Complement System in Cutaneous Squamous Cell Carcinoma


Abstract
1. Introduction
2. Cutaneous Squamous Cell Carcinoma (cSCC)
2.1. Epidemiology, Clinical Presentation, and Risk Factors of cSCC
2.2. Carcinogenesis and Molecular Alterations in cSCC
3. Complement System
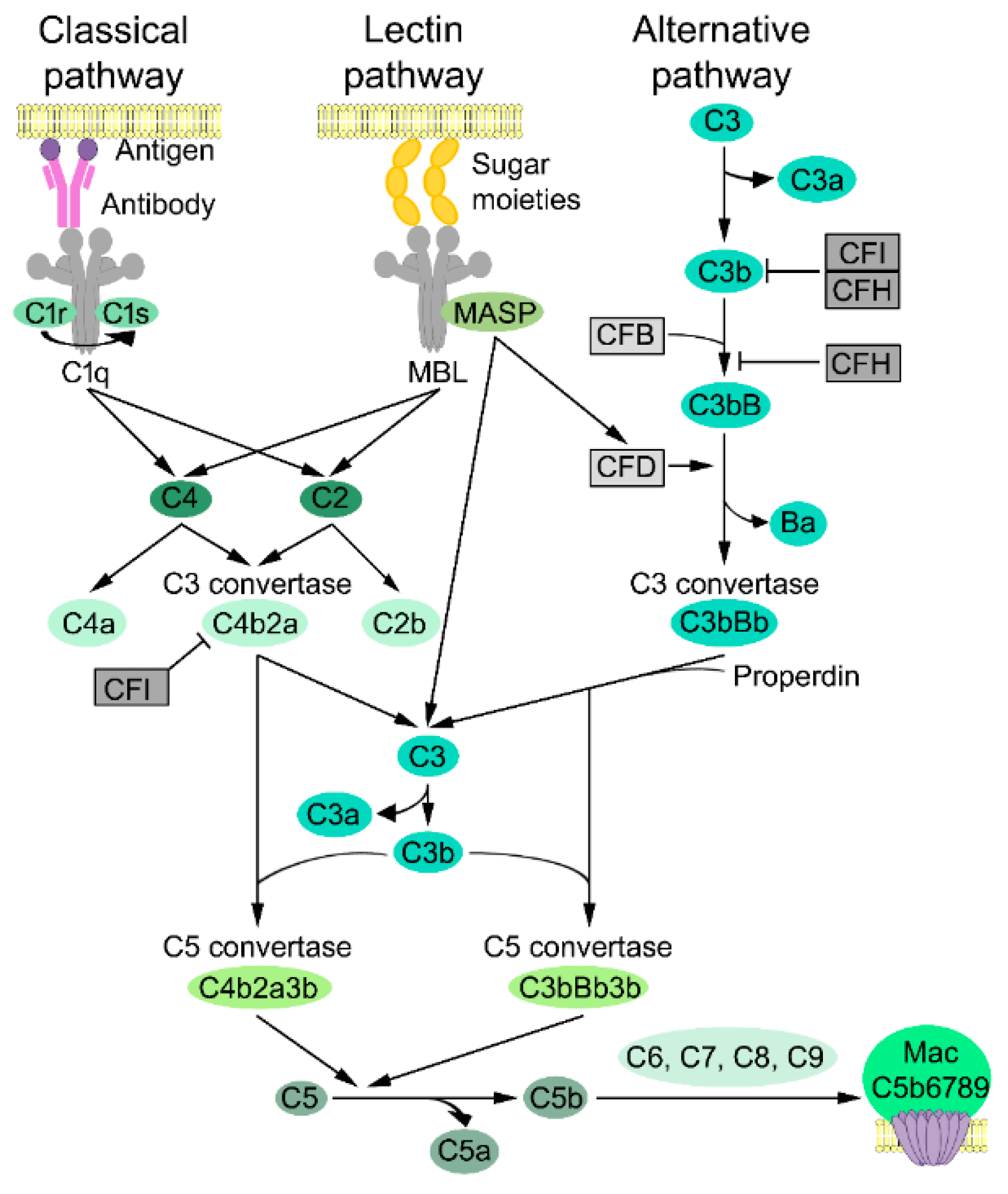
3.1. Complement Activation
3.2. Classical Pathway
3.3. Lectin Pathway
3.4. Alternative Pathway
3.5. Lytic Pathway
3.6. Complement Inhibitors
4. Complement System in cSCC
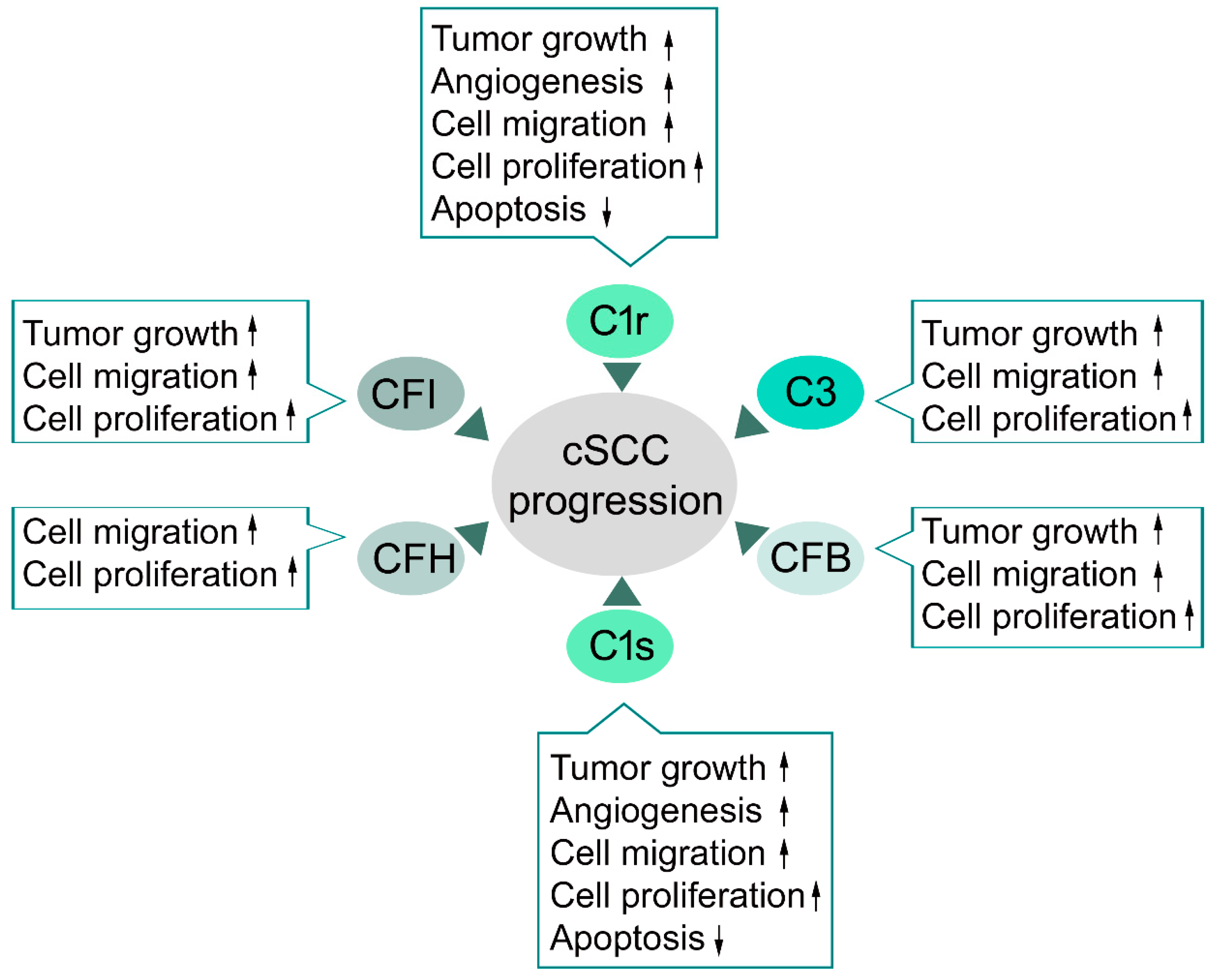
4.1. Complement Components in cSCC
4.2. Classical Pathway in cSCC
4.3. Alternative Pathway in cSCC
4.4. C3 and Terminal Pathway in cSCC
4.5. Complement Inhibitors in cSCC
5. Complement Targeted Cancer Therapy
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Nehal, K.S.; Bichakjian, C.K. Update on keratinocyte carcinomas. N. Engl. J. Med. 2018, 379, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Kivisaari, A.; Kähäri, V.M. Squamous cell carcinoma of the skin: Emerging need for novel biomarkers. World J. Clin. Oncol. 2013, 4, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.Y.; Toland, A.E. High risk cutaneous squamous cell carcinoma of the head and neck. World J. Otorhinolaryngol Head Neck Surg. 2016, 2, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Cho, R.J.; Alexandrov, L.B.; den Breems, N.Y.; Atanasova, V.S.; Farshchian, M.; Purdom, E.; Nguyen, T.N.; Coarfa, C.; Rajapakshe, K.; Prisco, M.; et al. APOBEC mutation drives early-onset squamous cell carcinomas in recessive dystrophic epidermolysis bullosa. Sci. Transl. Med. 2018, 10, eaas9668. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Inman, G.J.; Wang, J.; Nagano, A.; Alexandrov, L.B.; Purdie, K.J.; Taylor, R.G.; Sherwood, V.; Thomson, J.; Hogan, S.; Spender, L.C.; et al. The genomic landscape of cutaneous SCC reveals drivers and a novel azathioprine associated mutational signature. Nat. Commun. 2018, 9, 3667. [Google Scholar] [CrossRef]
- Durinck, S.; Ho, C.; Wang, N.J.; Liao, W.; Jakkula, L.R.; Collisson, E.A.; Pons, J.; Chan, S.W.; Lam, E.T.; Chu, C.; et al. Temporal dissection of tumorigenesis in primary cancers. Cancer Discov. 2011, 1, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Hanna, G.J.; Laga, A.C.; Haddad, R.I.; Lorch, J.H.; Hammerman, P.S. Genomic analysis of metastatic cutaneous squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 1447–1456. [Google Scholar] [CrossRef]
- Pickering, C.R.; Zhou, J.H.; Lee, J.J.; Drummond, J.A.; Peng, S.A.; Saade, R.E.; Tsai, K.Y.; Curry, J.L.; Tetzlaff, M.T.; Lai, S.Y.; et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin. Cancer Res. 2014, 20, 6582–6592. [Google Scholar] [CrossRef]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef]
- Nissinen, L.; Farshchian, M.; Riihilä, P.; Kähäri, V.M. New perspectives on role of tumor microenvironment in progression of cutaneous squamous cell carcinoma. Cell Tissue Res. 2016, 365, 691–702. [Google Scholar] [CrossRef]
- Pio, R.; Corrales, L.; Lambris, J.D. The role of complement in tumor growth. Adv. Exp. Med. Biol. 2014, 772, 229–262. [Google Scholar]
- Afshar-Kharghan, V. The role of the complement system in cancer. J. Clin. Investig. 2017, 127, 780–789. [Google Scholar] [CrossRef]
- Kochanek, D.M.; Ghouse, S.M.; Karbowniczek, M.M.; Markiewski, M.M. Complementing cancer metastasis. Front. Immunol. 2018, 9, 1629. [Google Scholar] [CrossRef]
- Cho, M.S.; Rupaimoole, R.; Choi, H.J.; Noh, K.; Chen, J.; Hu, Q.; Sood, A.K.; Afshar-Kharghan, V. Complement component 3 is regulated by TWIST1 and mediates epithelial-mesenchymal transition. J. Immunol. 2016, 196, 1412–1418. [Google Scholar] [CrossRef]
- Ajona, D.; Ortiz-Espinosa, S.; Pio, R. Complement anaphylatoxins C3a and C5a: Emerging roles in cancer progression and treatment. Semin. Cell Dev. Biol. 2019, 85, 153–163. [Google Scholar] [CrossRef]
- Rogers, H.W.; Weinstock, M.A.; Feldman, S.R.; Coldiron, B.M. Incidence Estimate of Nonmelanoma Skin Cancer (Keratinocyte Carcinomas) in the U.S. Population, 2012. JAMA Derm. 2015, 151, 1081–1086. [Google Scholar] [CrossRef]
- Green, A.C.; Olsen, C.M. Cutaneous squamous cell carcinoma: An epidemiological review. Br. J. Derm. 2017, 177, 373–381. [Google Scholar] [CrossRef]
- Muzic, J.G.; Schmitt, A.R.; Wright, A.C.; Alniemi, D.T.; Zubair, A.S.; Olazagasti Lourido, J.M.; Sosa Seda, I.M.; Weaver, A.L.; Baum, C.L. Incidence and trends of basal cell carcinoma and cutaneous squamous cell carcinoma: A population-based study in Olmsted County, Minnesota, 2000 to 2010. Mayo Clin. Proc. 2017, 92, 890–898. [Google Scholar] [CrossRef]
- Venables, Z.C.; Autier, P.; Nijsten, T.; Wong, K.F.; Langan, S.M.; Rous, B.; Broggio, J.; Harwood, C.; Henson, K.; Proby, C.M.; et al. Nationwide incidence of metastatic cutaneous squamous cell carcinoma in England. JAMA Derm. 2018. [Google Scholar] [CrossRef]
- Karia, P.S.; Han, J.; Schmults, C.D. Cutaneous squamous cell carcinoma: Estimated incidence of disease, nodal metastasis, and deaths from disease in the United States, 2012. J. Am. Acad Derm. 2013, 68, 957–966. [Google Scholar] [CrossRef]
- Schmults, C.D.; Karia, P.S.; Carter, J.B.; Han, J.; Qureshi, A.A. Factors predictive of recurrence and death from cutaneous squamous cell carcinoma: A 10-year, single-institution cohort study. JAMA Derm. 2013, 149, 541–547. [Google Scholar] [CrossRef]
- Nelson, T.G.; Ashton, R.E. Low incidence of metastasis and recurrence from cutaneous squamous cell carcinoma found in a UK population: Do we need to adjust our thinking on this rare but potentially fatal event? J. Surg. Oncol. 2017, 116, 783–788. [Google Scholar] [CrossRef]
- Rowe, D.E.; Carroll, R.J.; Day, C.L. Prognostic factors for local recurrence, metastasis, and survival rates in squamous cell carcinoma of the skin, ear, and lip. Implications for treatment modality selection. J. Am. Acad Derm. 1992, 26, 976–990. [Google Scholar] [CrossRef]
- Alam, M.; Ratner, D. Cutaneous squamous-cell carcinoma. N. Engl. J. Med. 2001, 344, 975–983. [Google Scholar] [CrossRef]
- Wehner, M.R.; Cidre Serrano, W.; Nosrati, A.; Schoen, P.M.; Chren, M.M.; Boscardin, J.; Linos, E. All-cause mortality in patients with basal and squamous cell carcinoma: A systematic review and meta-analysis. J. Am. Acad. Derm. 2018, 78, 663–672.e3. [Google Scholar] [CrossRef]
- Liang, S.B.; Ohtsuki, Y.; Furihata, M.; Takeuchi, T.; Iwata, J.; Chen, B.K.; Sonobe, H. Sun-exposure- and aging-dependent p53 protein accumulation results in growth advantage for tumour cells in carcinogenesis of nonmelanocytic skin cancer. Virchows Arch. 1999, 434, 193–199. [Google Scholar] [CrossRef]
- Ramos, J.; Villa, J.; Ruiz, A.; Armstrong, R.; Matta, J. UV dose determines key characteristics of nonmelanoma skin cancer. Cancer Epidemiol. Biomark. Prev. 2004, 13, 2006–2011. [Google Scholar]
- Lindelöf, B.; Sigurgeirsson, B.; Gäbel, H.; Stern, R.S. Incidence of skin cancer in 5356 patients following organ transplantation. Br. J. Derm. 2000, 143, 513–519. [Google Scholar]
- Velez, N.F.; Karia, P.S.; Vartanov, A.R.; Davids, M.S.; Brown, J.R.; Schmults, C.D. Association of advanced leukemic stage and skin cancer tumor stage with poor skin cancer outcomes in patients with chronic lymphocytic leukemia. JAMA Derm. 2014, 150, 280–287. [Google Scholar] [CrossRef]
- Brewer, J.D.; Shanafelt, T.D.; Khezri, F.; Sosa Seda, I.M.; Zubair, A.S.; Baum, C.L.; Arpey, C.J.; Cerhan, J.R.; Call, T.G.; Roenigk, R.K.; et al. Increased incidence and recurrence rates of nonmelanoma skin cancer in patients with non-Hodgkin lymphoma: A Rochester Epidemiology Project population-based study in Minnesota. J. Am. Acad. Derm. 2015, 72, 302–309. [Google Scholar] [CrossRef]
- Purdie, K.J.; Proby, C.M.; Rizvi, H.; Griffin, H.; Doorbar, J.; Sommerlad, M.; Feltkamp, M.C.; der Meijden, E.V.; Inman, G.J.; South, A.P.; et al. The role of human papillomaviruses and polyomaviruses in BRAF-Inhibitor induced cutaneous squamous cell carcinoma and benign squamoproliferative lesions. Front. Microbiol. 2018, 9, 1806. [Google Scholar] [CrossRef]
- Levine, D.E.; Karia, P.S.; Schmults, C.D. Outcomes of patients with multiple cutaneous squamous cell carcinomas: A 10-year single-institution cohort study. JAMA Derm. 2015, 151, 1220–1225. [Google Scholar] [CrossRef]
- Parekh, V.; Seykora, J.T. Cutaneous squamous cell carcinoma. Clin. Lab. Med. 2017, 37, 503–525. [Google Scholar] [CrossRef]
- Thompson, A.K.; Kelley, B.F.; Prokop, L.J.; Murad, M.H.; Baum, C.L. Risk factors for cutaneous squamous cell carcinoma recurrence, metastasis, and disease-specific death: A systematic review and meta-analysis. JAMA Derm. 2016, 152, 419–428. [Google Scholar] [CrossRef]
- Baum, C.L.; Wright, A.C.; Martinez, J.C.; Arpey, C.J.; Brewer, J.D.; Roenigk, R.K.; Otley, C.C. A new evidence-based risk stratification system for cutaneous squamous cell carcinoma into low, intermediate, and high risk groups with implications for management. J. Am. Acad. Derm. 2018, 78, 141–147. [Google Scholar] [CrossRef]
- Manyam, B.V.; Garsa, A.A.; Chin, R.I.; Reddy, C.A.; Gastman, B.; Thorstad, W.; Yom, S.S.; Nussenbaum, B.; Wang, S.J.; Vidimos, A.T.; et al. A multi-institutional comparison of outcomes of immunosuppressed and immunocompetent patients treated with surgery and radiation therapy for cutaneous squamous cell carcinoma of the head and neck. Cancer 2017, 123, 2054–2060. [Google Scholar] [CrossRef]
- Motley, R.; Kersey, P.; Lawrence, C. Multiprofessional guidelines for the management of the patient with primary cutaneous squamous cell carcinoma. Br. J. Derm. 2002, 146, 18–25. [Google Scholar] [CrossRef]
- Johnson, T.M.; Rowe, D.E.; Nelson, B.R.; Swanson, N.A. Squamous cell carcinoma of the skin (excluding lip and oral mucosa). J. Am. Acad. Derm. 1992, 26, 467–484. [Google Scholar] [CrossRef]
- Carucci, J.A.; Martinez, J.C.; Zeitouni, N.C.; Christenson, L.; Coldiron, B.; Zweibel, S.; Otley, C.C. In-transit metastasis from primary cutaneous squamous cell carcinoma in organ transplant recipients and nonimmunosuppressed patients: Clinical characteristics, management, and outcome in a series of 21 patients. Derm. Surg. 2004, 30, 651–655. [Google Scholar] [CrossRef]
- Tanvetyanon, T.; Padhya, T.; McCaffrey, J.; Kish, J.A.; Deconti, R.C.; Trotti, A.; Rao, N.G. Postoperative concurrent chemotherapy and radiotherapy for high-risk cutaneous squamous cell carcinoma of the head and neck. Head Neck 2015, 37, 840–845. [Google Scholar] [CrossRef]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with cemiplimab in advanced cutaneous squamous-cell carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef]
- South, A.P.; Purdie, K.J.; Watt, S.A.; Haldenby, S.; den Breems, N.Y.; Dimon, M.; Arron, S.T.; Kluk, M.J.; Aster, J.C.; McHugh, A.; et al. NOTCH1 mutations occur early during cutaneous squamous cell carcinogenesis. J. Investig. Derm. 2014, 134, 2630–2638. [Google Scholar] [CrossRef]
- Leffell, D.J. The scientific basis of skin cancer. J. Am. Acad. Derm. 2000, 42, 18–22. [Google Scholar] [CrossRef]
- Kolev, V.; Mandinova, A.; Guinea-Viniegra, J.; Hu, B.; Lefort, K.; Lambertini, C.; Neel, V.; Dummer, R.; Wagner, E.F.; Dotto, G.P. EGFR signalling as a negative regulator of Notch1 gene transcription and function in proliferating keratinocytes and cancer. Nat. Cell Biol. 2008, 10, 902–911. [Google Scholar] [CrossRef]
- Cammareri, P.; Rose, A.M.; Vincent, D.F.; Wang, J.; Nagano, A.; Libertini, S.; Ridgway, R.A.; Athineos, D.; Coates, P.J.; McHugh, A.; et al. Inactivation of TGFβ receptors in stem cells drives cutaneous squamous cell carcinoma. Nat. Commun. 2016, 7, 12493. [Google Scholar] [CrossRef]
- Karppinen, S.M.; Honkanen, H.K.; Heljasvaara, R.; Riihilä, P.; Autio-Harmainen, H.; Sormunen, R.; Harjunen, V.; Väisänen, M.R.; Väisänen, T.; Hurskainen, T.; et al. Collagens XV and XVIII show different expression and localisation in cutaneous squamous cell carcinoma: Type XV appears in tumor stroma, while XVIII becomes upregulated in tumor cells and lost from microvessels. Exp. Derm. 2016, 25, 348–354. [Google Scholar] [CrossRef]
- Martins, V.L.; Caley, M.P.; Moore, K.; Szentpetery, Z.; Marsh, S.T.; Murrell, D.F.; Kim, M.H.; Avari, M.; McGrath, J.A.; Cerio, R.; et al. Suppression of TGFβ and angiogenesis by type VII collagen in cutaneous SCC. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef]
- Rutkowski, M.J.; Sughrue, M.E.; Kane, A.J.; Ahn, B.J.; Fang, S.; Parsa, A.T. The complement cascade as a mediator of tissue growth and regeneration. Inflamm. Res. 2010, 59, 897–905. [Google Scholar] [CrossRef][Green Version]
- Navratil, J.S.; Watkins, S.C.; Wisnieski, J.J.; Ahearn, J.M. The globular heads of C1q specifically recognize surface blebs of apoptotic vascular endothelial cells. J. Immunol. 2001, 166, 3231–3239. [Google Scholar] [CrossRef]
- Forneris, F.; Wu, J.; Gros, P. The modular serine proteases of the complement cascade. Curr. Opin. Struct. Biol. 2012, 22, 333–341. [Google Scholar] [CrossRef]
- Sim, R.B.; Laich, A. Serine proteases of the complement system. Biochem. Soc. Trans. 2000, 28, 545–550. [Google Scholar] [CrossRef]
- Sim, R.B.; Tsiftsoglou, S.A. Proteases of the complement system. Biochem. Soc. Trans. 2004, 32, 21–27. [Google Scholar] [CrossRef]
- Dunkelberger, J.R.; Song, W.C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef]
- Merops database Release 12.1. Available online: https://www.ebi.ac.uk/merops/ (accessed on 26 April 2019).
- Morgan, B.P.; Gasque, P. Extrahepatic complement biosynthesis: Where, when and why? Clin. Exp. Immunol. 1997, 107, 1–7. [Google Scholar] [CrossRef]
- Janssen, B.J.; Gros, P. Conformational complexity of complement component C3. Adv. Exp. Med. Biol. 2006, 586, 291–312. [Google Scholar]
- Noris, M.; Remuzzi, G. Overview of complement activation and regulation. Semin. Nephrol. 2013, 33, 479–492. [Google Scholar] [CrossRef]
- Wang, H.; Wang, H.S.; Liu, Z.P. Agents that induce pseudo-allergic reaction. Drug Discov. Ther. 2011, 5, 211–219. [Google Scholar] [CrossRef]
- Gros, P.; Milder, F.J.; Janssen, B.J. Complement driven by conformational changes. Nat. Rev. Immunol. 2008, 8, 48–58. [Google Scholar] [CrossRef]
- Kemper, C.; Atkinson, J.P.; Hourcade, D.E. Properdin: Emerging roles of a pattern-recognition molecule. Annu. Rev. Immunol. 2010, 28, 131–155. [Google Scholar] [CrossRef]
- Coulthard, L.G.; Woodruff, T.M. Is the complement activation product C3a a proinflammatory molecule? Re-evaluating the evidence and the myth. J. Immunol. 2015, 194, 3542–3548. [Google Scholar] [CrossRef]
- Davis, A.E. Biological effects of C1 inhibitor. Drug News Perspect. 2004, 17, 439–446. [Google Scholar] [CrossRef]
- Rawal, N.; Pangburn, M.K. Role of the C3b-binding site on C4b-binding protein in regulating classical pathway C5 convertase. Mol. Immunol. 2007, 44, 1105–1114. [Google Scholar] [CrossRef]
- Mamidi, S.; Höne, S.; Kirschfink, M. The complement system in cancer: Ambivalence between tumour destruction and promotion. Immunobiology 2017, 222, 45–54. [Google Scholar] [CrossRef]
- Dovezenski, N.; Billetta, R.; Gigli, I. Expression and localization of proteins of the complement system in human skin. J. Clin. Investig. 1992, 90, 2000–2012. [Google Scholar] [CrossRef]
- Timar, K.K.; Pasch, M.C.; van den Bosch, N.H.; Jarva, H.; Junnikkala, S.; Meri, S.; Bos, J.D.; Asghar, S.S. Human keratinocytes produce the complement inhibitor factor H: Synthesis is regulated by interferon-γ. Mol. Immunol. 2006, 43, 317–325. [Google Scholar] [CrossRef]
- Timar, K.K.; Dallos, A.; Kiss, M.; Husz, S.; Bos, J.D.; Asghar, S.S. Expression of terminal complement components by human keratinocytes. Mol. Immunol. 2007, 44, 2578–2586. [Google Scholar] [CrossRef]
- Timar, K.K.; Junnikkala, S.; Dallos, A.; Jarva, H.; Bhuiyan, Z.A.; Meri, S.; Bos, J.D.; Asghar, S.S. Human keratinocytes produce the complement inhibitor factor I: Synthesis is regulated by interferon-γ. Mol. Immunol. 2007, 44, 2943–2949. [Google Scholar]
- Terui, T.; Ishii, K.; Ozawa, M.; Tabata, N.; Kato, T.; Tagami, H. C3 production of cultured human epidermal keratinocytes is enhanced by IFNγ and TNFα through different pathways. J. Investig. Derm. 1997, 108, 62–67. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Tufaro, A.P.; Chuang, J.C.; Prasad, N.; Chuang, A.; Chuang, T.C.; Fischer, A.C. Molecular markers in cutaneous squamous cell carcinoma. Int. J. Surg. Oncol. 2011, 2011, 231475. [Google Scholar] [CrossRef]
- Lujambio, A.; Akkari, L.; Simon, J.; Grace, D.; Tschaharganeh, D.F.; Bolden, J.E.; Zhao, Z.; Thapar, V.; Joyce, J.A.; Krizhanovsky, V.; et al. Non-cell-autonomous tumor suppression by p53. Cell 2013, 153, 449–460. [Google Scholar] [CrossRef]
- Trinchieri, G. Cancer and inflammation: An old intuition with rapidly evolving new concepts. Annu. Rev. Immunol. 2012, 30, 677–706. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Mantovani, A. Cancer-related inflammation: Common themes and therapeutic opportunities. Semin. Cancer Biol. 2012, 22, 33–40. [Google Scholar]
- Eder, K.; Kalman, B. The dynamics of interactions among immune and glioblastoma cells. Neuromolecular Med. 2015, 17, 335–352. [Google Scholar]
- Rutkowski, M.J.; Sughrue, M.E.; Kane, A.J.; Mills, S.A.; Parsa, A.T. Cancer and the complement cascade. Mol. Cancer Res. 2010, 8, 1453–1465. [Google Scholar] [CrossRef]
- Gunn, L.; Ding, C.; Liu, M.; Ma, Y.; Qi, C.; Cai, Y.; Hu, X.; Aggarwal, D.; Zhang, H.G.; Yan, J. Opposing roles for complement component C5a in tumor progression and the tumor microenvironment. J. Immunol. 2012, 189, 2985–2994. [Google Scholar]
- Reis, E.S.; Mastellos, D.C.; Ricklin, D.; Mantovani, A.; Lambris, J.D. Complement in cancer: Untangling an intricate relationship. Nat. Rev. Immunol. 2018, 18, 5–18. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hoste, E.; Arwert, E.N.; Lal, R.; South, A.P.; Salas-Alanis, J.C.; Murrell, D.F.; Donati, G.; Watt, F.M. Innate sensing of microbial products promotes wound-induced skin cancer. Nat. Commun. 2015, 6, 5932. [Google Scholar] [CrossRef]
- Riihilä, P.; Nissinen, L.; Ala-Aho, R.; Kallajoki, M.; Grénman, R.; Meri, S.; Peltonen, S.; Peltonen, J.; Kähäri, V.M. Complement Factor H - a biomarker for progression of cutaneous squamous cell carcinoma. J. Investig. Derm. 2014, 134, 498–506. [Google Scholar] [CrossRef]
- Riihilä, P.; Nissinen, L.; Farshchian, M.; Kivisaari, A.; Ala-Aho, R.; Kallajoki, M.; Grénman, R.; Meri, S.; Peltonen, S.; Peltonen, J.; et al. Complement factor I promotes progression of cutaneous squamous cell carcinoma. J. Investig. Derm. 2015, 135, 579–588. [Google Scholar] [CrossRef]
- Riihilä, P.; Nissinen, L.; Farshchian, M.; Kallajoki, M.; Kivisaari, A.; Meri, S.; Grénman, R.; Peltonen, S.; Peltonen, J.; Pihlajaniemi, T.; et al. Complement component C3 and complement factor B promote growth of cutaneous squamous cell carcinoma. Am. J. Pathol. 2017, 187, 1186–1197. [Google Scholar] [CrossRef]
- Riihilä, P.; Viiklepp, K.; Nissinen, L.; Farshchian, M.; Kallajoki, M.; Kivisaari, A.; Meri, S.; Peltonen, J.; Peltonen, S.; Kähäri, V.M. Tumor cell-derived complement components C1r and C1s promote growth of cutaneous squamous cell carcinoma. Br. J. Derm. 2019. [Google Scholar] [CrossRef]
- Bulla, R.; Tripodo, C.; Rami, D.; Ling, G.S.; Agostinis, C.; Guarnotta, C.; Zorzet, S.; Durigutto, P.; Botto, M.; Tedesco, F. C1q acts in the tumour microenvironment as a cancer-promoting factor independently of complement activation. Nat. Commun. 2016, 7, 10346. [Google Scholar] [CrossRef]
- Bareke, H.; Akbuga, J. Complement system’s role in cancer and its therapeutic potential in ovarian cancer. Scand. J. Immunol. 2018, 88, e12672. [Google Scholar] [CrossRef]
- Liszewski, M.K.; Kolev, M.; Le Friec, G.; Leung, M.; Bertram, P.G.; Fara, A.F.; Subias, M.; Pickering, M.C.; Drouet, C.; Meri, S.; et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity 2013, 39, 1143–1157. [Google Scholar] [CrossRef]
- Markiewski, M.M.; Lambris, J.D. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am. J. Pathol. 2007, 171, 715–727. [Google Scholar] [CrossRef]
- Nunez-Cruz, S.; Gimotty, P.A.; Guerra, M.W.; Connolly, D.C.; Wu, Y.Q.; DeAngelis, R.A.; Lambris, J.D.; Coukos, G.; Scholler, N. Genetic and pharmacologic inhibition of complement impairs endothelial cell function and ablates ovarian cancer neovascularization. Neoplasia 2012, 14, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Nitta, H.; Murakami, Y.; Wada, Y.; Eto, M.; Baba, H.; Imamura, T. Cancer cells release anaphylatoxin C5a from C5 by serine protease to enhance invasiveness. Oncol. Rep. 2014, 32, 1715–1719. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.S.; Vasquez, H.G.; Rupaimoole, R.; Pradeep, S.; Wu, S.; Zand, B.; Han, H.D.; Rodriguez-Aguayo, C.; Bottsford-Miller, J.; Huang, J.; et al. Autocrine effects of tumor-derived complement. Cell Rep. 2014, 6, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Piao, C.; Cai, L.; Qiu, S.; Jia, L.; Song, W.; Du, J. Complement 5a enhances hepatic metastases of colon cancer via monocyte chemoattractant protein-1-mediated inflammatory cell infiltration. J. Biol Chem 2015, 290, 10667–10676. [Google Scholar] [CrossRef] [PubMed]
- Boire, A.; Zou, Y.; Shieh, J.; Macalinao, D.G.; Pentsova, E.; Massagué, J. Complement Component 3 Adapts the Cerebrospinal Fluid for Leptomeningeal Metastasis. Cell 2017, 168, 1101–1113.e13. [Google Scholar] [CrossRef] [PubMed]
- Medler, T.R.; Murugan, D.; Horton, W.; Kumar, S.; Cotechini, T.; Forsyth, A.M.; Leyshock, P.; Leitenberger, J.J.; Kulesz-Martin, M.; Margolin, A.A.; et al. Complement C5a fosters fquamous carcinogenesis and limits T cell response to chemotherapy. Cancer Cell 2018, 34, 561–578.e6. [Google Scholar] [CrossRef] [PubMed]
- Sakiyama, H.; Inaba, N.; Toyoguchi, T.; Okada, Y.; Matsumoto, M.; Moriya, H.; Ohtsu, H. Immunolocalization of complement C1s and matrix metalloproteinase 9 (92kDa gelatinase/type IV collagenase) in the primary ossification center of the human femur. Cell Tissue Res. 1994, 277, 239–245. [Google Scholar] [CrossRef]
- Satyam, A.; Kannan, L.; Matsumoto, N.; Geha, M.; Lapchak, P.H.; Bosse, R.; Shi, G.P.; Dalle Lucca, J.J.; Tsokos, M.G.; Tsokos, G.C. Intracellular Activation of complement 3 is responsible for intestinal tissue damage during mesenteric ischemia. J. Immunol. 2017, 198, 788–797. [Google Scholar] [CrossRef]
- Ricklin, D.; Mastellos, D.C.; Reis, E.S.; Lambris, J.D. The renaissance of complement therapeutics. Nat. Rev. Nephrol. 2018, 14, 26–47. [Google Scholar] [CrossRef]
- Clinical Trials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03665129?term=STELLAR-001&rank=1 (accessed on 12 February 2019).
- Corrales, L.; Ajona, D.; Rafail, S.; Lasarte, J.J.; Riezu-Boj, J.I.; Lambris, J.D.; Rouzaut, A.; Pajares, M.J.; Montuenga, L.M.; Pio, R. Anaphylatoxin C5a creates a favorable microenvironment for lung cancer progression. J. Immunol. 2012, 189, 4674–4683. [Google Scholar] [CrossRef]
- Ajona, D.; Ortiz-Espinosa, S.; Moreno, H.; Lozano, T.; Pajares, M.J.; Agorreta, J.; Bértolo, C.; Lasarte, J.J.; Vicent, S.; Hoehlig, K.; et al. A combined PD-1/C5a blockade synergistically protects against lung cancer growth and metastasis. Cancer Discov. 2017, 7, 694–703. [Google Scholar] [CrossRef]
- Nabizadeh, J.A.; Manthey, H.D.; Steyn, F.J.; Chen, W.; Widiapradja, A.; Md Akhir, F.N.; Boyle, G.M.; Taylor, S.M.; Woodruff, T.M.; Rolfe, B.E. The complement C3a receptor contributes to melanoma tumorigenesis by inhibiting neutrophil and CD4+ T cell responses. J. Immunol. 2016, 196, 4783–4792. [Google Scholar] [CrossRef] [PubMed]
- Kwak, J.W.; Laskowski, J.; Li, H.Y.; McSharry, M.V.; Sippel, T.R.; Bullock, B.L.; Johnson, A.M.; Poczobutt, J.M.; Neuwelt, A.J.; Malkoski, S.P.; et al. Complement activation via a C3a receptor pathway alters CD4+ T lymphocytes and mediates lung cancer progression. Cancer Res. 2018, 78, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Pio, R.; Ajona, D.; Lambris, J.D. Complement inhibition in cancer therapy. Semin Immunol 2013, 25, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Bushey, R.T.; Moody, M.A.; Nicely, N.L.; Haynes, B.F.; Alam, S.M.; Keir, S.T.; Bentley, R.C.; Roy Choudhury, K.; Gottlin, E.B.; Campa, M.J.; et al. A therapeutic antibody for cancer, derived from single human B cells. Cell Rep. 2016, 15, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.L.; Pouw, R.B.; Kavanagh, D.; Sun, R.; Ricklin, D. Developments in anti-complement therapy; from disease to clinical trial. Mol. Immunol. 2018, 102, 89–119. [Google Scholar] [CrossRef] [PubMed]
- Morgan, B.P.; Harris, C.L. Complement, a target for therapy in inflammatory and degenerative diseases. Nat. Rev. Drug Discov. 2015, 14, 857–877. [Google Scholar] [CrossRef]
- Ricklin, D.; Barratt-Due, A.; Mollnes, T.E. Complement in clinical medicine: Clinical trials, case reports and therapy monitoring. Mol. Immunol 2017, 89, 10–21. [Google Scholar] [CrossRef]
- Derhaschnig, U.; Gilbert, J.; Jäger, U.; Böhmig, G.; Stingl, G.; Jilma, B. Combined integrated protocol/basket trial design for a first-in-human trial. Orphanet J. Rare Dis. 2016, 4, 134. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03488550 (accessed on 17 December 2018).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03010046 (accessed on 15 August 2018).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02222545 (accessed on 11 January 2019).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/show/NCT02682407 (accessed on 16 January 2018).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/show/NCT03205995 (accessed on 18 October 2018).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03500549 (accessed on 16 May 2019).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03531255 (accessed on 7 September 2018).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03525613 (accessed on 2 October 2018).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03525600 (accessed on 2 October 2018).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/show/NCT03226678 (accessed on 16 April 2019).
- Available online: http://investors.apellis.com/news-releases/news-release-details/apellis-pharmaceuticals-apl-2-receives-orphan-drug-designation (accessed on 20 December 2018).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03694444 (accessed on 29 January 2019).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03439839 (accessed on 1 March 2019).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03373461 (accessed on 24 May 2019).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03815825 (accessed on 3 July 2019).
- Sacks, S.H.; Zhou, W. The role of complement in the early immune response to transplantation. Nat. Rev. Immunol. 2012, 12, 431–442. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/show/NCT03053102 (accessed on 6 December 2018).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01835015 (accessed on 25 March 2016).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/show/NCT02946463 (accessed on 16 May 2019).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/show/NCT03056040 (accessed on 16 May 2019).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/show/NCT02949128 (accessed on 4 February 2019).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/show/NCT02763644 (accessed on 17 April 2019).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01527500 (accessed on 9 April 2019).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/show/NCT02515942 (accessed on 30 May 2019).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/show/NCT02534909 (accessed on 23 April 2019).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/show/NCT01526889 (accessed on 31 October 2018).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03157635 (accessed on 10 July 2019).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/show/NCT02591862 (accessed on 4 June 2018).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/show/NCT03030183 (accessed on 20 June 2019).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/show/NCT03078582 (accessed on 7 September 2018).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/show/NCT02397954 (accessed on 19 June 2019).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/show/NCT02686658 (accessed on 9 November 2018).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/show/NCT02352493 (accessed on 6 September 2017).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03303313 (accessed on 1 October 2018).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02245412 (accessed on 3 January 2019).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02128269 (accessed on 13 July 2017).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02246595 (accessed on 25 April 2016).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02866825 (accessed on 15 February 2017).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03001622 (accessed on 19 September 2017).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/show/NCT02464891 (accessed on 14 November 2017).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/show/NCT02384317 (accessed on 20 December 2016).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03301467 (accessed on 18 June 2019).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/show/NCT02994927 (accessed on 8 July 2019).
- Gallenkamp, J.; Spanier, G.; Wörle, E.; Englbrecht, M.; Kirschfink, M.; Greslechner, R.; Braun, R.; Schäfer, N.; Bauer, R.J.; Pauly, D. A novel multiplex detection array revealed systemic complement activation in oral squamous cell carcinoma. Oncotarget 2018, 9, 3001–3013. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Target | Drug (Company) | Entity | Approved Indication | Clinical Trial Phase [ID] [Ref] | Clinical Trial Indication [Ref] |
|---|---|---|---|---|---|
| C1r, C1s, MASPs | C1 inhibitor (Shire) | Protein | HAE | I [NCT02435732] [100] III [NCT02547220] [100] | Renal transplantation [100] |
| C1 inhibitor (Sanquin) | Protein | HAE | II [NCT02251041] [100] III [NCT01275976] [100] | Liver transplantation [100] Trauma, Sepsis, Multiple organ dysfunction syndrome [100] | |
| C1 inhibitor (CSL Behring) | Protein | HAE | I and II [NCT02134314] [100] II [NCT02936479] [100] | Renal transplantation [100] | |
| Conestat alpha (rhC1 inhibitor) (Pharming) | Protein | HAE | II [NCT02869347] [100] | Contrast-induced nephropathy [100] | |
| C1s | BIVV009/TNT009 (Bioverativ) | Antibody | N/A | I [NCT02502903] [100] | BP, CAD, WAIHA, ESRD [100,111] |
| C1q | ANX007 (Annexon) | Antibody | N/A | I [NCT03488550] [112] | Open-angle glaucoma [112] |
| ANX005 (Annexon) | Antibody | N/A | I [NCT03010046] [113] | Neurodegenerative and autoimmune diseases [113] | |
| MASP-2 1 | OMS721 (Omeros) | Antibody | N/A | II [NCT02222545] [114] II [NCT02682407] [115] III [NCT03205995] [116] | Thrombotic microangiopathies [114] IgA nephropathy, LN, MN, C3G [115] aHUS [116] |
| C3 | APL-2 (Apellis) | Peptide | N/A | I [NCT02588833] [100] I [NCT02264639] [100] | PNH [100,117,118] |
| I [NCT02461771] [100] | AMD [CNV] [100] | ||||
| II [NCT02503332] [100] | GA due to AMD [100,119,120] | ||||
| II [NCT03226678] [121] III [NCT03500549] [117] III [NCT03531255] [118] III [NCT03525613] [119] III [NCT03525600] [120] | WAIHA, CAD [121] IgA nephropathy [122] | ||||
| AMY-101/CP40 (Amyndas) | Peptide | N/A | I [NCT03316521] [100] II [ NCT03694444] [123] | Complement-mediated diseases [100] Periodontal disease [123] | |
| APL-9 (Apellis) | Peptide | N/A | I [ACTRN12616000862448] [100] | PNH [100] | |
| CFB 1 | LNP023 (Novartis) | Small-molecule | N/A | II [NCT03439839] [124] II [NCT03373461] [125] | PNH [124] IgA nephropathy [125] |
| IONIS-FB-LRx (Ionis) | Oligo-nucleotide | N/A | II [NCT03815825] [126] | AMD, GA [126] | |
| C3 convertase | Mirococept (MRC) | Protein | N/A | II [ISRCTN49958194] [127] | Transplantation [127] |
| CFD 1 | Lampalizumab (Genentech & Roche) | Antibody | II [NCT02288559] [100] III [NCT02745119] [100] III [NCT02247531] [100] III [NCT02247479] [100] | GA due to AMD [100] | |
| ACH-4471 (Achillion) | Small-molecule | N/A | II [NCT03053102] [128] | PNH [128] | |
| CFP 1 | CLG561 (Novartis) | Antibody | N/A | I [NCT01835015] [129] II [NCT02515942] {combination therapy with Tesidolumab} [100] | AMD [129] AMD, GA [100] |
| C5 | Eculizumab (Alexion) | Antibody | PNH, aHUS, gMG | II [NCT01303952] [100] | CAD [100] |
| II [NCT02093533] [100] | MPGN [100] | ||||
| II [NCT02493725] [100] | Guillain-Barre syndrome [100] | ||||
| II [NCT01567085] [100] II [NCT01919346] [100] II [NCT01895127] [100] II [NCT01399593] [100] I and II [NCT01106027] [100] II and III [NCT02145182] [100] | Renal transplantation [100] | ||||
| III [NCT02205541] [100] | STEC-HUS [100] | ||||
| III [NCT02301624] [100] III [NCT01997229] [100] | gMG [100] | ||||
| III [NCT01892345] [100] | Neuromyelitis optica [100] | ||||
| Ravulizumab/ALXN1210 (Alexion) | Antibody | N/A | I and II [NCT02598583] [100] II [NCT02605993] [100] III [NCT02946463] [130] III [NCT03056040] [131] III [NCT02949128] [132] | PNH 1 [100,130,131] aHUS 1 [132] | |
| Tesidolumab/LFG316 (Novartis, MorphoSys) | Antibody | N/A | I [NCT02878616] [100] II [NCT02763644] [133] II [NCT01527500] [134] II [NCT02515942] {combination therapy with CLG561} [135] II [NCT02534909] [136] II [NCT01526889] [137] | Renal transplantation [100] Transplant-associated microangiopathy [133] AMD 1 [134] GA 1 [135] PNH 1 [136] Uveitis [137] | |
| SKY59/RG6107/RO7112689 (Chugai, Roche) | Antibody | N/A | I and II [NCT03157635] [138] | PNH 1 [138] | |
| REGN3918 (Regeneron) | Antibody | N/A | I [NCT03115996] [100] | PNH 1 [100] | |
| Coversin (Akari) | Protein | N/A | II [NCT02591862] [139] | PNH 1 [139] | |
| RA101495 (Ra Pharma) | Peptide | N/A | II [NCT03030183] [140] II [NCT03078582] [141] | PNH 1 [140,141] | |
| Zimura/ARC1905 (Ophthotech) | Oligo-nucleotide | N/A | II [NCT02397954] [142] II to III [NCT02686658] [143] | IPCV 1 [142] AMD 1 [143] | |
| Cemdisiran */ALN-CC5 (Alnylam) | Oligo-nucleotide | N/A | I and II [NCT02352493] [144] II [NCT03303313] [145] | PNH 1 [144] aHUS 1 [145] | |
| C5a | ALXN1007 (Alexion) | Antibody | N/A | II [NCT02245412] [146] II [NCT02128269] [147] | GVHD 1 [146] APS 1 [147] |
| IFX-1 (InflaRx) | Antibody | N/A | II [NCT02246595] [148] II [NCT02866825] [149] II [NCT03001622] [150] | Sepsis, Septic shock [148] SIRS 1, Complex cardiac surgery [149] Hidradenitis suppurativa [150] | |
| C5aR1 1 | Avacopan/CCX168 (ChemoCentryx) | Small molecule | N/A | II [NCT02464891] [151] II [NCT02384317] [152] II [NCT03301467] [153] III [NCT02994927] [154] | aHUS 1 [151] IgA nephropathy [152] C3G 1 [153] AAV 1 [154] |
| IPH5401 (Innate Pharma) | Antibody | N/A | I [NCT03665129] {combination therapy with durvalumab} [101] | Advanced solid tumors [101] |
| Target | Drug (Company) | Entity | Phase of Development | Indication |
|---|---|---|---|---|
| C1s | BIVV020 (Bioverativ) | Antibody | Preclinical [100] | CAD 1 [100] |
| C2 | PRO-02 (Broteio/Argen-x) | Antibody | Preclinical [100] | Ischemia-reperfusion injury [100] |
| C3 | AMY-103 (Amyndas) | Peptide | Preclinical [100] | Not available |
| C3aR 1 | SB290157 | Small molecule | Preclinical | Melanoma [104], lung cancer [105] |
| CFB 1 | Bikaciomab (Novelmed) | Antibody | Preclinical [109] | AMD 1 [109] |
| C3 convertase | AMY-201/Mini-FH (Amyndas) | Protein | Preclinical [100] | Not available |
| CFD 1 | CFD inhibitor (Novartis) | Small molecule | Preclinical [100] | Not available |
| CFD inhibitor (Ra Pharma) | Peptide | Preclinical [100] | AMD 1, GA 1, Orphan renal diseases [100] | |
| ACH-5228 (Achillion) | Small molecule | Preclinical [100] | AMD, GA, C3G, IC-MPGN [100] | |
| MASP-3 1 | OMS906 (Omeros) | Antibody | Preclinical [100] | AP-mediated diseases [100] |
| CFP 1 | NM9401 (Novelmed) | Antibody | Preclinical [109] | PNH 1 [109] |
| CFH 1 | 5C6/Compsorbin (Amyndas) | Peptide | Preclinical [100] | Inflammation due to transplant and/or biomaterial [100] |
| C5 | SOBI005 (Sobi) | Protein | N/A Preclinical [100] | C5-mediated diseases [100] |
| C5 inhibitor (Ra Pharma) | Peptide | N/A Preclinical [100] | PNH 1, gMG 1, LN 1, CNS 1 diseases [100] | |
| Long-acting coversin (Akari) | Protein | N/A Preclinical [100] | Not available | |
| Mubodina (Adienne) | Antibody | N/A Preclinical [100] | Typical hemolytic uremic syndrome [100] | |
| C5a | IFX-2, IFX-3 (InflaRx) | Antibody | N/A Preclinical [100] | Not available |
| NOX-D19 to NOX-D21 | L-RNA Aptamer (Spiegelmer) | N/A Preclinical [109] | Sepsis and transplant rejection [109] | |
| C5aR1 1 | DF2593A (Dompe) | Small molecule | N/A Preclinical [100] | Inflammatory and neuropathic pain [100] |
| PMX-53 | Peptide | N/A Preclinical | SCC 1 [97] | |
| AcF-(OPdChaWR) | Peptide | N/A Preclinical | Cervical cancer [88] | |
| ALS-205 (Alsonex) | Peptide | N/A Preclinical [100] | ALS 1, Alzheimer disease, Huntington disease [100] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riihilä, P.; Nissinen, L.; Knuutila, J.; Rahmati Nezhad, P.; Viiklepp, K.; Kähäri, V.-M. Complement System in Cutaneous Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019, 20, 3550. https://doi.org/10.3390/ijms20143550
Riihilä P, Nissinen L, Knuutila J, Rahmati Nezhad P, Viiklepp K, Kähäri V-M. Complement System in Cutaneous Squamous Cell Carcinoma. International Journal of Molecular Sciences. 2019; 20(14):3550. https://doi.org/10.3390/ijms20143550
Chicago/Turabian StyleRiihilä, Pilvi, Liisa Nissinen, Jaakko Knuutila, Pegah Rahmati Nezhad, Kristina Viiklepp, and Veli-Matti Kähäri. 2019. "Complement System in Cutaneous Squamous Cell Carcinoma" International Journal of Molecular Sciences 20, no. 14: 3550. https://doi.org/10.3390/ijms20143550
APA StyleRiihilä, P., Nissinen, L., Knuutila, J., Rahmati Nezhad, P., Viiklepp, K., & Kähäri, V.-M. (2019). Complement System in Cutaneous Squamous Cell Carcinoma. International Journal of Molecular Sciences, 20(14), 3550. https://doi.org/10.3390/ijms20143550