Metabolic Syndrome and Salt-Sensitive Hypertension in Polygenic Obese TALLYHO/JngJ Mice: Role of Na/K-ATPase Signaling

 , , ,
, , , 



Abstract
1. Introduction
2. Results
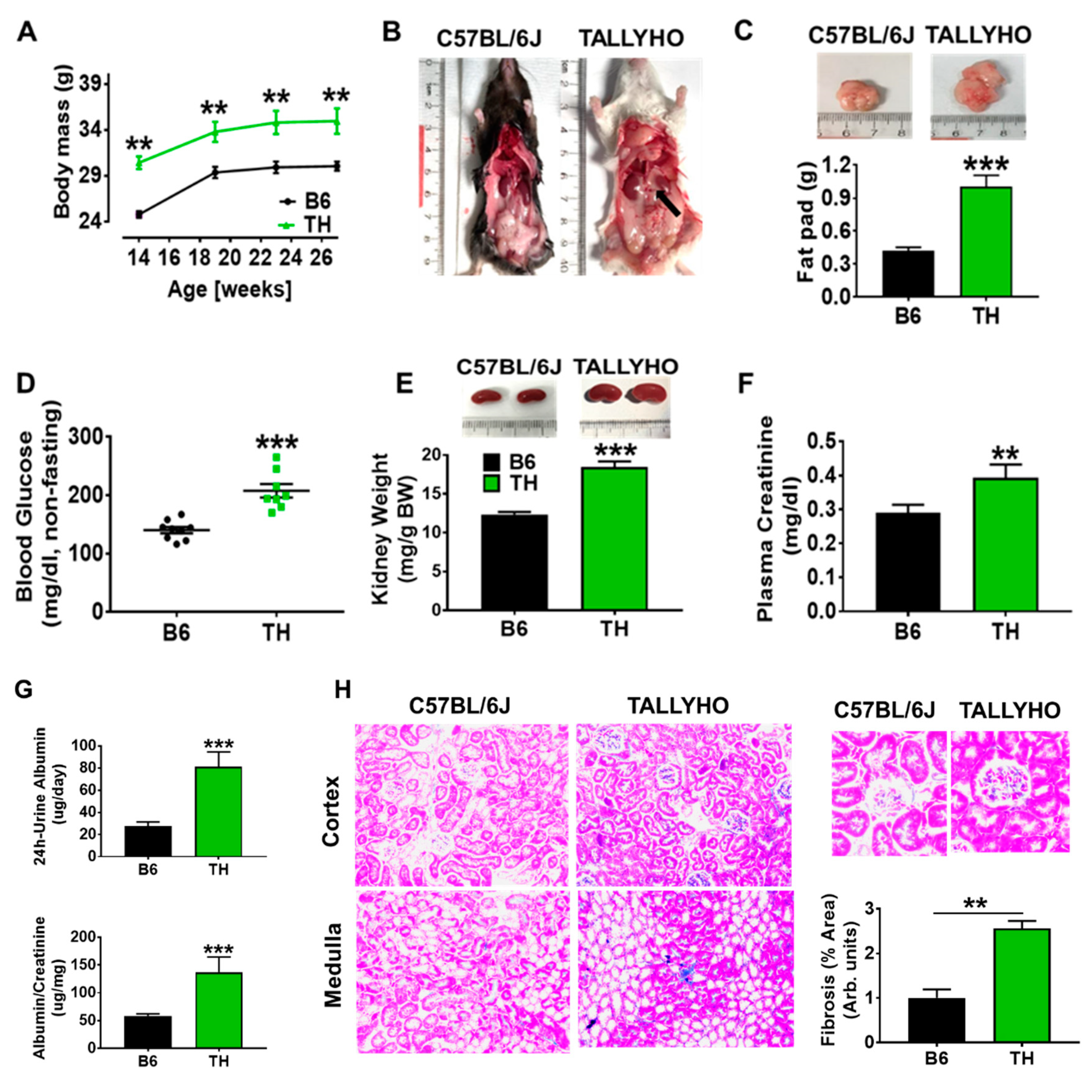
2.1. TH Mouse Characteristics
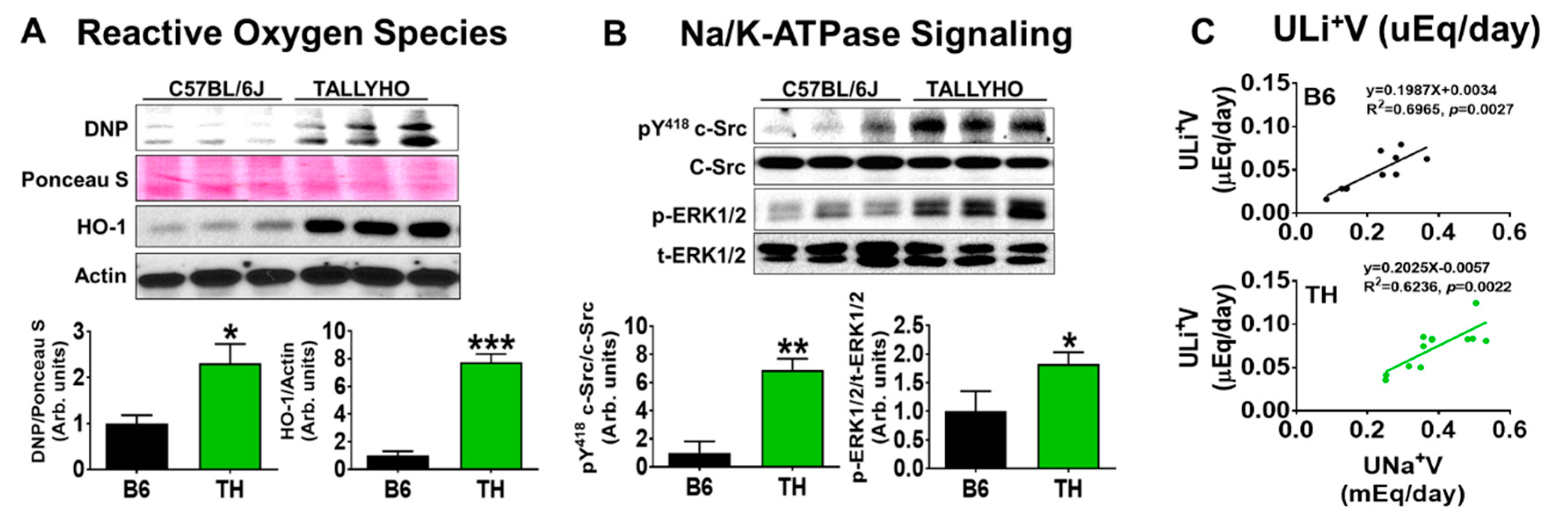
2.2. Systolic Blood Pressure, Renal Na+ Excretion, and Na/K-ATPase Signaling with Normal Chow in Obese TH Mice
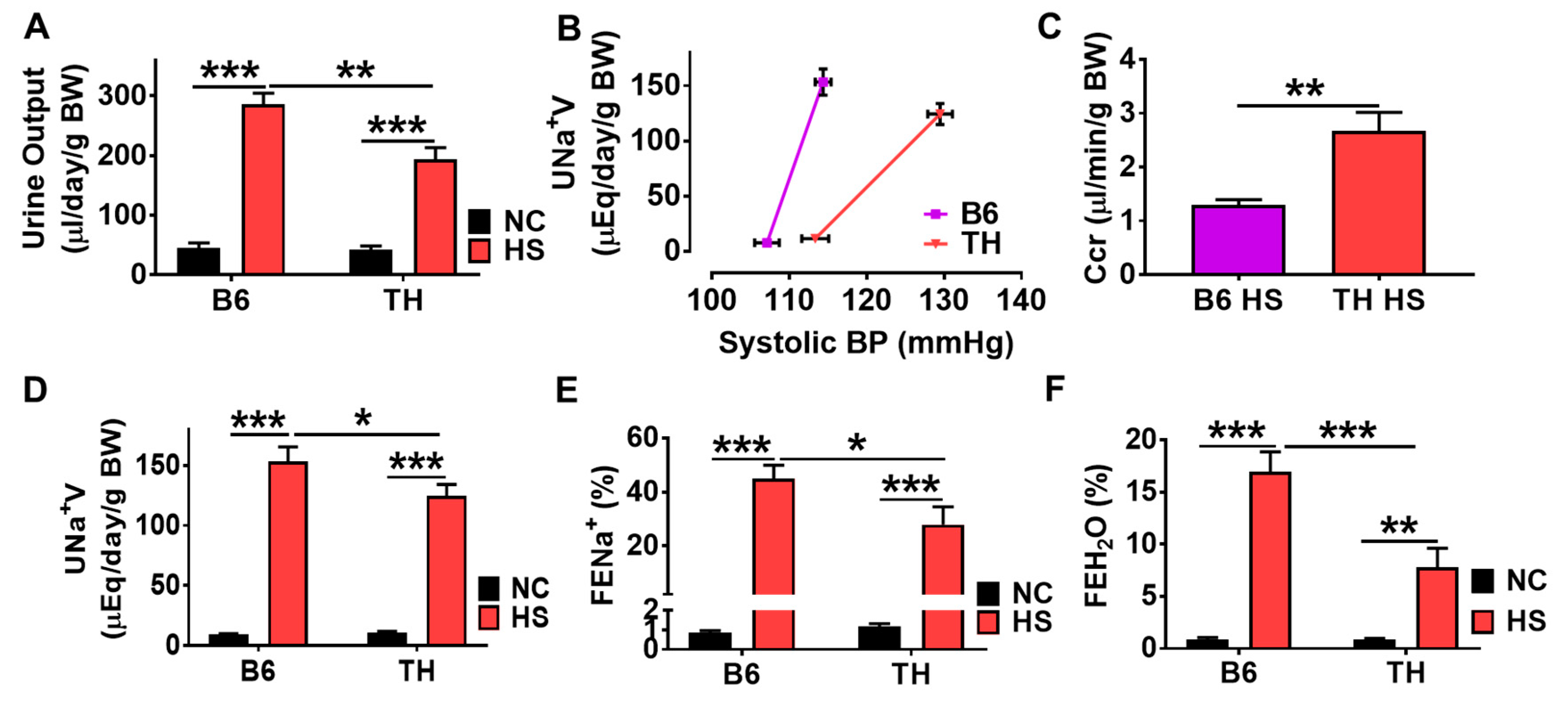
2.3. Systolic Blood Pressure and Fluid Retention with High Salt Diet in Obese TH Mice
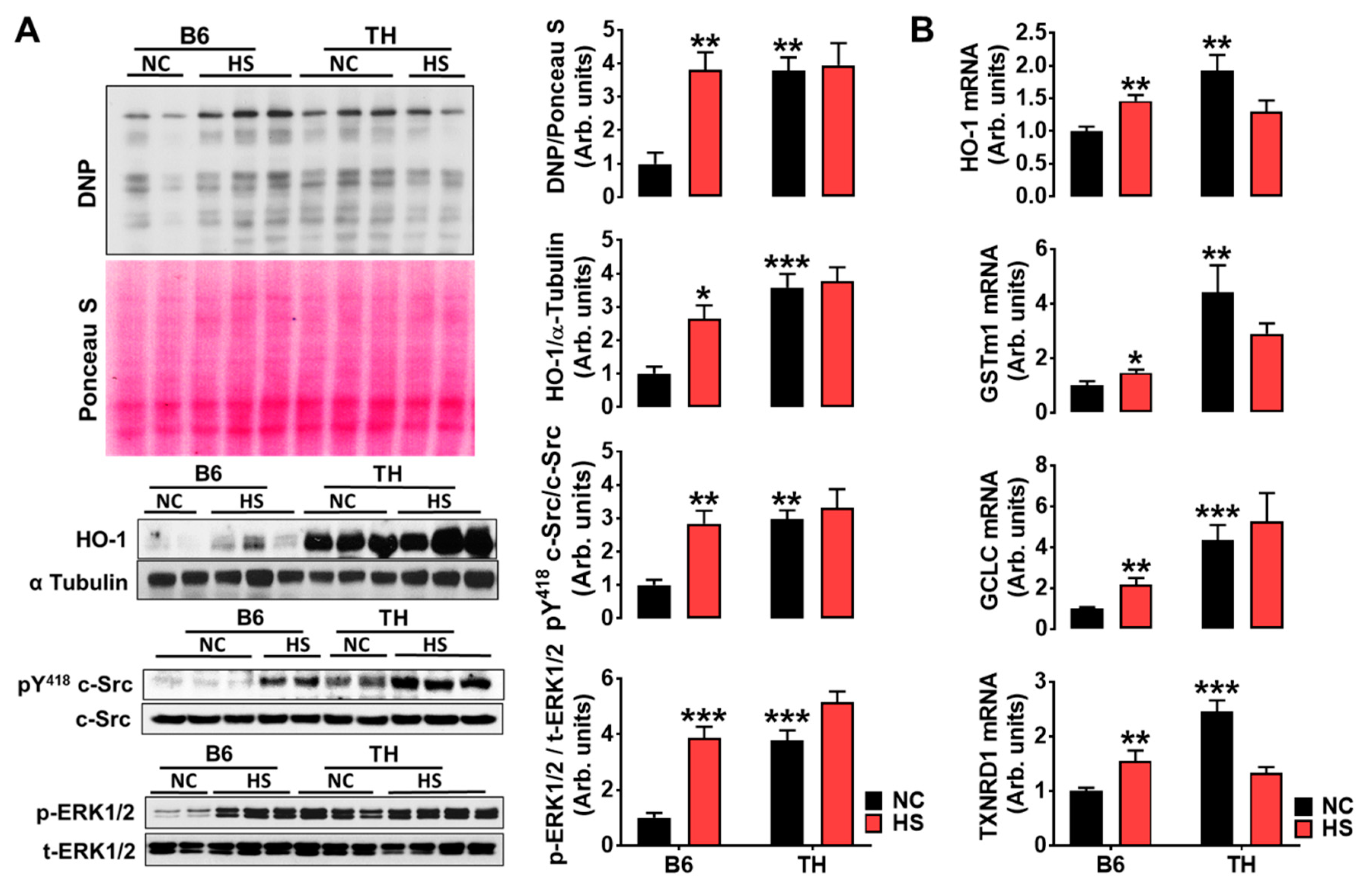
2.4. Impaired Renal Na+ Excretion and Na/K-ATPase Signaling with High Salt Diet in Obese TH Mice
3. Discussion
3.1. TH Mouse Appears to Be a Suitable Model to Research the Pathophysiology of Obesity and Metabolic Syndrome-Related Chronic Kidney Disease
3.2. Renal Na/K-ATPase Signaling Malfunction Response to Dietary Salt Contributes to Blunted Na+ and Water Excretion, Leading to Salt-Sensitive Hypertension in Obese TH Mice
3.3. Proximal Na/K-ATPase Signaling Might Be Implicated in Renal Na+ and Water Handling in Both B6 and TH Mice
Perspective
4. Materials and Methods
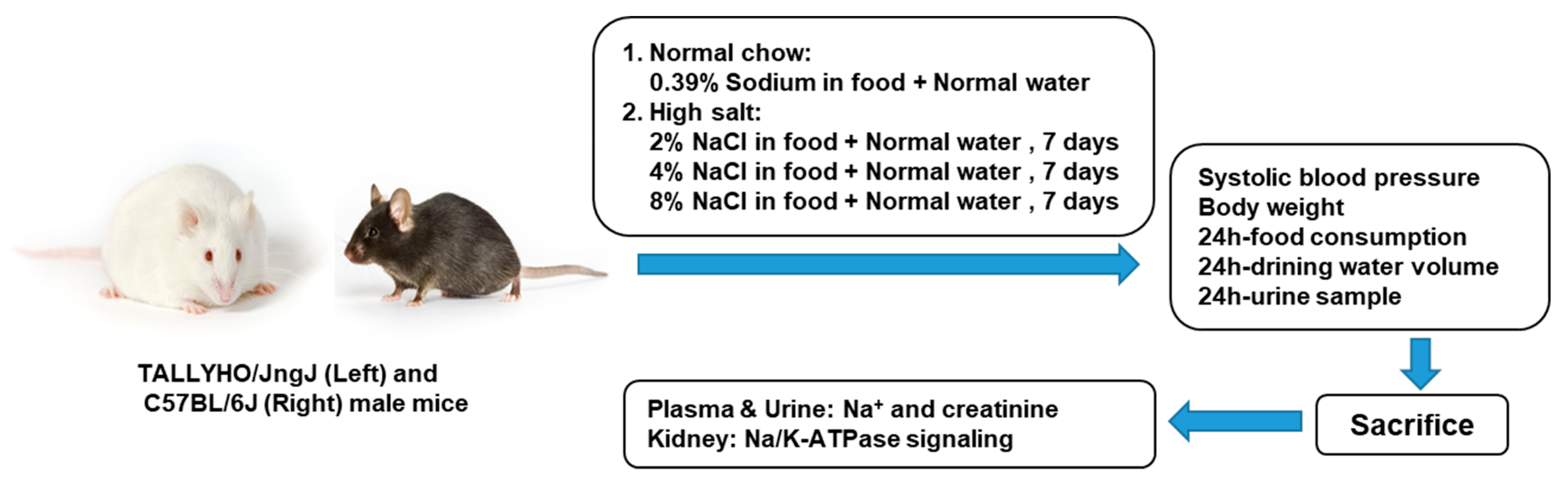
4.1. Experimental Protocol
4.2. Blood Pressure Measurement
4.3. Hematocrit (HCT) Measurement
4.4. Renal Na+ Handling Studies
- Ccr (mL/min) = (Ucr × 24-h urine volume)/(Pcr × 24 × 60)
- UNa+V (mEq/day) = UNa+ × urine flow rate × 24 × 60
- ULi+V (μEq/day) = ULi+ × urine flow rate × 24 × 60
- FENa+ (%) = 100 × [(UNa+ × Pcr)/(PNa+ × Ucr)]
- FEH2O (%) = 100 × [(24 h urine volume/24 × 60)/Ccr]
4.5. Western Blotting
4.6. Histopathological Staining
4.7. Measurements of mRNA Expression (Quantitative Real-Time qRT-PCR)
4.8. Statistics
5. Conclusions
- This study reports associations between renal Na/K-ATPase signaling-mediated Na+ excretion and blood pressure for the first time in polygenic obese TH mice.
- Obese TH mice have normal proximal tubule Na+ handling that is likely driven by activated proximal Na/K-ATPase signaling-mediated natriuresis.
- There is a relationship between dysfunctional Na/K-ATPase signaling and blunted pressure diuresis and natriuresis-induced hypertension in TH mice with a high salt diet.
- TH mice appear to be a suitable mouse model of obesity and metabolic syndrome, offering insight into predisposing factors among individuals in susceptibility to renal injury due to obesity. For example, activated Na/K-ATPase signaling might be a potential predisposing factor.
- When added to the evidence base from the obese TH mice, these results support the novel concept that Na/K-ATPase signaling-mediated Na+ handling is implicated in the pathophysiology of obesity and metabolic imbalance-associated hypertension.
- This novel concept would promote the establishment of a new field of investigation for the etiology of hypertension, explicitly targeting the mechanisms of obesity-induced changes in ROS signaling and renal tubular cell transport.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ROS | Reactive oxygen species |
| TH | TALLYHO/JngJ mouse |
| B6 | C57BL/6J mouse |
| HS | High salt diet |
| NC | Normal chow |
| BP | Blood pressure |
| DR | Dahl salt-resistant rat |
| DS | Dahl salt-sensitive rat |
| WT | Wild-type |
| ULi+V | Urinary lithium excretion rate |
| UNa+V & PNa+ | Urinary Na+ excretion rate and plasma Na+ concentration |
| FENa+ | Fractional Na+ excretion |
| Ccr | Creatinine clearance |
| HCT | Hematocrit |
| CKD | Chronic kidney disease |
| CTS | Cardiotonic steroids |
References
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F.; et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: A systematic analysis for the global burden of disease study 2013. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef]
- Chen, J.; Gu, D.; Huang, J.; Rao, D.C.; Jaquish, C.E.; Hixson, J.E.; Chen, C.S.; Chen, J.; Lu, F.; Hu, D.; et al. Metabolic syndrome and salt sensitivity of blood pressure in non-diabetic people in china: A dietary intervention study. Lancet 2009, 373, 829–835. [Google Scholar] [CrossRef]
- Hall, M.E.; do Carmo, J.M.; da Silva, A.A.; Juncos, L.A.; Wang, Z.; Hall, J.E. Obesity, hypertension, and chronic kidney disease. Int. J. Nephrol. Renov. Dis. 2014, 7, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Nizar, J.M.; Bhalla, V. Molecular mechanisms of sodium-sensitive hypertension in the metabolic syndrome. Curr. Hypertens. Rep. 2017, 19, 60. [Google Scholar] [CrossRef] [PubMed]
- Guyton, A.C. Physiologic regulation of arterial pressure. Am. J. Cardiol. 1961, 8, 401–407. [Google Scholar] [CrossRef]
- Guyton, A.C.; Coleman, T.G. Quantitative analysis of the pathophysiology of hypertension. 1969. J. Am. Soc. Nephrol. 1999, 10, 2248–2258. [Google Scholar]
- Guyton, A.C. Kidneys and fluids in pressure regulation. Small volume but large pressure changes. Hypertension 1992, 19, I2–I8. [Google Scholar] [CrossRef]
- Hall, J.E. Renal dysfunction, rather than nonrenal vascular dysfunction, mediates salt-induced hypertension. Circulation 2016, 133, 894–906. [Google Scholar] [CrossRef]
- Morris, R.C., Jr.; Schmidlin, O.; Sebastian, A.; Tanaka, M.; Kurtz, T.W. Vasodysfunction that involves renal vasodysfunction, not abnormally increased renal retention of sodium, accounts for the initiation of salt-induced hypertension. Circulation 2016, 133, 881–893. [Google Scholar] [CrossRef]
- Skou, J.C. The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim. Biophys. Acta 1957, 23, 394–401. [Google Scholar] [CrossRef]
- Yan, Y.; Shapiro, A.P.; Haller, S.; Katragadda, V.; Liu, L.; Tian, J.; Basrur, V.; Malhotra, D.; Xie, Z.J.; Abraham, N.G.; et al. Involvement of reactive oxygen species in a feed-forward mechanism of na/k-atpase-mediated signaling transduction. J. Biol. Chem. 2013, 288, 34249–34258. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yan, Y.; Liu, L.; Xie, Z.; Malhotra, D.; Joe, B.; Shapiro, J.I. Impairment of na/k-atpase signaling in renal proximal tubule contributes to dahl salt-sensitive hypertension. J. Biol. Chem. 2011, 286, 22806–22813. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Haller, S.; Shapiro, A.; Malhotra, N.; Tian, J.; Xie, Z.; Malhotra, D.; Shapiro, J.I.; Liu, J. Ouabain-stimulated trafficking regulation of the na/k-atpase and nhe3 in renal proximal tubule cells. Mol. Cell. Biochem. 2012, 367, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Saxton, A.M. The tallyho mouse as a model of human type 2 diabetes. Methods Mol. Biol. 2012, 933, 75–87. [Google Scholar] [PubMed]
- Periyasamy, S.M.; Liu, J.; Tanta, F.; Kabak, B.; Wakefield, B.; Malhotra, D.; Kennedy, D.J.; Nadoor, A.; Fedorova, O.V.; Gunning, W.; et al. Salt loading induces redistribution of the plasmalemmal na/k-atpase in proximal tubule cells. Kidney Int. 2005, 67, 1868–1877. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Shapiro, A.P.; Mopidevi, B.R.; Chaudhry, M.A.; Maxwell, K.; Haller, S.T.; Drummond, C.A.; Kennedy, D.J.; Tian, J.; Malhotra, D.; et al. Protein carbonylation of an amino acid residue of the na/k-atpase alpha1 subunit determines na/k-atpase signaling and sodium transport in renal proximal tubular cells. J Am. Heart. Assoc. 2016, 5, e003675. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Yan, Y.; Malhotra, D.; Liu, J.; Xie, Z.; Najjar, S.M.; Shapiro, J.I. Ouabain and insulin induce sodium pump endocytosis in renal epithelium. Hypertension 2012, 59, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Didion, S.P.; Lynch, C.M.; Faraci, F.M. Cerebral vascular dysfunction in tallyho mice: A new model of type ii diabetes. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1579–H1583. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, W.J. Normal sodium balance in dogs and in man. Cardiovasc. Res. 1977, 11, 375–408. [Google Scholar] [CrossRef]
- Gupta, B.N.; Linden, R.J.; Mary, D.A.; Weatherill, D. The influence of high and low sodium intake on blood volume in the dog. Q. J. Exp. Physiol. 1981, 66, 117–128. [Google Scholar] [CrossRef]
- Grant, H.; Reischsman, F. The effects of the ingestion of large amounts of sodium chloride on the arterial and venous pressures of normal subjects. Am. Heart J. 1946, 32, 704–712. [Google Scholar] [CrossRef]
- Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2005, 365, 1415–1428. [Google Scholar] [CrossRef]
- Mandal, R.; Loeffler, A.G.; Salamat, S.; Fritsch, M.K. Organ weight changes associated with body mass index determined from a medical autopsy population. Am. J. Forensic Med. Pathol. 2012, 33, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Kasiske, B.L.; Napier, J. Glomerular sclerosis in patients with massive obesity. Am. J. Nephrol. 1985, 5, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, N.; Okabayashi, Y.; Shimizu, A.; Yokoo, T. The renal pathology of obesity. Kidney Int. Rep. 2017, 2, 251–260. [Google Scholar] [CrossRef] [PubMed]
- National Chronic Kidney Disease Fact Sheet, 2017; US Department of Health and Human Services, Centers for Disease Control and Prevention: Atlanta, GA, USA, 2017.
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive summary of the third report of the national cholesterol education program (ncep) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (adult treatment panel iii). JAMA 2001, 285, 2486–2497. [Google Scholar] [CrossRef] [PubMed]
- Alberti, K.G.; Zimmet, P.; Shaw, J.; IDF Epidemiology Task Force Consensus Group. The metabolic syndrome—A new worldwide definition. Lancet 2005, 366, 1059–1062. [Google Scholar] [CrossRef]
- Foster, M.C.; Hwang, S.J.; Porter, S.A.; Massaro, J.M.; Hoffmann, U.; Fox, C.S. Fatty kidney, hypertension, and chronic kidney disease: The framingham heart study. Hypertension 2011, 58, 784–790. [Google Scholar] [CrossRef]
- Denvir, J.; Boskovic, G.; Fan, J.; Primerano, D.A.; Parkman, J.K.; Kim, J.H. Whole genome sequence analysis of the tallyho/jng mouse. BMC Genom. 2016, 17, 907. [Google Scholar] [CrossRef]
- Parkman, J.K.; Denvir, J.; Mao, X.; Dillon, K.D.; Romero, S.; Saxton, A.M.; Kim, J.H. Congenic mice demonstrate the presence of qtls conferring obesity and hypercholesterolemia on chromosome 1 in the tallyho mouse. Mamm. Genome 2017, 28, 487–497. [Google Scholar] [CrossRef]
- Creecy, A.; Uppuganti, S.; Unal, M.; Clay Bunn, R.; Voziyan, P.; Nyman, J.S. Low bone toughness in the tallyho model of juvenile type 2 diabetes does not worsen with age. Bone 2018, 110, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Trevisan, R.; Bruttomesso, D.; Vedovato, M.; Brocco, S.; Pianta, A.; Mazzon, C.; Girardi, C.; Jori, E.; Semplicini, A.; Tiengo, A.; et al. Enhanced responsiveness of blood pressure to sodium intake and to angiotensin ii is associated with insulin resistance in iddm patients with microalbuminuria. Diabetes 1998, 47, 1347–1353. [Google Scholar] [PubMed]
- Iuchi, H.; Sakamoto, M.; Suzuki, H.; Kayama, Y.; Ohashi, K.; Hayashi, T.; Ishizawa, S.; Yokota, T.; Tojo, K.; Yoshimura, M.; et al. Effect of one-week salt restriction on blood pressure variability in hypertensive patients with type 2 diabetes. PLoS ONE 2016, 11, e0144921. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.E. The kidney, hypertension, and obesity. Hypertension 2003, 41, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.E. Mechanisms of abnormal renal sodium handling in obesity hypertension. Am. J. Hypertens. 1997, 10, 49S–55S. [Google Scholar] [CrossRef]
- Kassab, S.; Kato, T.; Wilkins, F.C.; Chen, R.; Hall, J.E.; Granger, J.P. Renal denervation attenuates the sodium retention and hypertension associated with obesity. Hypertension 1995, 25, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Wehner, P.; Shapiro, J.I. Renal denervation to treat cardiac fibrosis? J. Am. Heart. Assoc. 2014, 3, e001556. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Kandzari, D.E.; O’Neill, W.W.; D’Agostino, R.; Flack, J.M.; Katzen, B.T.; Leon, M.B.; Liu, M.; Mauri, L.; Negoita, M.; et al. A controlled trial of renal denervation for resistant hypertension. N. Engl. J. Med. 2014, 370, 1393–1401. [Google Scholar] [CrossRef]
- Engholm, M.; Bertelsen, J.B.; Mathiassen, O.N.; Botker, H.E.; Vase, H.; Peters, C.D.; Bech, J.N.; Buus, N.H.; Schroeder, A.P.; Rickers, H.; et al. Effects of renal denervation on coronary flow reserve and forearm dilation capacity in patients with treatment-resistant hypertension. A randomized, double-blinded, sham-controlled clinical trial. Int. J. Cardiol. 2018, 250, 29–34. [Google Scholar] [CrossRef]
- Bhat, A.; Kuang, Y.M.; Gan, G.C.; Burgess, D.; Denniss, A.R. An update on renal artery denervation and its clinical impact on hypertensive disease. BioMed Res. Int. 2015, 2015, 607079. [Google Scholar] [CrossRef]
- Wilcox, C.S. Reactive oxygen species: Roles in blood pressure and kidney function. Curr. Hypertens. Rep. 2002, 4, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Ando, K. Increased salt sensitivity in obese hypertension: Role of the sympathetic nervous system. Curr. Hypertens. Rev. 2013, 9, 264–269. [Google Scholar] [CrossRef]
- Hubens, L.E.; Verloop, W.L.; Joles, J.A.; Blankestijn, P.J.; Voskuil, M. Ischemia and reactive oxygen species in sympathetic hyperactivity states: A vicious cycle that can be interrupted by renal denervation? Curr. Hypertens. Rep. 2013, 15, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tian, J.; Haas, M.; Shapiro, J.I.; Askari, A.; Xie, Z. Ouabain interaction with cardiac Na+/K+-ATPase initiates signal cascades independent of changes in intracellular na+ and ca2+ concentrations. J. Biol. Chem. 2000, 275, 27838–27844. [Google Scholar] [PubMed]
- Liu, L.; Li, J.; Liu, J.; Yuan, Z.; Pierre, S.V.; Qu, W.; Zhao, X.; Xie, Z. Involvement of Na+/K+-ATPase in hydrogen peroxide-induced hypertrophy in cardiac myocytes. Free Radic. Biol. Med. 2006, 41, 1548–1556. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Kometiani, P.; Liu, J.; Li, J.; Shapiro, J.I.; Askari, A. Intracellular reactive oxygen species mediate the linkage of Na+/K+-ATPase to hypertrophy and its marker genes in cardiac myocytes. J. Biol. Chem. 1999, 274, 19323–19328. [Google Scholar] [CrossRef]
- Tian, J.; Liu, J.; Garlid, K.D.; Shapiro, J.I.; Xie, Z. Involvement of mitogen-activated protein kinases and reactive oxygen species in the inotropic action of ouabain on cardiac myocytes. A potential role for mitochondrial k(atp) channels. Mol. Cell. Biochem. 2003, 242, 181–187. [Google Scholar] [CrossRef]
- Kennedy, D.J.; Vetteth, S.; Periyasamy, S.M.; Kanj, M.; Fedorova, L.; Khouri, S.; Kahaleh, M.B.; Xie, Z.; Malhotra, D.; Kolodkin, N.I.; et al. Central role for the cardiotonic steroid marinobufagenin in the pathogenesis of experimental uremic cardiomyopathy. Hypertension 2006, 47, 488–495. [Google Scholar] [CrossRef]
- Elkareh, J.; Kennedy, D.J.; Yashaswi, B.; Vetteth, S.; Shidyak, A.; Kim, E.G.; Smaili, S.; Periyasamy, S.M.; Hariri, I.M.; Fedorova, L.; et al. Marinobufagenin stimulates fibroblast collagen production and causes fibrosis in experimental uremic cardiomyopathy. Hypertension 2007, 49, 215–224. [Google Scholar] [CrossRef]
- Yan, Y.; Shapiro, J.I. The physiological and clinical importance of sodium potassium atpase in cardiovascular diseases. Curr. Opin. Pharmacol. 2016, 27, 43–49. [Google Scholar] [CrossRef]
- Sodhi, K.; Maxwell, K.; Yan, Y.; Liu, J.; Chaudhry, M.A.; Getty, M.; Xie, Z.; Abraham, N.G.; Shapiro, J.I. Pnaktide inhibits na/k-atpase reactive oxygen species amplification and attenuates adipogenesis. Sci. Adv. 2015, 1, e1500781. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, K.; Srikanthan, K.; Goguet-Rubio, P.; Nichols, A.; Mallick, A.; Nawab, A.; Martin, R.; Shah, P.T.; Chaudhry, M.; Sigdel, S.; et al. pNaKtide attenuates steatohepatitis and atherosclerosis by blocking Na/K-ATPase/ROS amplification in c57bl6 and apoe knockout mice fed a western diet. Sci. Rep. 2017, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tian, J.; Chaudhry, M.; Maxwell, K.; Yan, Y.; Wang, X.; Shah, P.T.; Khawaja, A.A.; Martin, R.; Robinette, T.J.; et al. Attenuation of Na/K-ATPase mediated oxidant amplification with pnaktide ameliorates experimental uremic cardiomyopathy. Sci. Rep. 2016, 6, 34592. [Google Scholar] [CrossRef] [PubMed]
- Petrushanko, I.Y.; Mitkevich, V.A.; Lakunina, V.A.; Anashkina, A.A.; Spirin, P.V.; Rubtsov, P.M.; Prassolov, V.S.; Bogdanov, N.B.; Hanggi, P.; Fuller, W.; et al. Cysteine residues 244 and 458-459 within the catalytic subunit of na,k-atpase control the enzyme’s hydrolytic and signaling function under hypoxic conditions. Redox Biol. 2017, 13, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Petrushanko, I.; Bogdanov, N.; Bulygina, E.; Grenacher, B.; Leinsoo, T.; Boldyrev, A.; Gassmann, M.; Bogdanova, A. Na-K-ATPase in rat cerebellar granule cells is redox sensitive. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R916–R925. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yan, Y.; Nie, Y.; Shapiro, J.I. Na/K-ATPase signaling and salt sensitivity: The role of oxidative stress. Antioxidants 2017, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Lakunina, V.A.; Burnysheva, K.M.; Mitkevich, V.A.; Makarov, A.A.; Petrushanko, I.Y. changes in the receptor function of na,k-atpase during hypoxia and ischemia. Mol. Biol. 2017, 51, 172–179. [Google Scholar] [CrossRef]
- Cwynar, M.; Stompor, T.; Barton, H.; Grodzicki, T. Endogenous lithium clearance: A diagnostic method of assessing sodium sensitivity in hypertension. Methodological and clinical implications. Kardiol. Pol. 2014, 72, 1–7. [Google Scholar] [CrossRef]
- Venezia, A.; Barba, G.; Russo, O.; Capasso, C.; De Luca, V.; Farinaro, E.; Cappuccio, F.P.; Galletti, F.; Rossi, G.; Strazzullo, P. Dietary sodium intake in a sample of adult male population in southern italy: Results of the olivetti heart study. Eur. J. Clin. Nutr. 2010, 64, 518–524. [Google Scholar] [CrossRef][Green Version]
- Strazzullo, P.; Barba, G.; Cappuccio, F.P.; Siani, A.; Trevisan, M.; Farinaro, E.; Pagano, E.; Barbato, A.; Iacone, R.; Galletti, F. Altered renal sodium handling in men with abdominal adiposity: A link to hypertension. J. Hypertens. 2001, 19, 2157–2164. [Google Scholar] [CrossRef]
- Bagrov, A.Y.; Shapiro, J.I.; Fedorova, O.V. Endogenous cardiotonic steroids: Physiology, pharmacology, and novel therapeutic targets. Pharm. Rev. 2009, 61, 9–38. [Google Scholar] [CrossRef] [PubMed]
- Koomans, H.A.; Boer, W.H.; Dorhout Mees, E.J. Evaluation of lithium clearance as a marker of proximal tubule sodium handling. Kidney Int. 1989, 36, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Cai, T.; Tian, J.; Qu, W.; Xie, Z.J. Functional characterization of src-interacting Na/K-ATPase using RNA interference assay. J. Biol. Chem. 2006, 281, 19709–19719. [Google Scholar] [CrossRef] [PubMed]
- Heppner, D.E.; Dustin, C.M.; Liao, C.; Hristova, M.; Veith, C.; Little, A.C.; Ahlers, B.A.; White, S.L.; Deng, B.; Lam, Y.W.; et al. Direct cysteine sulfenylation drives activation of the src kinase. Nat. Commun. 2018, 9, 4522. [Google Scholar] [CrossRef] [PubMed]
- Haller, S.T.; Yan, Y.; Drummond, C.A.; Xie, J.; Tian, J.; Kennedy, D.J.; Shilova, V.Y.; Xie, Z.; Liu, J.; Cooper, C.J.; et al. Rapamycin attenuates cardiac fibrosis in experimental uremic cardiomyopathy by reducing marinobufagenin levels and inhibiting downstream pro-fibrotic signaling. J. Am. Heart. Assoc. 2016, 5, e004106. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C57BL/6J (B6) | TALLYHO/JngJ (TH) | |
|---|---|---|
| Systolic blood pressure, (mmHg, n = 8–10) | 104.9 ± 1.6 | 110.5 ± 2.0 |
| Total Na+ excretion rate UNa+V, mEq/day, n = 9–10) | 0.619 ± 0.073 | 0.647 ± 0.065 |
| Fractional Na+ excretion (FENa+, %, n = 9–10) | 3.384 ± 0.324 | 3.875 ± 0.297 |
| Value = Mean ± SEM | ||
| Strain | Salt Diet | Group | Food Intake (g/mouse/day) | Na+ Intake (mEq/mouse/day) | Increases in 24 h Na+ IntakeΔ | Water Intake (mL/mouse/day) |
|---|---|---|---|---|---|---|
| C57BL/6J (B6) | Normal chow (NC) | NC n = 9 | 6.644 ± 0.370 | 1.127 ± 0.063 | 8.428 ± 0.598 | |
| HS n = 10 | 6.487 ± 0.431 | 1.100 ± 0.073 | 7.818 ± 0.388 | |||
| 2% NaCl (7 days) | NC n = 9 | 6.210 ± 0.429 | 1.053 ± 0.073 | −0.056 ± 0.175 | 8.357 ± 0.528 | |
| HS n = 10 | 6.569 ± 0.350 | 2.285 ± 0.122 * | 1.173 ± 0.627 | 8.424 ± 0.398 | ||
| 4% NaCl (7 days) | NC n = 9 | 7.204 ± 0.383 | 1.222 ± 0.065 | 0.203 ± 0.306 | 8.356 ± 0.757 | |
| HS n = 10 | 7.451 ± 0.555 | 5.183 ± 0.386 *** | 1.311 ± 0.613 | 12.481 ± 0.531 *** | ||
| 8% NaCl (7 days) | NC n = 9 | 7.819 ± 0.567 | 1.326 ± 0.096 | 0.082 ± 0.147 | 10.427 ± 1.054 | |
| HS n = 10 | 8.355 ± 0.596 | 11.624 ± 0.930 *** | 1.316 ± 0.562 | 21.548 ± 1.563 *** | ||
| TALLYHO/JngJ (TH) | Normal chow (NC) | NC n = 8 | 6.105 ± 0.692 | 1.035 ± 0.117 | 6.178 ± 0.565 | |
| HS n = 8 | 6.065 ± 0.431 | 1.028 ± 0.073 | 6.540 ± 0.318 | |||
| 2% NaCl (7 days) | NC n = 8 | 5.748 ± 0.263 | 0.975 ± 0.045 | 0.004 ± 0.250 | 7.218 ± 0.287 | |
| HS n = 8 | 4.588 ± 0.168 | 1.596 ± 0.058 | 0.607 ± 0.366 # | 5.553 ± 0.443 | ||
| 4% NaCl (7 days) | NC n = 8 | 5.816 ± 0.295 | 0.986 ± 0.050 | 0.203 ± 0.306 | 7.509 ± 0.279 | |
| HS n = 8 | 5.041 ± 0.113 | 3.507 ± 0.079 *** | 1.218 ± 0.260 | 7.826 ± 0.321 | ||
| 8% NaCl (7 days) | NC n = 8 | 7.265 ± 0.481 | 1.232 ± 0.082 | 0.262 ± 0.227 | 7.509 ± 0.279 | |
| HS n = 8 | 6.350 ± 0.620 | 8.835 ± 0.863 *** | 1.541 ± 0.759 | 16.474 ± 1.127 *** |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, Y.; Wang, J.; Chaudhry, M.A.; Nie, Y.; Sun, S.; Carmon, J.; Shah, P.T.; Bai, F.; Pratt, R.; Brickman, C.; et al. Metabolic Syndrome and Salt-Sensitive Hypertension in Polygenic Obese TALLYHO/JngJ Mice: Role of Na/K-ATPase Signaling. Int. J. Mol. Sci. 2019, 20, 3495. https://doi.org/10.3390/ijms20143495
Yan Y, Wang J, Chaudhry MA, Nie Y, Sun S, Carmon J, Shah PT, Bai F, Pratt R, Brickman C, et al. Metabolic Syndrome and Salt-Sensitive Hypertension in Polygenic Obese TALLYHO/JngJ Mice: Role of Na/K-ATPase Signaling. International Journal of Molecular Sciences. 2019; 20(14):3495. https://doi.org/10.3390/ijms20143495
Chicago/Turabian StyleYan, Yanling, Jiayan Wang, Muhammad A. Chaudhry, Ying Nie, Shuyan Sun, Jazmin Carmon, Preeya T. Shah, Fang Bai, Rebecca Pratt, Cameron Brickman, and et al. 2019. "Metabolic Syndrome and Salt-Sensitive Hypertension in Polygenic Obese TALLYHO/JngJ Mice: Role of Na/K-ATPase Signaling" International Journal of Molecular Sciences 20, no. 14: 3495. https://doi.org/10.3390/ijms20143495
APA StyleYan, Y., Wang, J., Chaudhry, M. A., Nie, Y., Sun, S., Carmon, J., Shah, P. T., Bai, F., Pratt, R., Brickman, C., Sodhi, K., Kim, J. H., Pierre, S., Malhotra, D., Rankin, G. O., Xie, Z.-j., Shapiro, J. I., & Liu, J. (2019). Metabolic Syndrome and Salt-Sensitive Hypertension in Polygenic Obese TALLYHO/JngJ Mice: Role of Na/K-ATPase Signaling. International Journal of Molecular Sciences, 20(14), 3495. https://doi.org/10.3390/ijms20143495