFP-like3 and
NFP-like4). For each paralog pair, we used two target sites to enhance CRISPR/Cas9 modifications. Three knockouts were used to determine which of the genes best supports mycorrhiza formation; two single knockouts of the paralog genes and a knockout of all
NFP-like genes. We found that the tested gRNAs differ in their modification rate. For the Paralog Pair 1 containing
NFP-like1 with two gRNAs, we only observed modifications in Target 1. Structural analyses were performed for all gRNAs used under study. Beneficial structures are displayed in
Table 2.
The gRNA with the highest editing efficiency was gRNA5 in
NFP-like1, the beneficial structures of gRNA5 are that it had a freely accessible seed region and the tracrRNA is unbound at position 51–53. Furthermore, at position 3 of gRNA5 is no cytosine and at position 16 no guanine, which are unfavorable. A guanine is located at position 20 of gRNA5, and three out of four nucleotides at the end of the gRNA sequence are purine residues. On the other hand, the gRNA that did not reveal any mutation in the analyzed regenerates was the second
NFP-like1-gRNA6. In this gRNA, one possibility that gRNA6 did not reveal in any modification, could be that the nucleotide 19 of the seed region is paired to nucleotide 51 of the tracrRNA. According Wong et al. [
18], gRNAs that have a paired seed region with nucleotides 51–53 of the tracrRNA were correlated with non-functional gRNAs. In case that gRNA9 had a paired seed region and generated modifications we cannot confirm this finding for the gRNAs used under study. In addition, gRNA6 had no purine residues at the last four nucleotides, so that this can be correlated with non-effective gRNA [
16] and the target strand is the transcribed strand. Although gRNA7 had a bound seed region to the tracrRNA too, modifications were observed at the target. This may be correlated with the target strand, that is the not transcribed strand and with the presence of two purine residues at the end of gRNA7. The used gRNA8 showed sporadic modifications even though it had a paired seed region and paired tracrRNA nucleotides 51–53. In addition, gRNA8 had a lower GC content of 30%, but it still generates modifications. According to gRNA6, it shows that gRNA8 had two purine residues at the end of gRNA sequence. These finding may indicate that we had problems generating a knockout mutant for
NFP-like3 and
NFP-like4.
For the editing of the aspen-sex marker gene
TOZ19, gRNA9 was transformed into poplar clone W52. From a total of 13 lines generated, five lines were homozygously edited, but a further five lines didn’t reveal any editing in the target region. Given the gRNA properties, this result is remarkable because the seed region was partially bound to the region of nucleotides 51–53. According to Wong et al. [
18], this could be an exclusion criterion for functional gRNAs. However, either possibly the inadequate binding of the seed region or the given positive properties of the gRNA (55% GC content plus two purine residues in the last four nucleotides) led to functional integrity.
The significance of the individual required nucleotides in the gRNA sequence proposed by Doench et al. [
17] could not be confirmed. On the contrary, e.g., the required cytosine at position 16 did not seem to have any influence. The gRNAs1, 3, and 5 worked very well but contained no cytosine here. The only gRNA with cytosine at this position was gRNA6, which did not work at all. It cannot be ruled out, however, that the functionality can be influenced by this nucleotide composition on a small scale if all other properties are suitable. In practice, CRISPR/Cas9 is a widely used tool, because apart from an NGG sequence as PAM, no essential requirements are placed on the target sequence.
The presence of gRNA stem loops that was highlighted by Liang et al. [
15] in regard to a cleavage effort could not be verified. All gRNAs contained the RAR stem loop classified as crucial. Differences in the gRNAs allowed conclusions to be drawn regarding the stem loops 1–3: Since gRNA1 worked well without stem loop 1 and gRNA3 without stem loop 2, they could not be crucial for function. The 3
rd stem loop was only absent in gRNA7 that didn’t work satisfactorily, but contained further adverse features, thus the importance of loop 3 could not be clearly determined.
Taking our results together, particular attention should be paid to the GC content of the gRNA, as also recommended by Liang et al. [
15] and Ren et al. [
56], and the need for four purine residues in the last four positions of the gRNA according to Wang et al. [
16], based on their findings in human cells. In agreement with the gRNA sequence, additional attention should then be paid to target the non-transcribed DNA strand and to possible bonds (for example to the seed region) when inserted into the tracrRNA that can be designed by the experimenter.






{kind=link}
{kind=link}