BRCA1 and BRCA2 Testing through Next Generation Sequencing in a Small Cohort of Italian Breast/Ovarian Cancer Patients: Novel Pathogenic and Unknown Clinical Significance Variants

 ,
,
Abstract
1. Introduction
2. Results
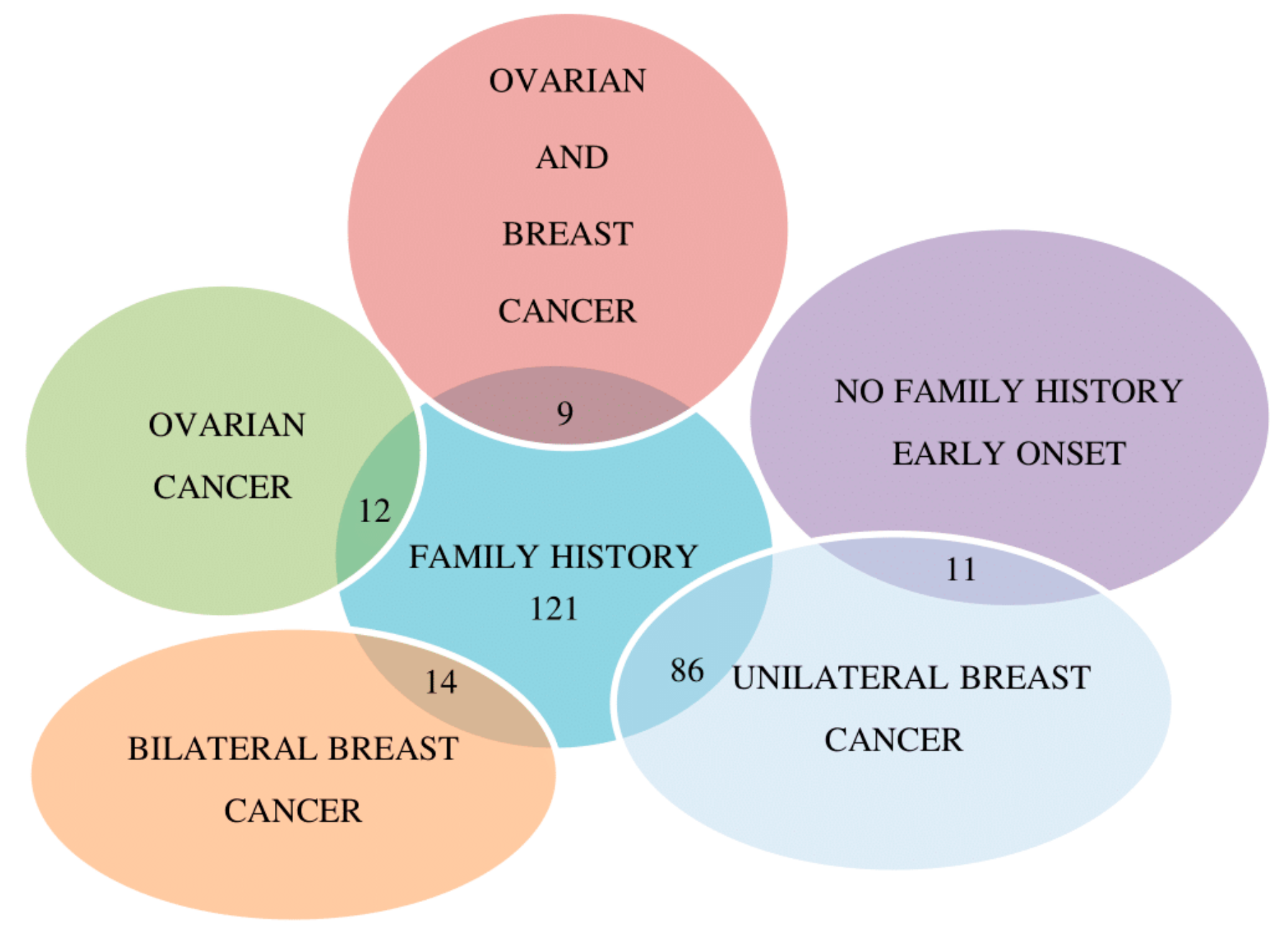
2.1. Molecular Analysis of BRCA1/2 Gene
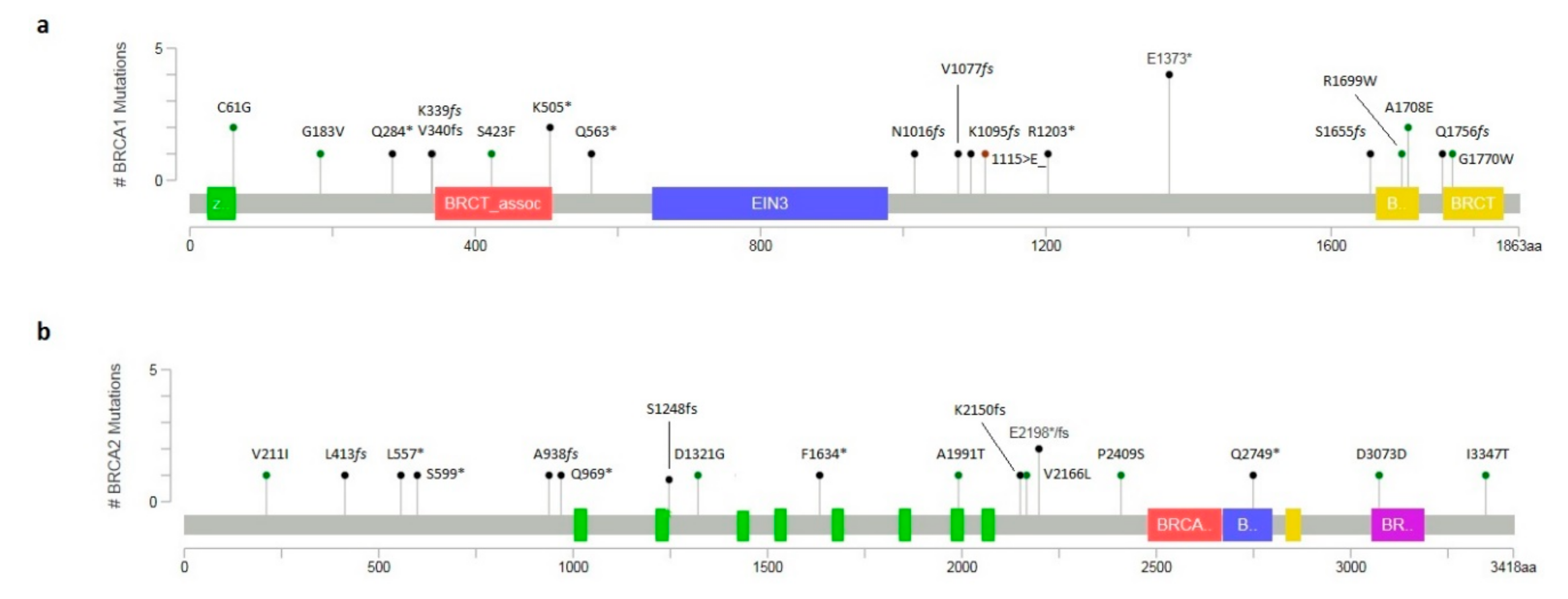
2.2. Novel Variants and In Silico Prediction Analysis
3. Discussion
4. Material and Methods
4.1. Patients
4.2. Molecular Analysis
4.3. In Silico Prediction Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rebbeck, T.R.; Friebel, T.M.; Friedman, E.; Huo, D.; Kwong, A.; Olah, E.; Olopade, O.I.; Solano, A.R.; Teo, S.H.; Thomassen, M.; et al. Mutational spectrum in a worldwide study of 29,700 families with BRCA1 or BRCA2 mutations. Hum. Mutat. 2018, 39, 593–620. [Google Scholar] [CrossRef] [PubMed]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. SOLO2/ENGOT-Ov21 investigators. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Toland, A.E.; Forman, A.; Couch, F.J.; Culver, J.O.; Eccles, D.M.; Foulkes, W.D.; Hogervorst, F.B.L.; Houdayer, C.; Levy-Lahad, E.; Monteiro, A.N.; et al. Clinical testing of BRCA1 and BRCA2: A worldwide snapshot of technological practices. NPJ Genom. Med. 2018, 15, 7. [Google Scholar] [CrossRef] [PubMed]
- Concolino, P.; Rizza, R.; Mignone, F.; Costella, A.; Guarino, D.; Carboni, I.; Capoluongo, E.; Santonocito, C.; Urbani, A.; Minucci, A. A comprehensive BRCA1/2 NGS pipeline for an immediate Copy Number Variation (CNV) detection in breast and ovarian cancer molecular diagnosis. Clin. Chim. Acta 2018, 480, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Minucci, A.; Concolino, P.; De Bonis, M.; Costella, A.; Paris, I.; Scambia, G.; Capoluongo, E. Preliminary molecular evidence associating a novel BRCA1 synonymous variant with hereditary ovarian cancer syndrome. Hum. Genome Var. 2018, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Minucci, A.; Lalle, M.; De Leo, R.; Mazzuccato, G.; Scambia, G.; Urbani, A.; Fagotti, A.; Concolino, P.; Capoluongo, E. Additional molecular and clinical evidence open the way to definitive IARC classification of the BRCA1 c.5332G > A (p.Asp1778Asn) variant. Clin. Biochem. 2019, 63, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Gelli, E.; Colombo, M.; Pinto, A.M.; De Vecchi, G.; Foglia, C.; Amitrano, S.; Morbidoni, V.; Imperatore, V.; Manoukian, S.; Baldassarri, M.; et al. Usefulness and Limitations of Comprehensive Characterization of mRNA Splicing Profiles in the Definition of the Clinical Relevance of BRCA1/2 Variants of Uncertain Significance. Cancers 2019, 11, 295. [Google Scholar] [CrossRef]
- Lindor, N.M.; Guidugli, L.; Wang, X.; Vallée, M.P.; Monteiro, A.N.; Tavtigian, S.; Goldgar, D.E.; Couch, F.J. A review of a multifactorial probability-based model for classification of BRCA1 and BRCA2 variants of uncertain significance (VUS). Hum. Mutat. 2012, 33, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Ernst, C.; Hahnen, E.; Engel, C.; Nothnagel, M.; Weber, J.; Schmutzler, R.K.; Hauke, J. Performance of in silico prediction tools for the classification of rare BRCA1/2 missense variants in clinical diagnostics. BMC Med. Genom. 2018, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Millot, G.A.; Carvalho, M.A.; Caputo, S.M.; Vreeswijk, M.P.; Brown, M.A.; Webb, M.; Rouleau, E.; Neuhausen, S.L.; Hansen, T.V.; Galli, A.; et al. A guide for functional analysis of BRCA1 variants of uncertain significance. Hum. Mutat. 2012, 33, 1526–1537. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, P.; Van der Gulden, H.; Van der Heijden, I.; Drost, R.; Klijn, C.N.; Prasetyanti, P.; Pieterse, M.; Wientjens, E.; Seibler, J.; Hogervorst, F.B.; et al. A high-throughput functional complementation assay for classification of BRCA1 missense variants. Cancer Discov. 2013, 3, 1142–1155. [Google Scholar] [CrossRef] [PubMed]
- Ferla, R.; Calò, V.; Cascio, S.; Rinaldi, G.; Badalamenti, G.; Carreca, I.; Surmacz, E.; Colucci, G.; Bazan, V.; Russo, A. Founder mutations in BRCA1 and BRCA2 genes. Ann. Oncol. 2007, 18, vi93–vi98. [Google Scholar] [CrossRef] [PubMed]
- Evidence-Based Network for the Interpretation of Germline Mutant Alleles (ENIGMA). Available online: https://enigmaconsortium.org/library/general-documents/enigma-classification-criteria/ (accessed on 6 May 2019).
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60, 706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Glusman, G.; Caballero, J.; Mauldin, D.E.; Hood, L.; Roach, J.C. Kaviar: An accessible system for testing SNV novelty. Bioinformatics 2011, 27, 3216–3217. [Google Scholar] [CrossRef] [PubMed]
- Hudson, T.J.; Anderson, W.; Artez, A.; Barker, A.D.; Bell, C.; Bernabé, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; Gerhard, D.S.; et al. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar] [PubMed]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Quang, D.; Chen, Y.; Xie, X. DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef] [PubMed]
- Capalbo, C.; Ricevuto, E.; Vestri, A.; Ristori, E.; Sidoni, T.; Buffone, O.; Adamo, B.; Cortesi, E.; Marchetti, P.; Scambia, G.; et al. BRCA1 and BRCA2 genetic testing in Italian breast and/or ovarian cancer families: Mutation spectrum and prevalence and analysis of mutation prediction models. Ann. Oncol. 2006, 17, vii34–vii40. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, M.; Marchetti, C.; De Leo, R.; Musella, A.; Capoluongo, E.; Paris, I.; Benedetti Panici, P.; Scambia, G.; Fagotti, A. BRCA mutational status, initial disease presentation, and clinical outcome in high-grade serous advanced ovarian cancer: A multicenter study. Am. J. Obstet. Gynecol. 2017, 217, 334.e1–334.e9. [Google Scholar] [CrossRef]
- Quiles, F.; Fernández-Rodríguez, J.; Mosca, R.; Feliubadaló, L.; Tornero, E.; Brunet, J.; Blanco, I.; Capellá, G.; Pujana, M.À.; Aloy, P.; et al. Functional and structural analysis of C-terminal BRCA1 missense variants. PLoS ONE 2013, 8, e61302. [Google Scholar] [CrossRef]
- Lavie, O.; Narod, S.; Lejbkowicz, F.; Dishon, S.; Goldberg, Y.; Gemer, O.; Rennert, G. Double heterozygosity in the BRCA1 and BRCA2 genes in the Jewish population. Ann. Oncol. 2011, 22, 964–966. [Google Scholar] [CrossRef] [PubMed]
- Musolino, A.; Naldi, N.; Michiara, M.; Bella, M.A.; Zanelli, P.; Bortesi, B.; Capelletti, M.; Savi, M.; Neri, T.M.; Ardizzoni, A. A breast cancer patient from Italy with germline mutations in both the BRCA1 and BRCA2 genes. Breast Cancer Res. Treat. 2005, 91, 203–205. [Google Scholar] [CrossRef] [PubMed]
- Pilato, B.; De Summa, S.; Danza, K.; Lambo, R.; Paradiso, A.; Tommasi, S. Maternal and paternal lineage double heterozygosity alteration in familial breast cancer: A first case report. Breast Cancer Res. Treat. 2010, 124, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Zuradelli, M.; Peissel, B.; Manoukian, S.; Zaffaroni, D.; Barile, M.; Pensotti, V.; Cavallari, U.; Masci, G.; Mariette, F.; Benski, A.C.; et al. Four new cases of double heterozygosity for BRCA1 and BRCA2 gene mutations: Clinical, pathological, and family characteristics. Breast Cancer Res. Treat. 2010, 124, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Vietri, M.T.; Molinari, A.M.; Caliendo, G.; De Paola, M.L.; Giovanna, D.; Gambardella, A.L.; Petronella, P.; Cioffi, M. Double heterozygosity in the BRCA1 and BRCA2 genes in Italian family. Clin. Chem. Lab. Med. 2013, 51, 2319–2324. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, T.R.; Friebel, T.M.; Mitra, N.; Wan, F.; Chen, S.; Andrulis, I.L.; Apostolou, P.; Arnold, N.; Arun, B.K. Inheritance of deleterious mutations at both BRCA1 and BRCA2 in an international sample of 32,295 women. Breast Cancer Res. 2016, 18, 112. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.A.; Iversen, E.S., Jr.; Gudbjartsson, D.F.; Hiller, E.H.; Garber, J.E.; Peshkin, B.N.; Lerman, C.; Watson, P.; Lynch, H.T.; Hilsenbeck, S.G.; et al. BRCAPRO validation, sensitivity of genetic testing of BRCA1/BRCA2, and prevalence of other breast cancer susceptibility genes. J. Clin. Oncol. 2002, 20, 2701–2712. [Google Scholar] [CrossRef]
- Concolino, P.; Mello, E.; Minucci, A.; Santonocito, C.; Scambia, G.; Giardina, B.; Capoluongo, E. Advanced tools for BRCA1/2 mutational screening: Comparison between two methods for large genomic rearrangements (LGRs) detection. Clin. Chem. Lab. Med. 2014, 52, 1119–1127. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The Human Genomic Variant Search Engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]






 : proband.
: proband.
: proband.
: proband.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Sex | Gene | Pathogenic Variant (HGVS Variant Sequence Δ) | Pathogenic Variant (HGVS Protein) | Database | Clinical Significance | Proband Cancer/Age of Onset (Relatives) |
|---|---|---|---|---|---|---|---|
| BF56-18 | F | BRCA1 | c.181T>G (rs28897672) | p.(Cys61Gly) | ENIGMA; Clin Var; LOVD | Pathogenic (Class 5) | Unilateral Breast 30 (Sister *: Breast 42; Mother *: Breast 48) |
| GU79-18 | M | BRCA2 | c.2808_2811delACAA (rs80359351) | p.(Ala938ProfsTer21) | ENIGMA; Clin Var; LOVD | Pathogenic (Class 5) | Bilateral Breast 65 (Maternal Aunt †: Breast 52, Ovarian Cancer 64; Daughter * asymptomatic 29; Daughter * asymptomatic 32) |
| CD97-18 | F | BRCA1 | c.5123C>A (rs28897696) | p.(Ala1708Glu) | ENIGMA; Clin Var; LOVD | Pathogenic (Class 5) | Unilateral Breast 51 (Sister twin *: Breast 27; Mother *: Breast 44) |
| SE53-18 | F | BRCA2 | c.1796_1800delCTTAT (rs276174813) | p.(Ser599Ter) | ENIGMA; Clin Var; LOVD | Pathogenic (Class 5) | Bilateral Breast 43, 49 (Sister *: Breast 37, Father †: Breast 62) |
| MC52-18 | F | BRCA1 | c.3044dupG (rs80357746) | p.(Asn1016LysfsTer2) | ENIGMA; Clin Var | Pathogenic (Class 5) | TN Breast 35 (Paternal Aunt †: Breast 51, Ovarian Cancer 66; Paternal Cousin *: Breast 36) |
| RS72-17 | F | BRCA1 | c.4117G>T (rs80357259) | p.(Glu1373Ter) | ENIGMA; Clin Var; LOVD | Pathogenic (Class 5) | TN Breast 40 (Mother *: Bilateral Breast 35, 39; Maternal Grandmother †: Ovarian Cancer 55) |
| NF20-18 | F | BRCA1 | c.1687C>T (rs80356898) | p.(Gln563Ter) | ENIGMA; Clin Var; LOVD | Pathogenic (Class 5) | TN Breast 36 (Sister *: Ovarian Cancer 44; Brother * asymptomatic 37) |
| RL94-18 | F | BRCA1 | c.3285delA (rs397509051) | p.(Lys1095AsnfsTer14) | ENIGMA; Clin Var; LOVD | Pathogenic (Class 5) | TN Breast 51 (Maternal Aunt †: Breast 60; Maternal Cousin †: Ovarian Cancer 48) |
| PV86-17 | F | BRCA2 ▪ | c.2905C>T (rs886038080) c.6447_6448dupTA (rs397507858) | p.(Gln969Ter) p.(Lys2150IlefsTer19) | ENIGMA; Clin Var; LOVD | Pathogenic (Class 5) | Unilateral Breast 49 (Father †: Prostate 57; Paternal Aunt †: Breast 51, Stomach 57; Paternal Uncle †: Lung 55) |
| PM48-17 | F | BRCA1 | c.850C>T (rs397509330) | p.(Gln284Ter) | ENIGMA; Clin Var; LOVD | Pathogenic (Class 5) | TN Breast 46 (Sister *: Ovarian Cancer 49; Daughter * 21asymptomatic) |
| AL78-17 | F | BRCA2 | c.8245C>T (rs1135401925) | p.(Gln2749Ter) | ENIGMA; Clin Var | Pathogenic (Class 5) | Unilateral Breast 64 (Daughter *: Ovarian Cancer 36; Son 32 * asymptomatic) |
| DPF54-18 | F | BRCA2 ▪ | c.631G>A (rs80358871) c.7008-2A>T (rs81002823) | p.(Val211Ile) p.? | ENIGMA; Clin Var; LOVD | Pathogenic (Class 5) | Unilateral Breast 36 (Father *: Prostate 54; Paternal Grandmother†: Ovarian Cancer 55; Brother * 44 asymptomatic) |
| CS25-18 | F | BRCA2 | c.3744_3747delTGAG (rs80359403) | p.(Ser1248ArgfsTer10) | ENIGMA; Clin Var; LOVD | Pathogenic (Class 5) | Bilateral Breast 44,45, Lung Cancer 53, Pancreas Cancer 66 (Sister *: Bilateral Breast 40,42, Sister *: Bilateral Breast 38,42) |
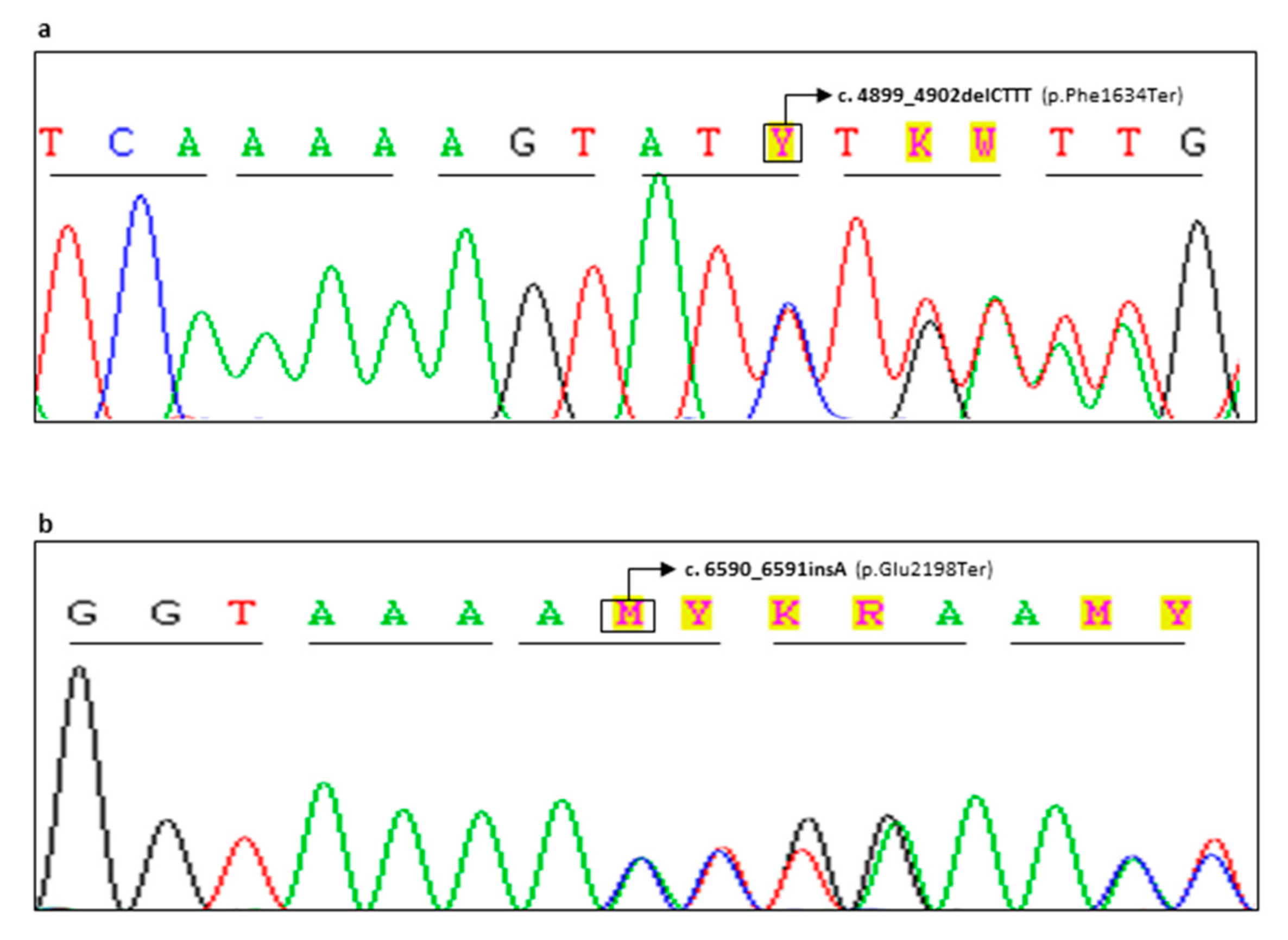
| CG98-18 | F | BRCA2 | c.4899_4902delCTTT | p.(Phe1634Ter) | Novel Variant | Pathogenic (Class 5) | Bilateral Breast 45,46 (Father†: Lung Cancer 66, Paternal Aunt†: Breast 52, Paternal Cousin*: Breast 36, Paternal Cousin: Breast 40) |
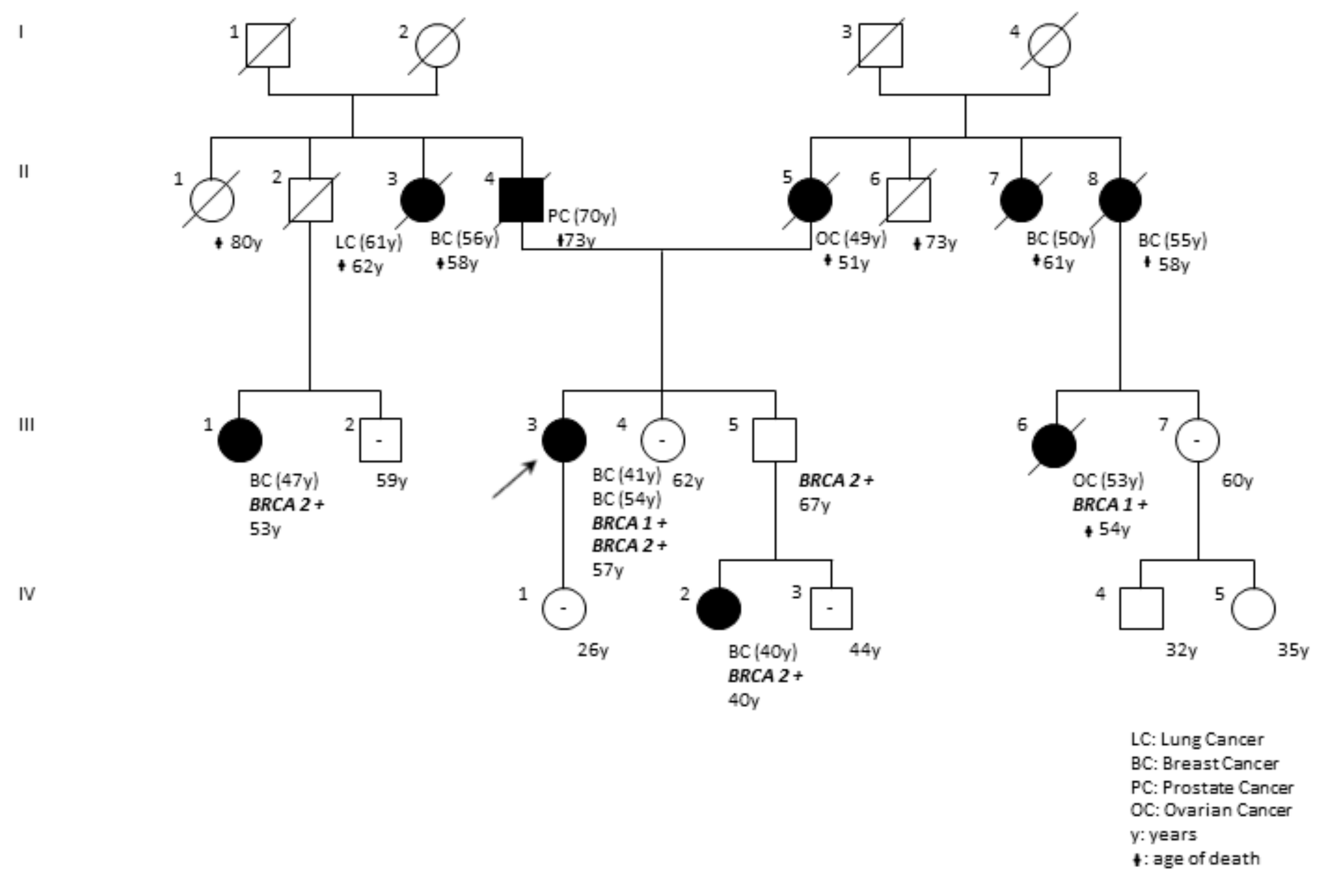
| DP97-18 | F | BRCA1 BRCA2 | c.5095C>T (rs55770810) c.1238delT (rs80359271) | p.(Arg1699Trp) p.(Leu413HisfsTer17) | ENIGMA; Clin Var; LOVD Clin Var | Pathogenic (Class 5) | The pedigree of the family is reported in Figure 6. |
| FG65-19 | F | BRCA2 | c.6590_6591insA | p.(Glu2198Ter) | Novel Variant | Pathogenic (Class 5) | Unilateral Breast 29 (Sister *: Breast 37, Sister *: Breast 38, Sister: Breast 40) |
| MR21-17 | F | BRCA1 | c.1016dupA | (p.Val340GlyfsTer6) | ENIGMA; Clin Var | Pathogenic (Class 5) | Ovarian 46 (Mother †: Breast 44, Maternal Uncle: Lung Cancer 55, Maternal Cousin *: Breast 33) |
| MC81-17 | F | BRCA1 | c.548G>T (rs1555594081) | p.(Gly183Val) | Clin Var | VUS | Unilateral Breast 48 (Father *: Breast 76) |
| FM54-17 | F | BRCA1 | c.5468-1G>A (rs80358048) | p.? | Clin Var; LOVD | Pathogenic (Class 5) | TN Breast 44 (Mother: Breast 64, Maternal Aunt: Breast 40; Pancreas Cancer:46) |
| TM34-17 | F | BRCA1 | c.1513A>T (rs397508877) | p.(Lys505Ter) | ENIGMA; Clin Var; LOVD | Pathogenic (Class 5) | Unilateral Breast 29 (Paternal Grandmother †: Ovarian Cancer 57) |
| SA22-18 | F | BRCA1 | c.3607C>T (rs62625308) | p.(Arg1203Ter) | ENIGMA; Clin Var; LOVD | Pathogenic (Class 5) | Unilateral Breast 30 |
| GV55-18 | F | BRCA1 | c.5266dupC (rs80357906) | p.(Gln1756ProfsTer74) | ENIGMA; Clin Var | Pathogenic (Class 5) | Ovarian cancer 47 (Mother: Bilateral breast 49,53; Maternal Aunt: Breast 60) |
| GM55-17 | F | BRCA1 | c.5123C>A (rs28897696) | p.(Ala1708Glu) | ENIGMA; Clin Var; LOVD | Pathogenic (Class 5) | Unilateral Breast 44 (Sister: Breast 46; Father †: Breast 62, Lung Cancer 73) |
| LA89-17 | F | BRCA2 | c.1670T>G (rs80358452) | p.(Leu557Ter) | Clin Var | Pathogenic (Class 5) | Unilateral Breast 32 |
| LR22-17 | F | BRCA1 | c.(?_-1387-1)_(80+1_81-1)del | p.0? | LOVD | Pathogenic (Class 5) | Unilateral Breast 36 (Mother *: Breast 44; Sister *: Breast 33; Maternal Cousin: Breast 47) |
| OM41-18 | F | BRCA1 | c.(5193+1_5194-1)_(5277+1_5278-1)del | p.0? | LOVD | Pathogenic (Class 5) | Unilateral Breast 38 (Mother: Breast 56, Colorectal Cancer 48) |
| QS25-17 | F | BRCA1 | c.181T>G (rs28897672) | p.(Cys61Gly) | ENIGMA; Clin Var; LOVD | Pathogenic (Class 5) | Unilateral Breast 51 (Sister: Breast 44; Paternal Aunt: Breast 40; Paternal Aunt: Breast 46) |
| SC36-17 | F | BRCA1 | c.1513A>T (rs397508877) | p.(Lys505Ter) | ENIGMA; Clin Var; LOVD | Pathogenic (Class 5) | Bilateral Breast 42,57 (Paternal Aunt: Breast 48; Paternal Cousin: 32) |
| TG12-17 | F | BRCA1 | c.3228_3229delAG (rs80357635) | p.(Val1077CysfsTer3) | ENIGMA; Clin Var | Pathogenic (Class 5) | TN Breast 45 (Mother: Ovarian Cancer51; Maternal Grandmother †: Breast 58) |
| KL11-16 | F | BRCA1 | c.4117G>T (rs80357259) | p.(Glu1373Ter) | ENIGMA; Clin Var; LOVD | Pathogenic (Class 5) | Ovarian Cancer 41 (Mother †: Breast 47, Ovarian 62) |
| VF31-17 | F | BRCA1 | c.4964_4982delctggcctgaccccagaaga (rs80359876) | p.(Ser1655TyrfsTer16) | ENIGMA; Clin Var | Pathogenic (Class 5) | Unilateral Breast 44, Ovarian 48 (Maternal Aunt: Breast 51; Maternal Aunt: Ovarian 55) |
| PS99-18 | F | BRCA1 | c.4117G>T (rs80357259) | p.(Glu1373Ter) | ENIGMA; Clin Var; LOVD | Pathogenic (Class 5) | Ovarian Cancer 48 (Mother: Ovarian Cancer 52; Sister: Bilateral Breast 36,42) |
| MV55-17 | F | BRCA1 | c.1016delA (rs80357569) | p.(Lys339ArgfsTer2) | ENIGMA; Clin Var; LOVD | Pathogenic (Class 5) | Unilateral Breast 29 |
| ID38-56 | F | BRCA2 | c.6591_6592delTG (rs80359605) | p.(Glu2198AsnfsTer4) | ENIGMA; Clin Var; LOVD | Pathogenic (Class 5) | Ovarian Cancer 45 (Paternal Grandmother †: Breast 55, Ovarian Cancer 62) |
| PV22-16 | F | BRCA1 | c.4117G>T (rs80357259) | p.(Glu1373Ter) | ENIGMA; Clin Var; LOVD | Pathogenic (Class 5) | Unilateral Breast 47 (Paternal Cousin: Breast 32; Paternal Cousin: Breast 36) |
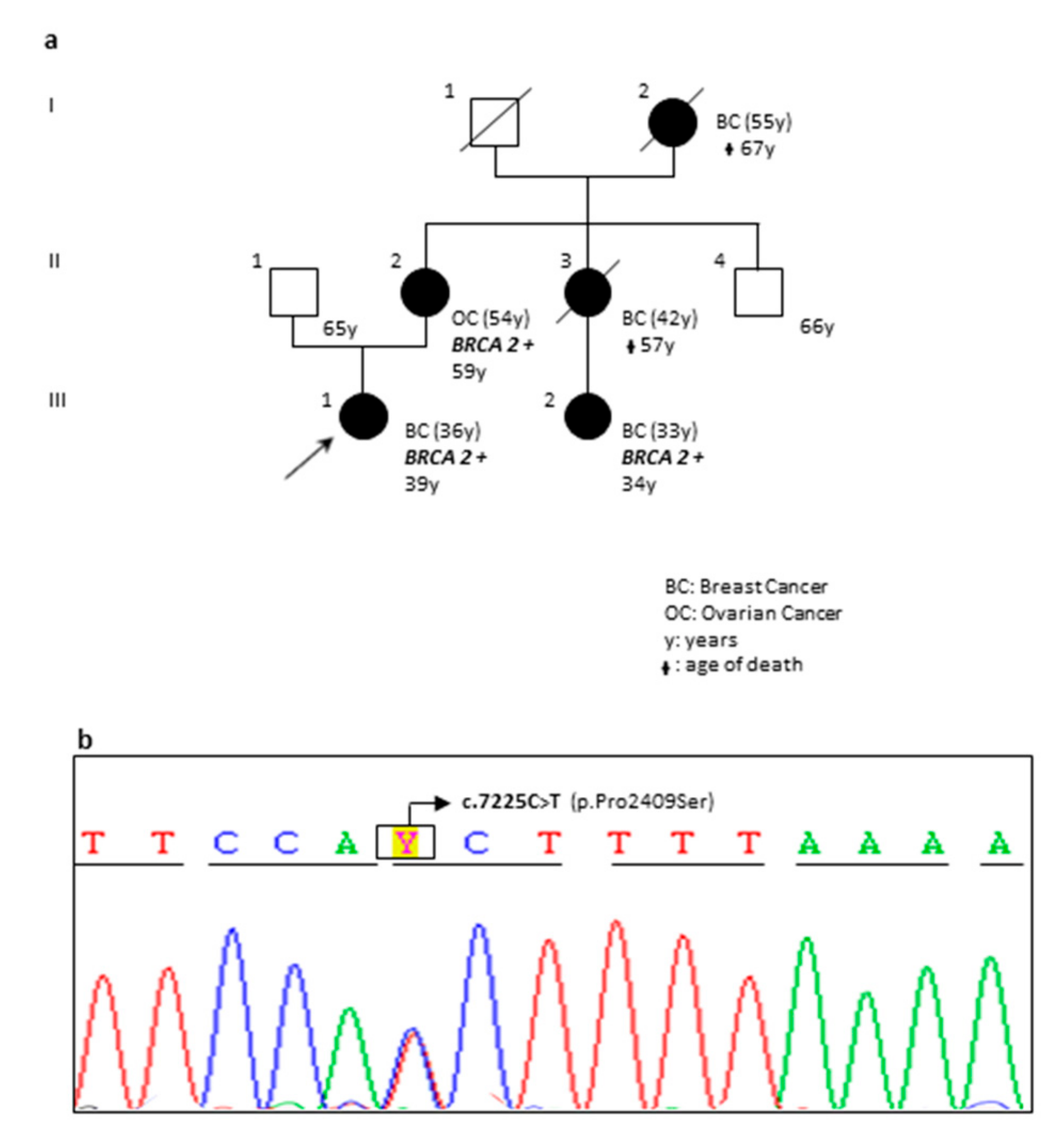
| OA78-18 | F | BRCA2 | c.7225C>T | p.(Pro2409Ser) | Novel Variant | VUS | The pedigree of the family is reported in Figure 2. |
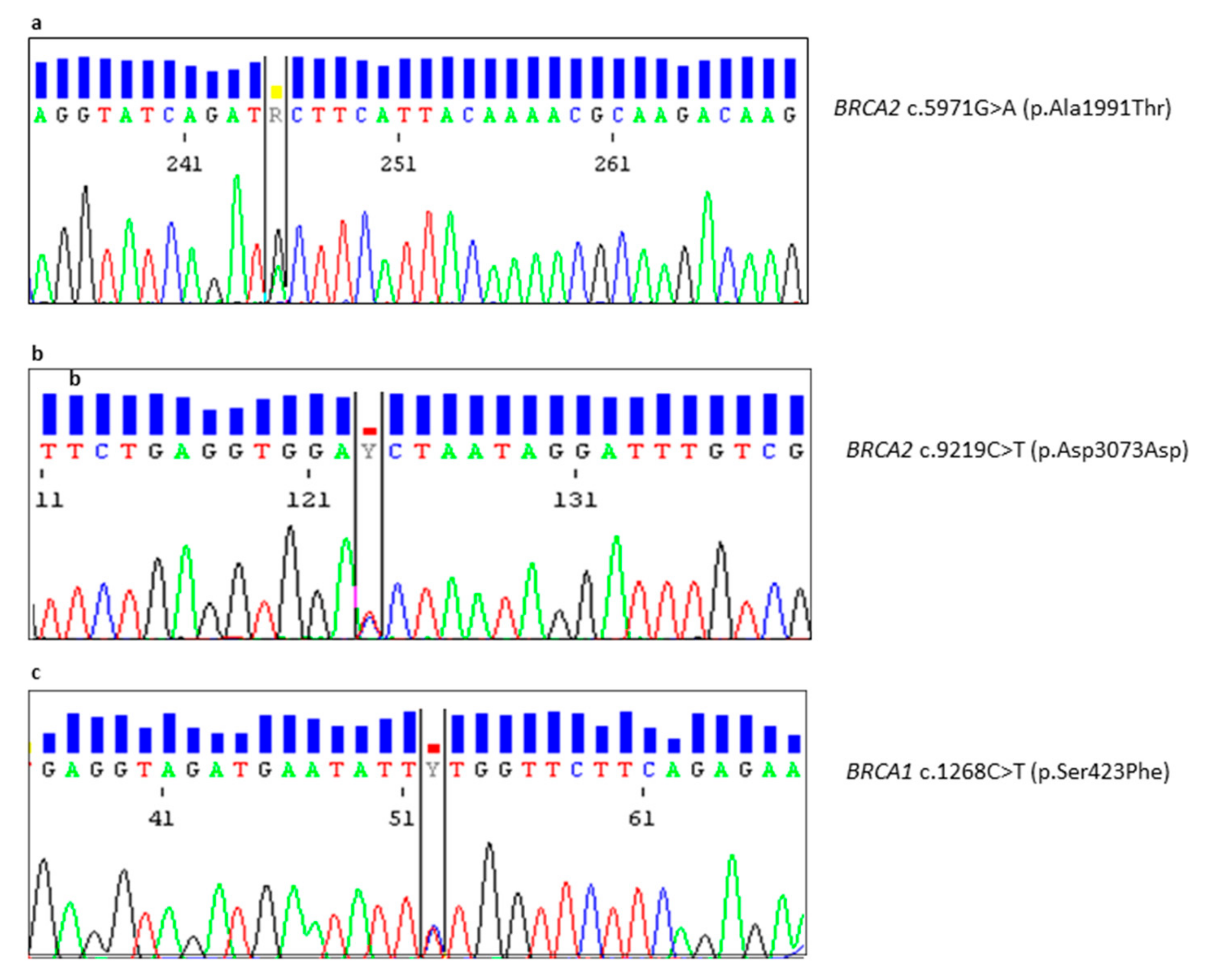
| PM55-16 | M | BRCA2 | c.5971G>A | p.(Ala1991Thr) | Novel Variant | VUS | Unilateral Breast 50 (Paternal Aunt: Breast 49; Paternal Aunt: Breast 42) |
| CA66-17 | F | BRCA2 | c.3962A>G (rs80358645) | p.(Asp1321Gly) | Clin Var | VUS | TN Breast 51 (Mother: Breast 46; Maternal Cousin: Breast 44) |
| AG55-17 | F | BRCA2 | c.6496G>T (rs750084851) | p.(Val2166Leu) | Clin Var | VUS | Bilateral Breast 45,46 (Maternal Grandmother: Breast 71) |
| AM11-17 | F | BRCA2 | c.10040T>C (rs587782373) | p.(Ile3347Thr) | Clin Var | VUS | Unilateral Breast 39 |
| TC22-17 | F | BRCA1 | c.3344_3346delAAG | p.(Glu1115del) | Clin Var | VUS | Unilateral Breast 34 |
| MR52-18 | F | BRCA2 | c.9219c>T | p.(Asp3073Asp) | Novel Variant | VUS | Unilateral Breast 48 (Mother: Breast 55; Maternal Aunt: Breast 48) |
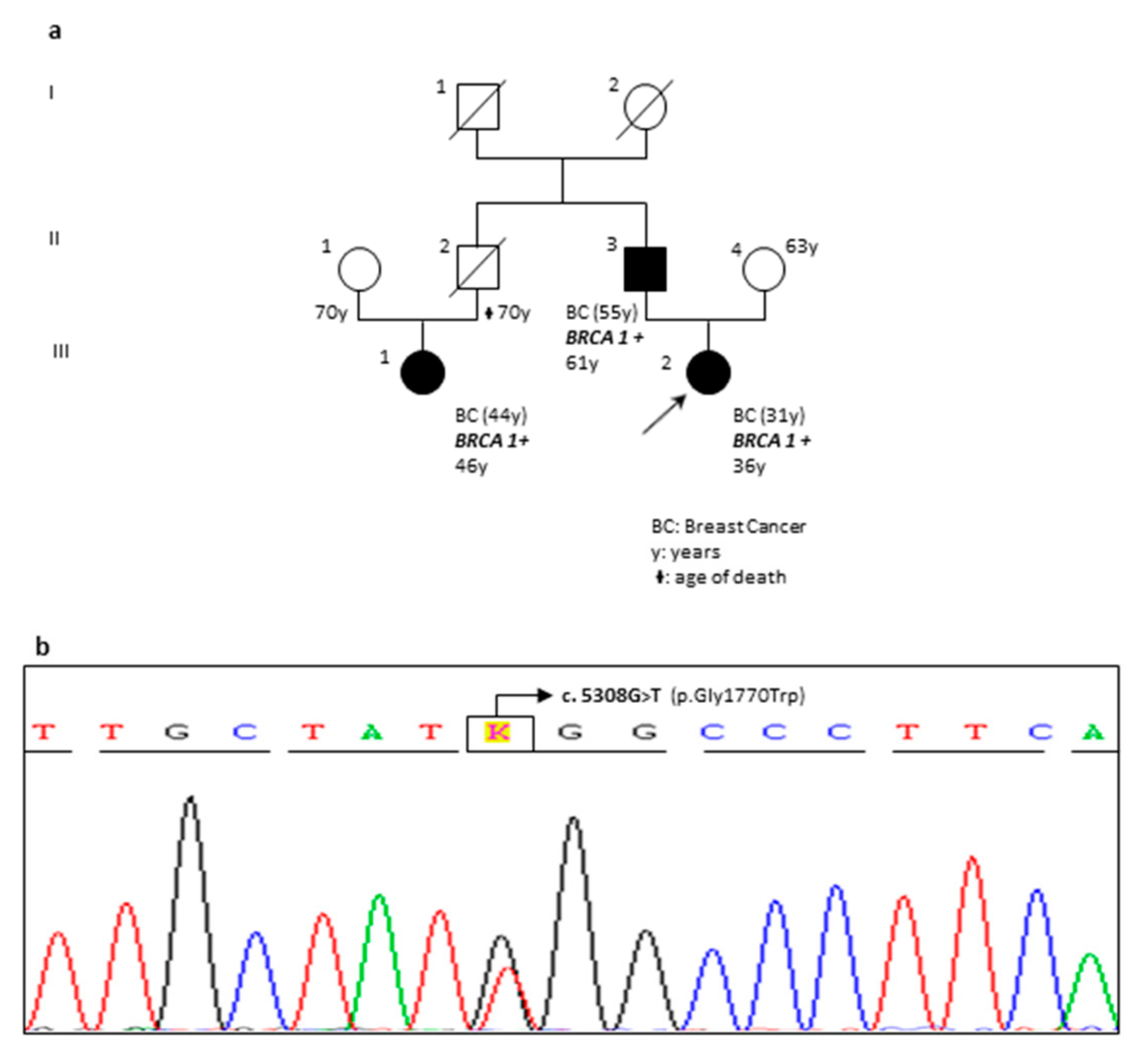
| RR56-18 | F | BRCA1 | c.5308G>T | p.(Gly1770Trp) | Novel Variant | VUS | The pedigree of the family is reported in Figure 4. |
| GH09-19 | F | BRCA1 | c.1268C>T | p.(Ser423Phe) | Novel Variant | VUS | Unilateral Breast 48 (Sister: Breast 49; Sister: Breast 52) |
| BRCA1 Pathogenic Variant (HGVS Sequence Δ/Protein) | BRCA2 Pathogenic Variant (HGVS Sequence Δ/Protein) | Sex | Inheritance | Proband Cancer (Age at Diagnosis) | Reference | |
|---|---|---|---|---|---|---|
| Mother | Father | |||||
| c.4285_4286insG; p.(Tyr1429Ter) | c.7738C>T; p.(Gln2580Ter) | F | ND | ND | Breast37 | [14] |
| c.5266dupC; p.(Gln1756ProfsTer74) | c.5796_5797delTA; p.(His1932GlnfsTer12) | F | BRCA1 | BRCA2 | Breast38, Ovarian42 | [15] |
| c.835delC; p.(His279MetfsTer19) | c.8195T>G; p.(Leu2732Ter) | F | ND | ND | Breast43 | [16] |
| c.3916_3917delTT; p.(Leu1306AspfsTer23) | c.5379delT; p.(Asn1793LysfsTer2) | F | WT | ND | Breast30, Ovarian36 | [15] |
| c.1687C>T; p.(Gln563Ter) | c.6469C>T; p.(Gln2157Ter) | F | ND; ND | ND | Breast46, Ovarian58 | [16] |
| c.2405_2406delTG; p.(Val802GlufsTer7) | c.4284dupT; p.(Gln1429SerfsTer9) | F | ND | ND | Breast and Ovarian52 | [16] |
| c.547+2T>A | c.2830A>T; p.(Lys944Ter) | F | WT | BRCA1/BRCA2 | Breast35 | [17] |
| c.213-12A>G | c.7180A>T; p.(Arg2394Ter) | F | - | - | Breast- | [18] |
| c.3228_3229delAG; p.(Gly1077AlafsTer8) | c.9253dupA; p.(Thr3085AsnfsTer26) | F | - | - | Breast- | [18] |
| c.3477_3480delAAAG; p.(Ile1159MetfsTer50) | c.9401delG; p.(Gly3134AlafsTer29) | F | - | - | Ovarian- | [18] |
| c.5095C>T; p.(Arg1699Trp) | c.1238delT; p.(Leu413HisfsTer17) | F | BRCA1; BRCA2 | BRCA2 | Bilateral Breast41,54 | This report |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Concolino, P.; Gelli, G.; Rizza, R.; Costella, A.; Scambia, G.; Capoluongo, E. BRCA1 and BRCA2 Testing through Next Generation Sequencing in a Small Cohort of Italian Breast/Ovarian Cancer Patients: Novel Pathogenic and Unknown Clinical Significance Variants. Int. J. Mol. Sci. 2019, 20, 3442. https://doi.org/10.3390/ijms20143442
Concolino P, Gelli G, Rizza R, Costella A, Scambia G, Capoluongo E. BRCA1 and BRCA2 Testing through Next Generation Sequencing in a Small Cohort of Italian Breast/Ovarian Cancer Patients: Novel Pathogenic and Unknown Clinical Significance Variants. International Journal of Molecular Sciences. 2019; 20(14):3442. https://doi.org/10.3390/ijms20143442
Chicago/Turabian StyleConcolino, Paola, Gianfranco Gelli, Roberta Rizza, Alessandra Costella, Giovanni Scambia, and Ettore Capoluongo. 2019. "BRCA1 and BRCA2 Testing through Next Generation Sequencing in a Small Cohort of Italian Breast/Ovarian Cancer Patients: Novel Pathogenic and Unknown Clinical Significance Variants" International Journal of Molecular Sciences 20, no. 14: 3442. https://doi.org/10.3390/ijms20143442
APA StyleConcolino, P., Gelli, G., Rizza, R., Costella, A., Scambia, G., & Capoluongo, E. (2019). BRCA1 and BRCA2 Testing through Next Generation Sequencing in a Small Cohort of Italian Breast/Ovarian Cancer Patients: Novel Pathogenic and Unknown Clinical Significance Variants. International Journal of Molecular Sciences, 20(14), 3442. https://doi.org/10.3390/ijms20143442