Human DNA Virus Exploitation of the MAPK-ERK Cascade

Abstract
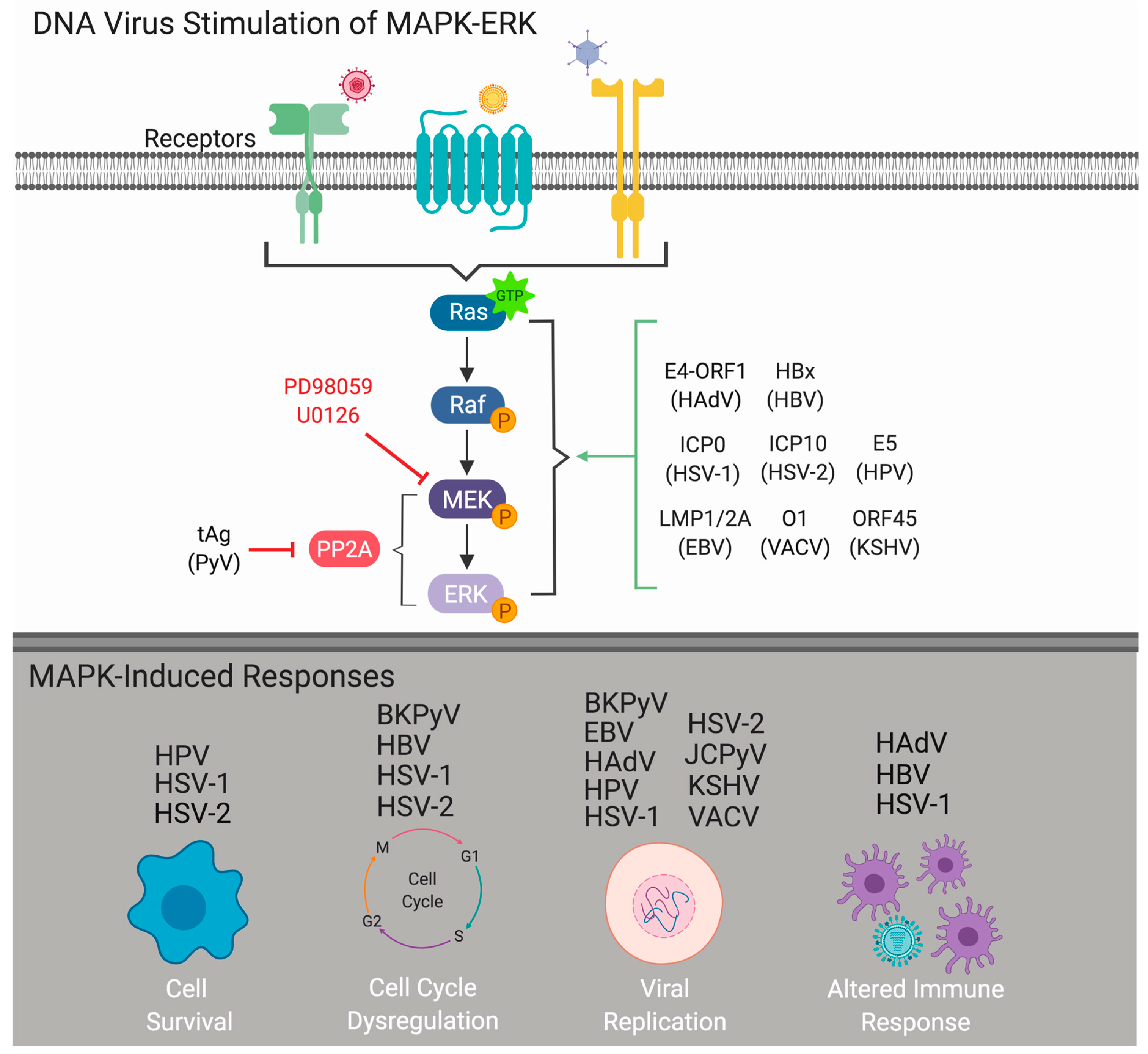
1. Introduction
2. MAPK-ERK Cascade
3. DNA Viruses
3.1. Adenoviridae
Adenovirus
3.2. Poxviridae
Vaccinia Virus
3.3. Polyomaviridae
3.3.1. JC Polyomavirus
3.3.2. BK Polyomavirus
3.3.3. Other Polyomaviruses
3.4. Papillomaviridae
Human Papillomaviruses
3.5. Herpesviridae
3.5.1. Herpes Simplex Virus
3.5.2. Kaposi’s Sarcoma-Associated Herpesvirus
3.5.3. Epstein-Barr Virus
3.6. Hepadnaviridae
Hepatitis B Virus
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shaul, Y.D.; Seger, R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. Biophys. Acta 2007, 1773, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Pleschka, S. RNA viruses and the mitogenic Raf/MEK/ERK signal transduction cascade. Biol. Chem. 2008, 389, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Panteva, M.; Korkaya, H.; Jameel, S. Hepatitis viruses and the MAPK pathway: Is this a survival strategy? Virus Res. 2003, 92, 131–140. [Google Scholar] [CrossRef]
- Bonjardim, C.A. Viral exploitation of the MEK/ERK pathway—A tale of vaccinia virus and other viruses. Virology 2017, 507, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef]
- Maik-Rachline, G.; Hacohen-Lev-Ran, A.; Seger, R. Nuclear ERK: Mechanism of Translocation, Substrates, and Role in Cancer. Int. J. Mol. Sci. 2019, 20, 1194. [Google Scholar] [CrossRef]
- Wortzel, I.; Seger, R. The ERK Cascade: Distinct Functions within Various Subcellular Organelles. Genes Cancer 2011, 2, 195–209. [Google Scholar] [CrossRef]
- MacCorkle, R.A.; Tan, T.H. Mitogen-activated protein kinases in cell-cycle control. Cell Biochem. Biophys. 2005, 43, 451–461. [Google Scholar] [CrossRef]
- Wlodarchak, N.; Xing, Y. PP2A as a master regulator of the cell cycle. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 162–184. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Khandelwal, N.; Thachamvally, R.; Tripathi, B.N.; Barua, S.; Kashyap, S.K.; Maherchandani, S.; Kumar, N. Role of MAPK/MNK1 signaling in virus replication. Virus Res. 2018, 253, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Dudley, D.T.; Pang, L.; Decker, S.J.; Bridges, A.J.; Saltiel, A.R. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc. Natl. Acad. Sci. USA 1995, 92, 7686–7689. [Google Scholar] [CrossRef] [PubMed]
- Favata, M.F.; Horiuchi, K.Y.; Manos, E.J.; Daulerio, A.J.; Stradley, D.A.; Feeser, W.S.; Van Dyk, D.E.; Pitts, W.J.; Earl, R.A.; Hobbs, F.; et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 1998, 273, 18623–18632. [Google Scholar] [CrossRef]
- DuShane, J.K.; Wilczek, M.P.; Crocker, M.A.; Maginnis, M.S. High-Throughput Characterization of Viral and Cellular Protein Expression Patterns During JC Polyomavirus Infection. Front. Microbiol. 2019, 10, 783. [Google Scholar] [CrossRef] [PubMed]
- Ghebremedhin, B. Human adenovirus: Viral pathogen with increasing importance. Eur. J. Microbiol. Immunol. (Bp) 2014, 4, 26–33. [Google Scholar] [CrossRef]
- Lion, T. Adenovirus infections in immunocompetent and immunocompromised patients. Clin. Microbiol. Rev. 2014, 27, 441–462. [Google Scholar] [CrossRef]
- Saha, B.; Wong, C.M.; Parks, R.J. The adenovirus genome contributes to the structural stability of the virion. Viruses 2014, 6, 3563–3583. [Google Scholar] [CrossRef]
- Tamanini, A.; Rolfini, R.; Nicolis, E.; Melotti, P.; Cabrini, G. MAP kinases and NF-kappaB collaborate to induce ICAM-1 gene expression in the early phase of adenovirus infection. Virology 2003, 307, 228–242. [Google Scholar] [CrossRef]
- Rajaiya, J.; Xiao, J.; Rajala, R.V.; Chodosh, J. Human adenovirus type 19 infection of corneal cells induces p38 MAPK-dependent interleukin-8 expression. Virol. J. 2008, 5, 17. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Suomalainen, M.; Nakano, M.Y.; Boucke, K.; Keller, S.; Greber, U.F. Adenovirus-activated PKA and p38/MAPK pathways boost microtubule-mediated nuclear targeting of virus. EMBO J. 2001, 20, 1310–1319. [Google Scholar] [CrossRef] [PubMed]
- Bruder, J.T.; Kovesdi, I. Adenovirus infection stimulates the Raf/MAPK signaling pathway and induces interleukin-8 expression. J. Virol. 1997, 71, 398–404. [Google Scholar] [PubMed]
- Alcorn, M.J.; Booth, J.L.; Coggeshall, K.M.; Metcalf, J.P. Adenovirus type 7 induces interleukin-8 production via activation of extracellular regulated kinase 1/2. J. Virol. 2001, 75, 6450–6459. [Google Scholar] [CrossRef] [PubMed]
- Booth, J.L.; Coggeshall, K.M.; Gordon, B.E.; Metcalf, J.P. Adenovirus type 7 induces interleukin-8 in a lung slice model and requires activation of Erk. J. Virol. 2004, 78, 4156–4164. [Google Scholar] [CrossRef]
- Schumann, M.; Dobbelstein, M. Adenovirus-induced extracellular signal-regulated kinase phosphorylation during the late phase of infection enhances viral protein levels and virus progeny. Cancer Res. 2006, 66, 1282–1288. [Google Scholar] [CrossRef]
- Wu, W.; Booth, J.L.; Coggeshall, K.M.; Metcalf, J.P. Calcium-dependent viral internalization is required for adenovirus type 7 induction of IL-8 protein. Virology 2006, 355, 18–29. [Google Scholar] [CrossRef][Green Version]
- Wold, W.S.; Toth, K. Adenovirus vectors for gene therapy, vaccination and cancer gene therapy. Curr. Gene Ther. 2013, 13, 421–433. [Google Scholar] [CrossRef]
- Mingozzi, F.; High, K.A. Immune responses to AAV vectors: Overcoming barriers to successful gene therapy. Blood 2013, 122, 23–36. [Google Scholar] [CrossRef]
- Taipale, K.; Tahtinen, S.; Havunen, R.; Koski, A.; Liikanen, I.; Pakarinen, P.; Koivisto-Korander, R.; Kankainen, M.; Joensuu, T.; Kanerva, A.; et al. Interleukin 8 activity influences the efficacy of adenoviral oncolytic immunotherapy in cancer patients. Oncotarget 2018, 9, 6320–6335. [Google Scholar] [CrossRef]
- Kotha, P.L.; Sharma, P.; Kolawole, A.O.; Yan, R.; Alghamri, M.S.; Brockman, T.L.; Gomez-Cambronero, J.; Excoffon, K.J. Adenovirus entry from the apical surface of polarized epithelia is facilitated by the host innate immune response. PLoS Pathog. 2015, 11, e1004696. [Google Scholar] [CrossRef] [PubMed]
- Bhat, N.R.; Fan, F. Adenovirus infection induces microglial activation: Involvement of mitogen-activated protein kinase pathways. Brain Res. 2002, 948, 93–101. [Google Scholar] [CrossRef]
- Si, J.; Kim, M.; Lim, M.Y.; Ko, G. Enhancement of enteric adenovirus cultivation in a novel Ras-overexpressing cell line. J. Gen. Virol. 2014, 95, 171–178. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kong, K.; Kumar, M.; Taruishi, M.; Javier, R.T. Adenovirus E4-ORF1 Dysregulates Epidermal Growth Factor and Insulin/Insulin-Like Growth Factor Receptors To Mediate Constitutive Myc Expression. J. Virol. 2015, 89, 10774–10785. [Google Scholar] [CrossRef] [PubMed]
- Moss, B. Poxvirus DNA replication. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Belongia, E.A.; Naleway, A.L. Smallpox vaccine: The good, the bad, and the ugly. Clin. Med. Res. 2003, 1, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Keinath, K.; Church, T.; Kurth, B.; Hulten, E. Myocarditis secondary to smallpox vaccination. BMJ Case Rep. 2018, 2018. [Google Scholar] [CrossRef]
- Grabenstein, J.D.; Winkenwerder, W., Jr. US military smallpox vaccination program experience. JAMA 2003, 289, 3278–3282. [Google Scholar] [CrossRef]
- Twardzik, D.R.; Brown, J.P.; Ranchalis, J.E.; Todaro, G.J.; Moss, B. Vaccinia virus-infected cells release a novel polypeptide functionally related to transforming and epidermal growth factors. Proc. Natl. Acad. Sci. USA 1985, 82, 5300–5304. [Google Scholar] [CrossRef]
- Yarden, Y. The EGFR family and its ligands in human cancer. signalling mechanisms and therapeutic opportunities. Eur. J. Cancer 2001, 37 (Suppl. 4), S3–S8. [Google Scholar] [CrossRef]
- Andrade, A.A.; Silva, P.N.; Pereira, A.C.; De Sousa, L.P.; Ferreira, P.C.; Gazzinelli, R.T.; Kroon, E.G.; Ropert, C.; Bonjardim, C.A. The vaccinia virus-stimulated mitogen-activated protein kinase (MAPK) pathway is required for virus multiplication. Biochem. J. 2004, 381, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Hruby, D.E.; Ball, L.A. Mapping and identification of the vaccinia virus thymidine kinase gene. J. Virol. 1982, 43, 403–409. [Google Scholar] [PubMed]
- de Magalhaes, J.C.; Andrade, A.A.; Silva, P.N.; Sousa, L.P.; Ropert, C.; Ferreira, P.C.; Kroon, E.G.; Gazzinelli, R.T.; Bonjardim, C.A. A mitogenic signal triggered at an early stage of vaccinia virus infection: Implication of MEK/ERK and protein kinase A in virus multiplication. J. Biol. Chem. 2001, 276, 38353–38360. [Google Scholar] [CrossRef] [PubMed]
- Schweneker, M.; Lukassen, S.; Spath, M.; Wolferstatter, M.; Babel, E.; Brinkmann, K.; Wielert, U.; Chaplin, P.; Suter, M.; Hausmann, J. The vaccinia virus O1 protein is required for sustained activation of extracellular signal-regulated kinase 1/2 and promotes viral virulence. J. Virol. 2012, 86, 2323–2336. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.N.; Soares, J.A.; Brasil, B.S.; Nogueira, S.V.; Andrade, A.A.; de Magalhaes, J.C.; Bonjardim, M.B.; Ferreira, P.C.; Kroon, E.G.; Bruna-Romero, O.; et al. Differential role played by the MEK/ERK/EGR-1 pathway in orthopoxviruses vaccinia and cowpox biology. Biochem. J. 2006, 398, 83–95. [Google Scholar] [CrossRef]
- Leite, F.G.G.; Torres, A.A.; De Oliveira, L.C.; Da Cruz, A.F.P.; Soares-Martins, J.A.P.; Pereira, A.; Trindade, G.S.; Abrahao, J.S.; Kroon, E.G.; Ferreira, P.C.P.; et al. c-Jun integrates signals from both MEK/ERK and MKK/JNK pathways upon vaccinia virus infection. Arch. Virol. 2017, 162, 2971–2981. [Google Scholar] [CrossRef]
- Allander, T.; Andreasson, K.; Gupta, S.; Bjerkner, A.; Bogdanovic, G.; Persson, M.A.; Dalianis, T.; Ramqvist, T.; Andersson, B. Identification of a third human polyomavirus. J. Virol. 2007, 81, 4130–4136. [Google Scholar] [CrossRef]
- Gaynor, A.M.; Nissen, M.D.; Whiley, D.M.; Mackay, I.M.; Lambert, S.B.; Wu, G.; Brennan, D.C.; Storch, G.A.; Sloots, T.P.; Wang, D. Identification of a novel polyomavirus from patients with acute respiratory tract infections. PLoS Pathog. 2007, 3, e64. [Google Scholar] [CrossRef]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef]
- Ho, J.; Jedrych, J.J.; Feng, H.; Natalie, A.A.; Grandinetti, L.; Mirvish, E.; Crespo, M.M.; Yadav, D.; Fasanella, K.E.; Proksell, S.; et al. Human polyomavirus 7-associated pruritic rash and viremia in transplant recipients. J. Infect. Dis. 2015, 211, 1560–1565. [Google Scholar] [CrossRef]
- van der Meijden, E.; Janssens, R.W.; Lauber, C.; Bouwes Bavinck, J.N.; Gorbalenya, A.E.; Feltkamp, M.C. Discovery of a new human polyomavirus associated with trichodysplasia spinulosa in an immunocompromized patient. PLoS Pathog. 2010, 6, e1001024. [Google Scholar] [CrossRef] [PubMed]
- Scuda, N.; Hofmann, J.; Calvignac-Spencer, S.; Ruprecht, K.; Liman, P.; Kuhn, J.; Hengel, H.; Ehlers, B. A novel human polyomavirus closely related to the african green monkey-derived lymphotropic polyomavirus. J. Virol. 2011, 85, 4586–4590. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Phan, G.Q.; Raiji, M.T.; Murphy, P.M.; McDermott, D.H.; McBride, A.A. Complete genome sequence of a tenth human polyomavirus. J. Virol. 2012, 86, 10887. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.S.; Reyes, A.; Antonio, M.; Saha, D.; Ikumapayi, U.N.; Adeyemi, M.; Stine, O.C.; Skelton, R.; Brennan, D.C.; Mkakosya, R.S.; et al. Discovery of STL polyomavirus, a polyomavirus of ancestral recombinant origin that encodes a unique T antigen by alternative splicing. Virology 2013, 436, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Korup, S.; Rietscher, J.; Calvignac-Spencer, S.; Trusch, F.; Hofmann, J.; Moens, U.; Sauer, I.; Voigt, S.; Schmuck, R.; Ehlers, B. Identification of a novel human polyomavirus in organs of the gastrointestinal tract. PLoS ONE 2013, 8, e58021. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.; Pereira, M.; Rhodes, R.H.; An, P.; Pipas, J.M.; Jain, K.; Kapoor, A.; Briese, T.; Faust, P.L.; Lipkin, W.I. Identification of a novel polyomavirus in a pancreatic transplant recipient with retinal blindness and vasculitic myopathy. J. Infect. Dis. 2014, 210, 1595–1599. [Google Scholar] [CrossRef] [PubMed]
- Schowalter, R.M.; Pastrana, D.V.; Pumphrey, K.A.; Moyer, A.L.; Buck, C.B. Merkel cell polyomavirus and two previously unknown polyomaviruses are chronically shed from human skin. Cell Host Microbe 2010, 7, 509–515. [Google Scholar] [CrossRef]
- Hirsch, H.H.; Kardas, P.; Kranz, D.; Leboeuf, C. The human JC polyomavirus (JCPyV): Virological background and clinical implications. APMIS 2013, 121, 685–727. [Google Scholar] [CrossRef]
- Hirsch, H.H. BK virus: Opportunity makes a pathogen. Clin. Infect. Dis. 2005, 41, 354–360. [Google Scholar] [CrossRef]
- Hirsch, H.H.; Brennan, D.C.; Drachenberg, C.B.; Ginevri, F.; Gordon, J.; Limaye, A.P.; Mihatsch, M.J.; Nickeleit, V.; Ramos, E.; Randhawa, P.; et al. Polyomavirus-associated nephropathy in renal transplantation: Interdisciplinary analyses and recommendations. Transplantation 2005, 79, 1277–1286. [Google Scholar] [CrossRef]
- Comoli, P.; Binggeli, S.; Ginevri, F.; Hirsch, H.H. Polyomavirus-associated nephropathy: Update on BK virus-specific immunity. Transpl. Infect. Dis. 2006, 8, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Funk, G.A.; Steiger, J.; Hirsch, H.H. Rapid dynamics of polyomavirus type BK in renal transplant recipients. J. Infect. Dis. 2006, 193, 80–87. [Google Scholar] [CrossRef] [PubMed]
- DeCaprio, J.A.; Garcea, R.L. A cornucopia of human polyomaviruses. Nat. Rev. Microbiol. 2013, 11, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Assetta, B.; Atwood, W.J. The biology of JC polyomavirus. Biol. Chem. 2017, 398, 839–855. [Google Scholar] [CrossRef] [PubMed]
- Padgett, B.L.; Walker, D.L.; ZuRhein, G.M.; Eckroade, R.J.; Dessel, B.H. Cultivation of papova-like virus from human brain with progressive multifocal leucoencephalopathy. Lancet 1971, 1, 1257–1260. [Google Scholar] [CrossRef]
- Kean, J.M.; Rao, S.; Wang, M.; Garcea, R.L. Seroepidemiology of human polyomaviruses. PLoS Pathog. 2009, 5, e1000363. [Google Scholar] [CrossRef] [PubMed]
- Egli, A.; Infanti, L.; Dumoulin, A.; Buser, A.; Samaridis, J.; Stebler, C.; Gosert, R.; Hirsch, H.H. Prevalence of polyomavirus BK and JC infection and replication in 400 healthy blood donors. J. Infect. Dis. 2009, 199, 837–846. [Google Scholar] [CrossRef]
- Aksamit, A.J.; Major, E.O.; Ghatak, N.R.; Sidhu, G.S.; Parisi, J.E.; Guccion, J.G. Diagnosis of progressive multifocal leukoencephalopathy by brain biopsy with biotin labeled DNA:DNA in situ hybridization. J. Neuropathol. Exp. Neurol. 1987, 46, 556–566. [Google Scholar] [CrossRef]
- Houff, S.A.; Katz, D.; Kufta, C.V.; Major, E.O. A rapid method for in situ hybridization for viral DNA in brain biopsies from patients with AIDS. AIDS 1989, 3, 843–845. [Google Scholar] [CrossRef]
- Major, E.O. Progressive multifocal leukoencephalopathy in patients on immunomodulatory therapies. Annu. Rev. Med. 2010, 61, 35–47. [Google Scholar] [CrossRef]
- Friedman-Kien, A.E. Disseminated Kaposi’s sarcoma syndrome in young homosexual men. J. Am. Acad. Dermatol. 1981, 5, 468–471. [Google Scholar] [CrossRef]
- Molloy, E.S.; Calabrese, L.H. Progressive multifocal leukoencephalopathy: A national estimate of frequency in systemic lupus erythematosus and other rheumatic diseases. Arthritis Rheum. 2009, 60, 3761–3765. [Google Scholar] [CrossRef] [PubMed]
- Assetta, B.; Maginnis, M.S.; Gracia Ahufinger, I.; Haley, S.A.; Gee, G.V.; Nelson, C.D.; O’Hara, B.A.; Allen Ramdial, S.A.; Atwood, W.J. 5-HT2 receptors facilitate JC polyomavirus entry. J. Virol. 2013, 87, 13490–13498. [Google Scholar] [CrossRef] [PubMed]
- Roth, B.L. Irving Page Lecture: 5-HT(2A) serotonin receptor biology: Interacting proteins, kinases and paradoxical regulation. Neuropharmacology 2011, 61, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Querbes, W.; Benmerah, A.; Tosoni, D.; Di Fiore, P.P.; Atwood, W.J. A JC virus-induced signal is required for infection of glial cells by a clathrin- and eps15-dependent pathway. J. Virol. 2004, 78, 250–256. [Google Scholar] [CrossRef] [PubMed]
- DuShane, J.K.; Wilczek, M.P.; Mayberry, C.L.; Maginnis, M.S. ERK Is a Critical Regulator of JC Polyomavirus Infection. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Safak, M.; Gallia, G.L.; Khalili, K. A 23-bp sequence element from human neurotropic JC virus is responsive to NF-kappa B subunits. Virology 1999, 262, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, V.; Jensen, P.N.; Major, E.O. MEK1/2 inhibitors block basal and transforming growth factor 1beta1-stimulated JC virus multiplication. J. Virol. 2007, 81, 6412–6418. [Google Scholar] [CrossRef]
- Bollag, B.; Hofstetter, C.A.; Reviriego-Mendoza, M.M.; Frisque, R.J. JC virus small T antigen binds phosphatase PP2A and Rb family proteins and is required for efficient viral DNA replication activity. PLoS ONE 2010, 5, e10606. [Google Scholar] [CrossRef]
- Enam, S.; Sweet, T.M.; Amini, S.; Khalili, K.; Del Valle, L. Evidence for involvement of transforming growth factor beta1 signaling pathway in activation of JC virus in human immunodeficiency virus 1-associated progressive multifocal leukoencephalopathy. Arch. Pathol. Lab Med. 2004, 128, 282–291. [Google Scholar] [CrossRef]
- Ripple, M.J.; Parker Struckhoff, A.; Trillo-Tinoco, J.; Li, L.; Margolin, D.A.; McGoey, R.; Del Valle, L. Activation of c-Myc and Cyclin D1 by JCV T-Antigen and beta-catenin in colon cancer. PLoS ONE 2014, 9, e106257. [Google Scholar] [CrossRef] [PubMed]
- Gan, D.D.; Khalili, K. Interaction between JCV large T-antigen and beta-catenin. Oncogene 2004, 23, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Manley, K.; O’Hara, B.A.; Gee, G.V.; Simkevich, C.P.; Sedivy, J.M.; Atwood, W.J. NFAT4 is required for JC virus infection of glial cells. J. Virol. 2006, 80, 12079–12085. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Woolridge, S.; Biffi, R.; Borghi, E.; Lassak, A.; Ferrante, P.; Amini, S.; Khalili, K.; Safak, M. Members of the AP-1 family, c-Jun and c-Fos, functionally interact with JC virus early regulatory protein large T antigen. J. Virol. 2003, 77, 5241–5252. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Tang, W.J.; Bu, X.; Bermudez, V.; Martin, M.; Folk, W.R. AP1 enhances polyomavirus DNA replication by promoting T-antigen-mediated unwinding of DNA. J. Virol. 1996, 70, 4914–4918. [Google Scholar] [PubMed]
- Hirsch, H.H.; Randhawa, P.; AST Infectious Diseases Community of Practice. BK polyomavirus in solid organ transplantation. Am. J. Transplant 2013, 13 (Suppl. 4), 179–188. [Google Scholar] [CrossRef]
- Gardner, S.D.; Field, A.M.; Coleman, D.V.; Hulme, B. New human papovavirus (B.K.) isolated from urine after renal transplantation. Lancet 1971, 1, 1253–1257. [Google Scholar] [CrossRef]
- Hirsch, H.H.; Steiger, J. Polyomavirus BK. Lancet Infect. Dis. 2003, 3, 611–623. [Google Scholar] [CrossRef]
- Cubitt, C.L. Molecular genetics of the BK virus. Adv. Exp. Med. Biol. 2006, 577, 85–95. [Google Scholar] [CrossRef]
- Rinaldo, C.H.; Tylden, G.D.; Sharma, B.N. The human polyomavirus BK (BKPyV): Virological background and clinical implications. APMIS 2013, 121, 728–745. [Google Scholar] [CrossRef]
- Rundell, K.; Major, E.O.; Lampert, M. Association of cellular 56,000- and 32,000-molecular-weight protein with BK virus and polyoma virus t-antigens. J. Virol. 1981, 37, 1090–1093. [Google Scholar] [PubMed]
- Tognon, M.; Corallini, A.; Martini, F.; Negrini, M.; Barbanti-Brodano, G. Oncogenic transformation by BK virus and association with human tumors. Oncogene 2003, 22, 5192–5200. [Google Scholar] [CrossRef] [PubMed]
- Levican, J.; Acevedo, M.; Leon, O.; Gaggero, A.; Aguayo, F. Role of BK human polyomavirus in cancer. Infect. Agent Cancer 2018, 13, 12. [Google Scholar] [CrossRef] [PubMed]
- Seamone, M.E.; Wang, W.; Acott, P.; Beck, P.L.; Tibbles, L.A.; Muruve, D.A. MAP kinase activation increases BK polyomavirus replication and facilitates viral propagation in vitro. J. Virol. Methods 2010, 170, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.D.; Lee, E.E.; Yue, Y.; Stork, J.; Pock, L.; North, J.P.; Vandergriff, T.; Cockerell, C.; Hosler, G.A.; Pastrana, D.V.; et al. Human polyomavirus 6 and 7 are associated with pruritic and dyskeratotic dermatoses. J. Am. Acad. Dermatol. 2017, 76, 932–940.e3. [Google Scholar] [CrossRef]
- Wu, J.H.; Simonette, R.A.; Nguyen, H.P.; Rady, P.L.; Tyring, S.K. Molecular mechanisms supporting a pathogenic role for human polyomavirus 6 small T antigen: Protein phosphatase 2A targeting and MAPK cascade activation. J. Med. Virol. 2017, 89, 742–747. [Google Scholar] [CrossRef] [PubMed]
- Kazem, S.; van der Meijden, E.; Feltkamp, M.C. The trichodysplasia spinulosa-associated polyomavirus: Virological background and clinical implications. APMIS 2013, 121, 770–782. [Google Scholar] [CrossRef]
- Wu, J.H.; Narayanan, D.; Simonette, R.A.; Rady, P.L.; Tyring, S.K. Human polyomavirus 7 (HPyV7)-associated dermatoses: Novel molecular mechanism driven by viral activation of 4E-BP1 and MEK-ERK-cJun. Int. J. Dermatol. 2019, 58, 383–387. [Google Scholar] [CrossRef]
- Wu, J.H.; Simonette, R.A.; Nguyen, H.P.; Rady, P.L.; Tyring, S.K. Small T-antigen of the TS-associated polyomavirus activates factors implicated in the MAPK pathway. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 1061–1062. [Google Scholar] [CrossRef]
- Wu, J.H.; Simonette, R.A.; Nguyen, H.P.; Rady, P.L.; Tyring, S.K. Merkel cell polyomavirus in Merkel cell carcinogenesis: Small T antigen-mediates c-Jun phosphorylation. Virus Genes 2016, 52, 397–399. [Google Scholar] [CrossRef]
- Van Doorslaer, K.; Li, Z.; Xirasagar, S.; Maes, P.; Kaminsky, D.; Liou, D.; Sun, Q.; Kaur, R.; Huyen, Y.; McBride, A.A. The Papillomavirus Episteme: A major update to the papillomavirus sequence database. Nucleic Acids Res. 2017, 45, D499–D506. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.V. Human papillomavirus: Gene expression, regulation and prospects for novel diagnostic methods and antiviral therapies. Future Microbiol. 2010, 5, 1493–1506. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 2015, 25 (Suppl. 1), 2–23. [Google Scholar] [CrossRef]
- Graham, S.V. The human papillomavirus replication cycle, and its links to cancer progression: A comprehensive review. Clin. Sci. (Lond.) 2017, 131, 2201–2221. [Google Scholar] [CrossRef] [PubMed]
- de Sanjose, S.; Quint, W.G.; Alemany, L.; Geraets, D.T.; Klaustermeier, J.E.; Lloveras, B.; Tous, S.; Felix, A.; Bravo, L.E.; Shin, H.R.; et al. Human papillomavirus genotype attribution in invasive cervical cancer: A retrospective cross-sectional worldwide study. Lancet Oncol. 2010, 11, 1048–1056. [Google Scholar] [CrossRef]
- Bruni, L.; Diaz, M.; Castellsague, X.; Ferrer, E.; Bosch, F.X.; de Sanjose, S. Cervical human papillomavirus prevalence in 5 continents: Meta-analysis of 1 million women with normal cytological findings. J. Infect. Dis. 2010, 202, 1789–1799. [Google Scholar] [CrossRef] [PubMed]
- Narisawa-Saito, M.; Kiyono, T. Basic mechanisms of high-risk human papillomavirus-induced carcinogenesis: Roles of E6 and E7 proteins. Cancer Sci. 2007, 98, 1505–1511. [Google Scholar] [CrossRef]
- Tomaic, V. Functional Roles of E6 and E7 Oncoproteins in HPV-Induced Malignancies at Diverse Anatomical Sites. Cancers (Basel) 2016, 8, 95. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Hoover, A.C.; Strand, G.L.; Nowicki, P.N.; Anderson, M.E.; Vermeer, P.D.; Klingelhutz, A.J.; Bossler, A.D.; Pottala, J.V.; Hendriks, W.J.; Lee, J.H. Impaired PTPN13 phosphatase activity in spontaneous or HPV-induced squamous cell carcinomas potentiates oncogene signaling through the MAP kinase pathway. Oncogene 2009, 28, 3960–3970. [Google Scholar] [CrossRef] [PubMed]
- Rong, C.; Muller, M.; Flechtenmacher, C.; Holzinger, D.; Dyckhoff, G.; Bulut, O.C.; Horn, D.; Plinkert, P.; Hess, J.; Affolter, A. Differential Activation of ERK Signaling in HPV-Related Oropharyngeal Squamous Cell Carcinoma. Cancers (Basel) 2019, 11, 584. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Chaturvedi, A.K.; Anderson, W.F.; Fakhry, C. Epidemiology of Human Papillomavirus-Positive Head and Neck Squamous Cell Carcinoma. J. Clin. Oncol. 2015, 33, 3235–3242. [Google Scholar] [CrossRef] [PubMed]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tan, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Spangle, J.M.; Munger, K. The HPV16 E6 oncoprotein causes prolonged receptor protein tyrosine kinase signaling and enhances internalization of phosphorylated receptor species. PLoS Pathog. 2013, 9, e1003237. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zhang, Q.; Nishitani, J.; Brown, J.; Shi, S.; Le, A.D. Overexpression of human papillomavirus type 16 oncoproteins enhances hypoxia-inducible factor 1 alpha protein accumulation and vascular endothelial growth factor expression in human cervical carcinoma cells. Clin. Cancer Res. 2007, 13, 2568–2576. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lin, B.; Liu, X.; Zhang, W.; Zhang, E.; Hu, L.; Ma, Y.; Li, X.; Tang, X. ERK Signaling Pathway Is Involved in HPV-16 E6 but not E7 Oncoprotein-Induced HIF-1alpha Protein Accumulation in NSCLC Cells. Oncol. Res. 2016, 23, 109–118. [Google Scholar] [CrossRef]
- Pim, D.; Collins, M.; Banks, L. Human papillomavirus type 16 E5 gene stimulates the transforming activity of the epidermal growth factor receptor. Oncogene 1992, 7, 27–32. [Google Scholar]
- Straight, S.W.; Hinkle, P.M.; Jewers, R.J.; McCance, D.J. The E5 oncoprotein of human papillomavirus type 16 transforms fibroblasts and effects the downregulation of the epidermal growth factor receptor in keratinocytes. J. Virol. 1993, 67, 4521–4532. [Google Scholar]
- Kim, S.H.; Juhnn, Y.S.; Kang, S.; Park, S.W.; Sung, M.W.; Bang, Y.J.; Song, Y.S. Human papillomavirus 16 E5 up-regulates the expression of vascular endothelial growth factor through the activation of epidermal growth factor receptor, MEK/ ERK1,2 and PI3K/Akt. Cell Mol. Life Sci. 2006, 63, 930–938. [Google Scholar] [CrossRef]
- Kodama, J.; Seki, N.; Tokumo, K.; Hongo, A.; Miyagi, Y.; Yoshinouchi, M.; Okuda, H.; Kudo, T. Vascular endothelial growth factor is implicated in early invasion in cervical cancer. Eur. J. Cancer 1999, 35, 485–489. [Google Scholar] [CrossRef]
- Cheng, W.F.; Chen, C.A.; Lee, C.N.; Wei, L.H.; Hsieh, F.J.; Hsieh, C.Y. Vascular endothelial growth factor and prognosis of cervical carcinoma. Obstet. Gynecol. 2000, 96, 721–726. [Google Scholar] [PubMed]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30 (Suppl. 5), F55–F70. [Google Scholar] [CrossRef]
- Zhang, B.; Spandau, D.F.; Roman, A. E5 protein of human papillomavirus type 16 protects human foreskin keratinocytes from UV B-irradiation-induced apoptosis. J. Virol. 2002, 76, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Surviladze, Z.; Sterk, R.T.; DeHaro, S.A.; Ozbun, M.A. Cellular entry of human papillomavirus type 16 involves activation of the phosphatidylinositol 3-kinase/Akt/mTOR pathway and inhibition of autophagy. J. Virol. 2013, 87, 2508–2517. [Google Scholar] [CrossRef] [PubMed]
- Fothergill, T.; McMillan, N.A. Papillomavirus virus-like particles activate the PI3-kinase pathway via alpha-6 beta-4 integrin upon binding. Virology 2006, 352, 319–328. [Google Scholar] [CrossRef]
- Crusius, K.; Auvinen, E.; Alonso, A. Enhancement of EGF- and PMA-mediated MAP kinase activation in cells expressing the human papillomavirus type 16 E5 protein. Oncogene 1997, 15, 1437–1444. [Google Scholar] [CrossRef]
- Surviladze, Z.; Dziduszko, A.; Ozbun, M.A. Essential roles for soluble virion-associated heparan sulfonated proteoglycans and growth factors in human papillomavirus infections. PLoS Pathog. 2012, 8, e1002519. [Google Scholar] [CrossRef]
- Howie, H.L.; Koop, J.I.; Weese, J.; Robinson, K.; Wipf, G.; Kim, L.; Galloway, D.A. Beta-HPV 5 and 8 E6 promote p300 degradation by blocking AKT/p300 association. PLoS Pathog. 2011, 7, e1002211. [Google Scholar] [CrossRef]
- Rosenberger, S.; De-Castro Arce, J.; Langbein, L.; Steenbergen, R.D.; Rosl, F. Alternative splicing of human papillomavirus type-16 E6/E6* early mRNA is coupled to EGF signaling via Erk1/2 activation. Proc. Natl. Acad. Sci. USA 2010, 107, 7006–7011. [Google Scholar] [CrossRef]
- Sharma, V.; Mobeen, F.; Prakash, T. Comparative Genomics of Herpesviridae Family to Look for Potential Signatures of Human Infecting Strains. Int. J. Genomics 2016, 2016, 9543274. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Shenk, T. Human cytomegalovirus genome. Curr. Top. Microbiol. Immunol. 2008, 325, 1–19. [Google Scholar] [PubMed]
- Davison, A.J. Comparative analysis of the genomes. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007; pp. 10–26. [Google Scholar]
- Whitley, R.J. Herpesviruses. In Medical Microbiology; Baron, S., Ed.; Available online: https://www.ncbi.nlm.nih.gov/books/NBK7627/ (accessed on 12 July 2019).
- Whitley, R.J.; Roizman, B. Herpes simplex virus infections. Lancet 2001, 357, 1513–1518. [Google Scholar] [CrossRef]
- Grunewald, K.; Desai, P.; Winkler, D.C.; Heymann, J.B.; Belnap, D.M.; Baumeister, W.; Steven, A.C. Three-dimensional structure of herpes simplex virus from cryo-electron tomography. Science 2003, 302, 1396–1398. [Google Scholar] [CrossRef] [PubMed]
- Laine, R.F.; Albecka, A.; van de Linde, S.; Rees, E.J.; Crump, C.M.; Kaminski, C.F. Structural analysis of herpes simplex virus by optical super-resolution imaging. Nat. Commun. 2015, 6, 5980. [Google Scholar] [CrossRef]
- Whitley, R.; Kimberlin, D.W.; Prober, C.G. HSV-1 AND 2: Pathogenesis and disease. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007; pp. 589–601. [Google Scholar]
- Xu, F.; Schillinger, J.A.; Sternberg, M.R.; Johnson, R.E.; Lee, F.K.; Nahmias, A.J.; Markowitz, L.E. Seroprevalence and coinfection with herpes simplex virus type 1 and type 2 in the United States, 1988-1994. J. Infect. Dis. 2002, 185, 1019–1024. [Google Scholar] [CrossRef] [PubMed]
- Wald, A.; Corey, L. HSV: Persistence in the population: Epidemiology, transmission. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007; pp. 656–671. [Google Scholar]
- Filippakis, H.; Spandidos, D.A.; Sourvinos, G. Herpesviruses: Hijacking the Ras signaling pathway. Biochim. Biophys. Acta 2010, 1803, 777–785. [Google Scholar] [CrossRef]
- Nicoll, M.P.; Proenca, J.T.; Efstathiou, S. The molecular basis of herpes simplex virus latency. FEMS Microbiol. Rev. 2012, 36, 684–705. [Google Scholar] [CrossRef]
- He, B.; Gross, M.; Roizman, B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 843–848. [Google Scholar] [CrossRef]
- Talloczy, Z.; Virgin, H.W.t.; Levine, B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2006, 2, 24–29. [Google Scholar] [CrossRef]
- Colao, I.; Pennisi, R.; Venuti, A.; Nygardas, M.; Heikkila, O.; Hukkanen, V.; Sciortino, M.T. The ERK-1 function is required for HSV-1-mediated G1/S progression in HEP-2 cells and contributes to virus growth. Sci. Rep. 2017, 7, 9176. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Van Sant, C.; Roizman, B. Herpes simplex virus 1 alpha regulatory protein ICP0 interacts with and stabilizes the cell cycle regulator cyclin D3. J. Virol. 1997, 71, 7328–7336. [Google Scholar] [PubMed]
- Kalamvoki, M.; Roizman, B. ICP0 enables and monitors the function of D cyclins in herpes simplex virus 1 infected cells. Proc. Natl. Acad. Sci. USA 2009, 106, 14576–14580. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, H.H.; van Loben Sels, J.M.; Davido, D.J. Herpes simplex virus 1 upregulates p35, alters CDK-5 localization, and stimulates CDK-5 kinase activity during acute infection in neurons. J. Virol. 2015, 89, 5171–5175. [Google Scholar] [CrossRef] [PubMed]
- Man, A.; Slevin, M.; Petcu, E.; Fraefel, C. The Cyclin-Dependent Kinase 5 Inhibitor Peptide Inhibits Herpes Simplex Virus Type 1 Replication. Sci. Rep. 2019, 9, 1260. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.C.; Smith, C.C.; Bose, D.; Kulka, M.; Broderick, R.; Aurelian, L. Intracellular internalization and signaling pathways triggered by the large subunit of HSV-2 ribonucleotide reductase (ICP10). Virology 1995, 210, 345–360. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Smith, R.L.; Escudero, J.M.; Wilcox, C.L. Regulation of the herpes simplex virus latency-associated transcripts during establishment of latency in sensory neurons in vitro. Virology 1994, 202, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.D.; Mezhir, J.J.; Bickenbach, K.; Veerapong, J.; Charron, J.; Posner, M.C.; Roizman, B.; Weichselbaum, R.R. Activated MEK suppresses activation of PKR and enables efficient replication and in vivo oncolysis by Deltagamma(1)34.5 mutants of herpes simplex virus 1. J. Virol. 2006, 80, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Leopardi, R.; Van Sant, C.; Roizman, B. The herpes simplex virus 1 protein kinase US3 is required for protection from apoptosis induced by the virus. Proc. Natl. Acad. Sci. USA 1997, 94, 7891–7896. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; He, S. The interplay between human herpes simplex virus infection and the apoptosis and necroptosis cell death pathways. Virol. J. 2016, 13, 77. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.; Roizman, B. The US3 protein kinase of herpes simplex virus 1 mediates the posttranslational modification of BAD and prevents BAD-induced programmed cell death in the absence of other viral proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 10410–10415. [Google Scholar] [CrossRef] [PubMed]
- Chuluunbaatar, U.; Roller, R.; Mohr, I. Suppression of extracellular signal-regulated kinase activity in herpes simplex virus 1-infected cells by the Us3 protein kinase. J. Virol. 2012, 86, 7771–7776. [Google Scholar] [CrossRef] [PubMed]
- Perkins, D.; Pereira, E.F.; Aurelian, L. The herpes simplex virus type 2 R1 protein kinase (ICP10 PK) functions as a dominant regulator of apoptosis in hippocampal neurons involving activation of the ERK survival pathway and upregulation of the antiapoptotic protein Bag-1. J. Virol. 2003, 77, 1292–1305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Feng, H.; Luo, L.; Zhou, Q.; Luo, Z.; Peng, Y. Distinct effects of knocking down MEK1 and MEK2 on replication of herpes simplex virus type 2. Virus Res. 2010, 150, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Nelson, J.; Aurelian, L.; Gober, M.; Goswami, B.B. Ras-GAP binding and phosphorylation by herpes simplex virus type 2 RR1 PK (ICP10) and activation of the Ras/MEK/MAPK mitogenic pathway are required for timely onset of virus growth. J. Virol. 2000, 74, 10417–10429. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Whitby, D.; Howard, M.R.; Tenant-Flowers, M.; Brink, N.S.; Copas, A.; Boshoff, C.; Hatzioannou, T.; Suggett, F.E.; Aldam, D.M.; Denton, A.S.; et al. Detection of Kaposi sarcoma associated herpesvirus in peripheral blood of HIV-infected individuals and progression to Kaposi’s sarcoma. Lancet 1995, 346, 799–802. [Google Scholar] [CrossRef]
- Arias, C.; Weisburd, B.; Stern-Ginossar, N.; Mercier, A.; Madrid, A.S.; Bellare, P.; Holdorf, M.; Weissman, J.S.; Ganem, D. KSHV 2.0: A comprehensive annotation of the Kaposi’s sarcoma-associated herpesvirus genome using next-generation sequencing reveals novel genomic and functional features. PLoS Pathog. 2014, 10, e1003847. [Google Scholar] [CrossRef] [PubMed]
- Ganem, D. KSHV infection and the pathogenesis of Kaposi’s sarcoma. Annu. Rev. Pathol. 2006, 1, 273–296. [Google Scholar] [CrossRef]
- Gao, X.; Ikuta, K.; Tajima, M.; Sairenji, T. 12-O-tetradecanoylphorbol-13-acetate induces Epstein-Barr virus reactivation via NF-kappaB and AP-1 as regulated by protein kinase C and mitogen-activated protein kinase. Virology 2001, 286, 91–99. [Google Scholar] [CrossRef]
- Cohen, A.; Brodie, C.; Sarid, R. An essential role of ERK signalling in TPA-induced reactivation of Kaposi’s sarcoma-associated herpesvirus. J. Gen. Virol. 2006, 87, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Ajibade, A.O.; Ye, F.; Kuhne, K.; Gao, S.J. Reactivation of Kaposi’s sarcoma-associated herpesvirus from latency requires MEK/ERK, JNK and p38 multiple mitogen-activated protein kinase pathways. Virology 2008, 371, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Harada, J.N.; Brown, H.J.; Deng, H.; Song, M.J.; Wu, T.T.; Kato-Stankiewicz, J.; Nelson, C.G.; Vieira, J.; Tamanoi, F.; et al. Systematic identification of cellular signals reactivating Kaposi sarcoma-associated herpesvirus. PLoS Pathog. 2007, 3, e44. [Google Scholar] [CrossRef] [PubMed]
- Ford, P.W.; Bryan, B.A.; Dyson, O.F.; Weidner, D.A.; Chintalgattu, V.; Akula, S.M. Raf/MEK/ERK signalling triggers reactivation of Kaposi’s sarcoma-associated herpesvirus latency. J. Gen. Virol. 2006, 87, 1139–1144. [Google Scholar] [CrossRef] [PubMed]
- Sarid, R.; Wiezorek, J.S.; Moore, P.S.; Chang, Y. Characterization and cell cycle regulation of the major Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) latent genes and their promoter. J. Virol. 1999, 73, 1438–1446. [Google Scholar] [PubMed]
- Akula, S.M.; Ford, P.W.; Whitman, A.G.; Hamden, K.E.; Shelton, J.G.; McCubrey, J.A. Raf promotes human herpesvirus-8 (HHV-8/KSHV) infection. Oncogene 2004, 23, 5227–5241. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pan, H.; Xie, J.; Ye, F.; Gao, S.J. Modulation of Kaposi’s sarcoma-associated herpesvirus infection and replication by MEK/ERK, JNK, and p38 multiple mitogen-activated protein kinase pathways during primary infection. J. Virol. 2006, 80, 5371–5382. [Google Scholar] [CrossRef]
- Naranatt, P.P.; Akula, S.M.; Zien, C.A.; Krishnan, H.H.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus induces the phosphatidylinositol 3-kinase-PKC-zeta-MEK-ERK signaling pathway in target cells early during infection: Implications for infectivity. J. Virol. 2003, 77, 1524–1539. [Google Scholar] [CrossRef]
- Sharma-Walia, N.; Krishnan, H.H.; Naranatt, P.P.; Zeng, L.; Smith, M.S.; Chandran, B. ERK1/2 and MEK1/2 induced by Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) early during infection of target cells are essential for expression of viral genes and for establishment of infection. J. Virol. 2005, 79, 10308–10329. [Google Scholar] [CrossRef]
- Li, X.; Du, S.; Avey, D.; Li, Y.; Zhu, F.; Kuang, E. ORF45-Mediated Prolonged c-Fos Accumulation Accelerates Viral Transcription during the Late Stage of Lytic Replication of Kaposi’s Sarcoma-Associated Herpesvirus. J. Virol. 2015, 89, 6895–6906. [Google Scholar] [CrossRef]
- Avey, D.; Tepper, S.; Li, W.; Turpin, Z.; Zhu, F. Phosphoproteomic Analysis of KSHV-Infected Cells Reveals Roles of ORF45-Activated RSK during Lytic Replication. PLoS Pathog. 2015, 11, e1004993. [Google Scholar] [CrossRef] [PubMed]
- Kuang, E.; Wu, F.; Zhu, F. Mechanism of sustained activation of ribosomal S6 kinase (RSK) and ERK by kaposi sarcoma-associated herpesvirus ORF45: Multiprotein complexes retain active phosphorylated ERK AND RSK and protect them from dephosphorylation. J. Biol. Chem. 2009, 284, 13958–13968. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y. EBV based cancer prevention and therapy in nasopharyngeal carcinoma. NPJ Precis Oncol. 2017, 1, 10. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J. The Burkitt Lymphoma: An African Enigma. McGill Med. J. 1964, 33, 130–133. [Google Scholar] [PubMed]
- Thorley-Lawson, D.A.; Babcock, G.J. A model for persistent infection with Epstein-Barr virus: The stealth virus of human B cells. Life Sci. 1999, 65, 1433–1453. [Google Scholar] [CrossRef]
- Babcock, G.J.; Hochberg, D.; Thorley-Lawson, A.D. The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity 2000, 13, 497–506. [Google Scholar] [CrossRef]
- Young, L.S.; Arrand, J.R.; Murray, P.G. EBV gene expression and regulation. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007; pp. 461–489. [Google Scholar]
- Roberts, M.L.; Cooper, N.R. Activation of a ras-MAPK-dependent pathway by Epstein-Barr virus latent membrane protein 1 is essential for cellular transformation. Virology 1998, 240, 93–99. [Google Scholar] [CrossRef]
- Vaysberg, M.; Hatton, O.; Lambert, S.L.; Snow, A.L.; Wong, B.; Krams, S.M.; Martinez, O.M. Tumor-derived variants of Epstein-Barr virus latent membrane protein 1 induce sustained Erk activation and c-Fos. J. Biol. Chem. 2008, 283, 36573–36585. [Google Scholar] [CrossRef]
- Fenton, M.; Sinclair, A.J. Divergent requirements for the MAPK(ERK) signal transduction pathway during initial virus infection of quiescent primary B cells and disruption of Epstein-Barr virus latency by phorbol esters. J. Virol. 1999, 73, 8913–8916. [Google Scholar]
- Mavromatidis, V.; Varga, Z.; Waczek, F.; Orfi, Z.; Orfi, L.; Keri, G.; Mosialos, G. Identification of protein kinase inhibitors with a selective negative effect on the viability of Epstein-Barr virus infected B cell lines. PLoS ONE 2014, 9, e95688. [Google Scholar] [CrossRef]
- Dawson, C.W.; Laverick, L.; Morris, M.A.; Tramoutanis, G.; Young, L.S. Epstein-Barr virus-encoded LMP1 regulates epithelial cell motility and invasion via the ERK-MAPK pathway. J. Virol. 2008, 82, 3654–3664. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, M.M.; Schulz, T.F. Regulation of intracellular signalling by the terminal membrane proteins of members of the Gammaherpesvirinae. J. Gen. Virol. 2006, 87, 1047–1074. [Google Scholar] [CrossRef] [PubMed]
- Panousis, C.G.; Rowe, D.T. Epstein-Barr virus latent membrane protein 2 associates with and is a substrate for mitogen-activated protein kinase. J. Virol. 1997, 71, 4752–4760. [Google Scholar] [PubMed]
- Chen, S.Y.; Lu, J.; Shih, Y.C.; Tsai, C.H. Epstein-Barr virus latent membrane protein 2A regulates c-Jun protein through extracellular signal-regulated kinase. J. Virol. 2002, 76, 9556–9561. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.J.; Longnecker, R. EBV LMP2A provides a surrogate pre-B cell receptor signal through constitutive activation of the ERK/MAPK pathway. J. Gen. Virol. 2008, 89, 1563–1568. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.B.; Blumberg, B.S. Viral hepatitis, postnecrotic cirrhosis, and hepatocellular carcinoma. Lancet 1969, 2, 953. [Google Scholar] [CrossRef]
- Edmondson, H.A.; Steiner, P.E. Primary carcinoma of the liver: A study of 100 cases among 48,900 necropsies. Cancer 1954, 7, 462–503. [Google Scholar] [CrossRef]
- Sherlock, S.; Fox, R.A.; Niazi, S.P.; Scheuer, P.J. Chronic liver disease and primary liver-cell cancer with hepatitis-associated (Australia) antigen in serum. Lancet 1970, 1, 1243–1247. [Google Scholar] [CrossRef]
- Sutnick, A.I.; London, W.T.; Gerstley, B.J.; Cronlund, M.M.; Blumberg, B.S. Anicteric hepatitis associated with Australia antigen. Occurrence in patients with Down’s syndrome. JAMA 1968, 205, 670–674. [Google Scholar] [CrossRef]
- Seeger, C.; Mason, W.S. Molecular biology of hepatitis B virus infection. Virology 2015, 479–480, 672–686. [Google Scholar] [CrossRef]
- Kaplan, P.M.; Greenman, R.L.; Gerin, J.L.; Purcell, R.H.; Robinson, W.S. DNA polymerase associated with human hepatitis B antigen. J. Virol. 1973, 12, 995–1005. [Google Scholar] [PubMed]
- Robinson, W.S.; Greenman, R.L. DNA polymerase in the core of the human hepatitis B virus candidate. J. Virol. 1974, 13, 1231–1236. [Google Scholar] [PubMed]
- Chung, T.W.; Lee, Y.C.; Kim, C.H. Hepatitis B viral HBx induces matrix metalloproteinase-9 gene expression through activation of ERK and PI-3K/AKT pathways: Involvement of invasive potential. FASEB J. 2004, 18, 1123–1125. [Google Scholar] [CrossRef] [PubMed]
- Chin, R.; Earnest-Silveira, L.; Koeberlein, B.; Franz, S.; Zentgraf, H.; Dong, X.; Gowans, E.; Bock, C.T.; Torresi, J. Modulation of MAPK pathways and cell cycle by replicating hepatitis B virus: Factors contributing to hepatocarcinogenesis. J. Hepatol. 2007, 47, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Benn, J.; Schneider, R.J. Hepatitis B virus HBx protein deregulates cell cycle checkpoint controls. Proc. Natl. Acad. Sci. USA 1995, 92, 11215–11219. [Google Scholar] [CrossRef]
- Wang, H.Y.; Yang, S.L.; Liang, H.F.; Li, C.H. HBx protein promotes oval cell proliferation by up-regulation of cyclin D1 via activation of the MEK/ERK and PI3K/Akt pathways. Int. J. Mol. Sci. 2014, 15, 3507–3518. [Google Scholar] [CrossRef]
- Henkler, F.; Lopes, A.R.; Jones, M.; Koshy, R. Erk-independent partial activation of AP-1 sites by the hepatitis B virus HBx protein. J. Gen. Virol. 1998, 79, 2737–2742. [Google Scholar] [CrossRef]
- Hildt, E.; Munz, B.; Saher, G.; Reifenberg, K.; Hofschneider, P.H. The PreS2 activator MHBs(t) of hepatitis B virus activates c-raf-1/Erk2 signaling in transgenic mice. EMBO J. 2002, 21, 525–535. [Google Scholar] [CrossRef]
- Chen, Z.; Li, Y.X.; Fu, H.J.; Ren, Y.L.; Zou, L.; Shen, S.Z.; Chen, P.; Sun, T.; Huang, C.H. Hepatitis B Virus Core Antigen Stimulates IL-6 Expression via p38, ERK and NF-kappaB Pathways in Hepatocytes. Cell Physiol. Biochem. 2017, 41, 91–100. [Google Scholar] [CrossRef]
- Nijhara, R.; Jana, S.S.; Goswami, S.K.; Rana, A.; Majumdar, S.S.; Kumar, V.; Sarkar, D.P. Sustained activation of mitogen-activated protein kinases and activator protein 1 by the hepatitis B virus X protein in mouse hepatocytes in vivo. J. Virol. 2001, 75, 10348–10358. [Google Scholar] [CrossRef]
- Moore, P.S.; Chang, Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 2010, 10, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part B: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- Holderfield, M.; Deuker, M.M.; McCormick, F.; McMahon, M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat. Rev. Cancer 2014, 14, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Kinch, M.S.; Hoyer, D.; Patridge, E.; Plummer, M. Target selection for FDA-approved medicines. Drug Discov. Today 2015, 20, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [PubMed]
- Bollag, G.; Tsai, J.; Zhang, J.; Zhang, C.; Ibrahim, P.; Nolop, K.; Hirth, P. Vemurafenib: The first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discov. 2012, 11, 873–886. [Google Scholar] [CrossRef]
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 2006, 5, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Ott, P.A.; Shapiro, G.I. The biology and clinical development of MEK inhibitors for cancer. Drugs 2014, 74, 2111–2128. [Google Scholar] [CrossRef]
- Delire, B.; Starkel, P. The Ras/MAPK pathway and hepatocarcinoma: Pathogenesis and therapeutic implications. Eur. J. Clin. Invest. 2015, 45, 609–623. [Google Scholar] [CrossRef]
- Bracarda, S.; Caserta, C.; Sordini, L.; Rossi, M.; Hamzay, A.; Crino, L. Protein kinase inhibitors in the treatment of renal cell carcinoma: Sorafenib. Ann. Oncol. 2007, 18 (Suppl. 6), vi22–vi25. [Google Scholar] [CrossRef]
- Muntane, J.; De la Rosa, A.J.; Docobo, F.; Garcia-Carbonero, R.; Padillo, F.J. Targeting tyrosine kinase receptors in hepatocellular carcinoma. Curr. Cancer Drug Targets 2013, 13, 300–312. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Viral Family | Virus | MAPK-ERK Role in Infectious Lifecycle | |||
|---|---|---|---|---|---|
| Enhances Viral Replication | Cell Cycle Dysregulation | Altered Immune Response | Promote Cell Survival | ||
| Adenoviridae | HAdV | + | + | ||
| (E4-ORF1) | |||||
| Poxviridae | VACV | + | |||
| (O1) | |||||
| Polyomaviridae | JCPyV | + | |||
| (tAg) | |||||
| BKPyV | + | + | |||
| (tAg) | |||||
| Papillomaviridae | HPV | + | + | ||
| (E5) | |||||
| Herpesviridae | HSV-1 | + | + | + | + |
| (ICP0) | (ICP34.5) | (US3) | |||
| HSV-2 | + | + | + | ||
| (ICP10) | (ICP10) | ||||
| KSHV | + | ||||
| (ORF45) | |||||
| EBV | + | ||||
| (LMP1/2A) | |||||
| Hepadnaviridae | HBV | + | + | + | |
| (HBx) | (MHBs(t)) | (HBcAg) | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
DuShane, J.K.; Maginnis, M.S. Human DNA Virus Exploitation of the MAPK-ERK Cascade. Int. J. Mol. Sci. 2019, 20, 3427. https://doi.org/10.3390/ijms20143427
DuShane JK, Maginnis MS. Human DNA Virus Exploitation of the MAPK-ERK Cascade. International Journal of Molecular Sciences. 2019; 20(14):3427. https://doi.org/10.3390/ijms20143427
Chicago/Turabian StyleDuShane, Jeanne K., and Melissa S. Maginnis. 2019. "Human DNA Virus Exploitation of the MAPK-ERK Cascade" International Journal of Molecular Sciences 20, no. 14: 3427. https://doi.org/10.3390/ijms20143427
APA StyleDuShane, J. K., & Maginnis, M. S. (2019). Human DNA Virus Exploitation of the MAPK-ERK Cascade. International Journal of Molecular Sciences, 20(14), 3427. https://doi.org/10.3390/ijms20143427