Protective Effects of Euthyroidism Restoration on Mitochondria Function and Quality Control in Cardiac Pathophysiology





Abstract
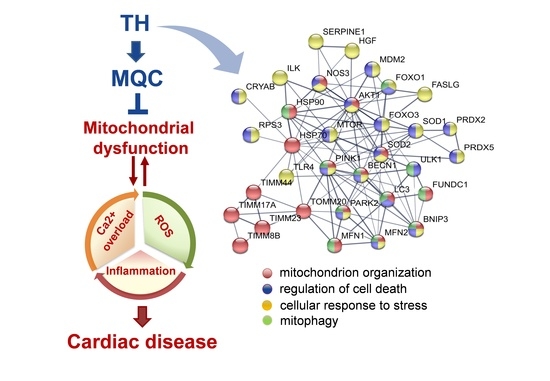
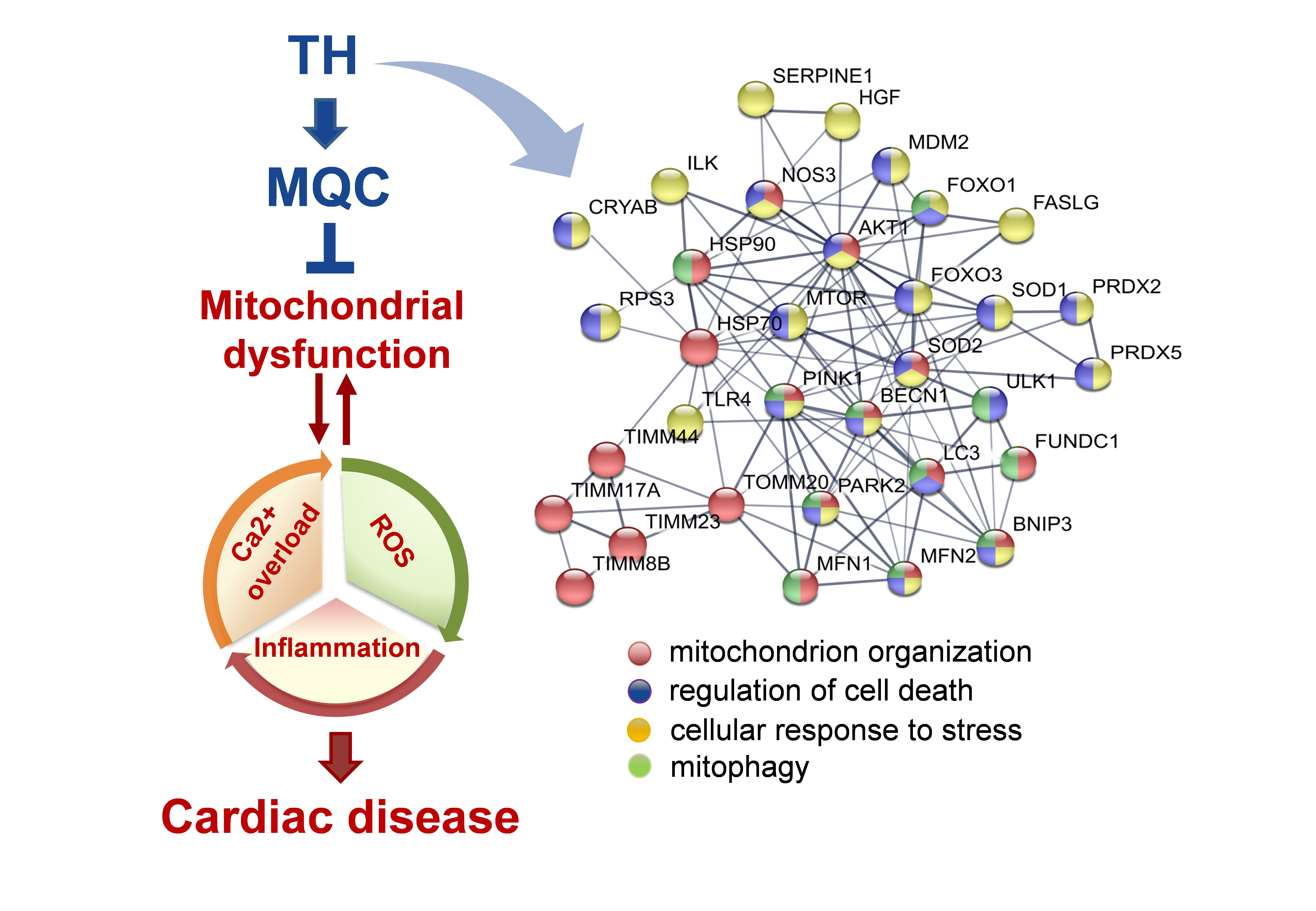{kind=link}
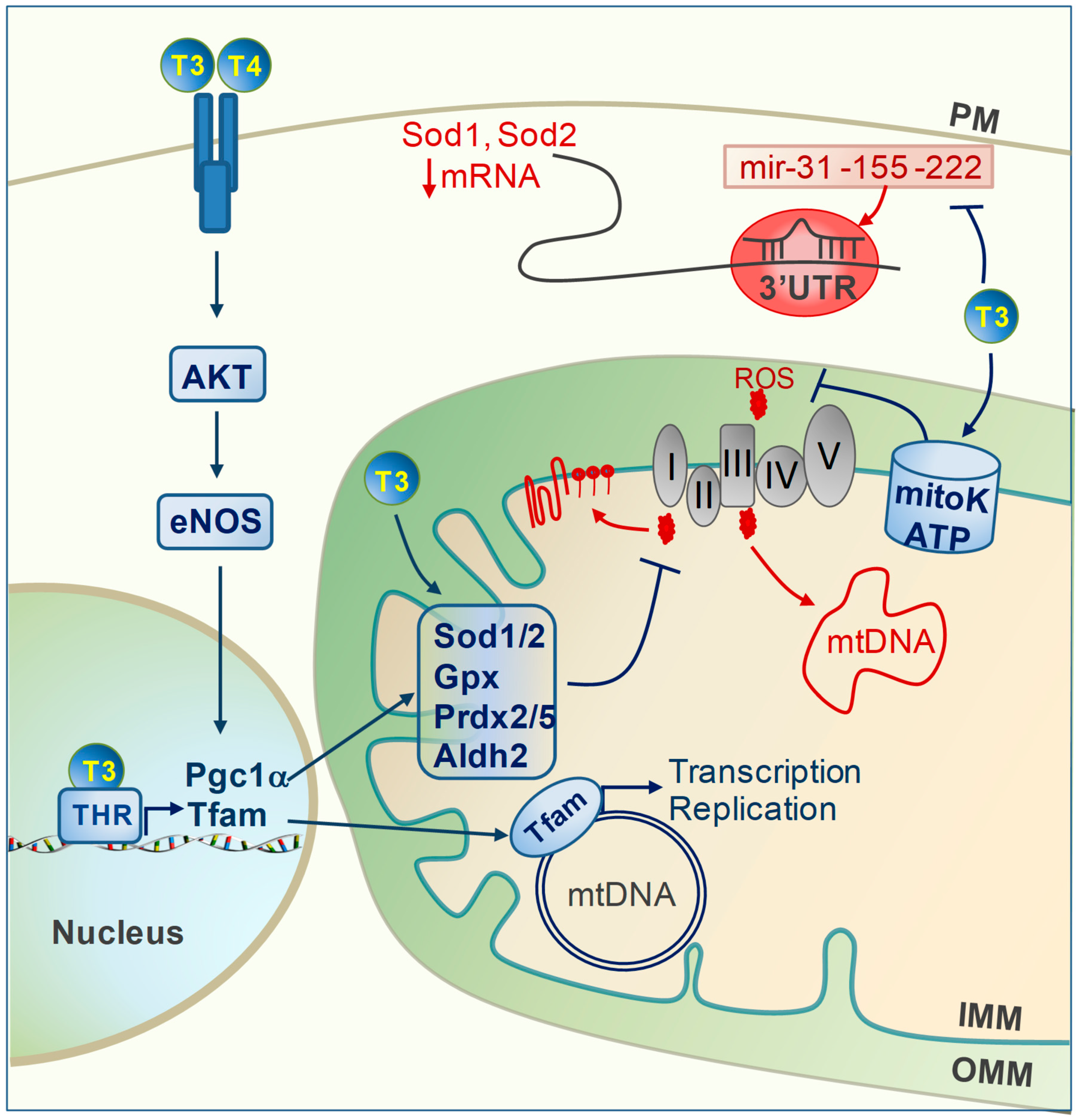{kind=link}
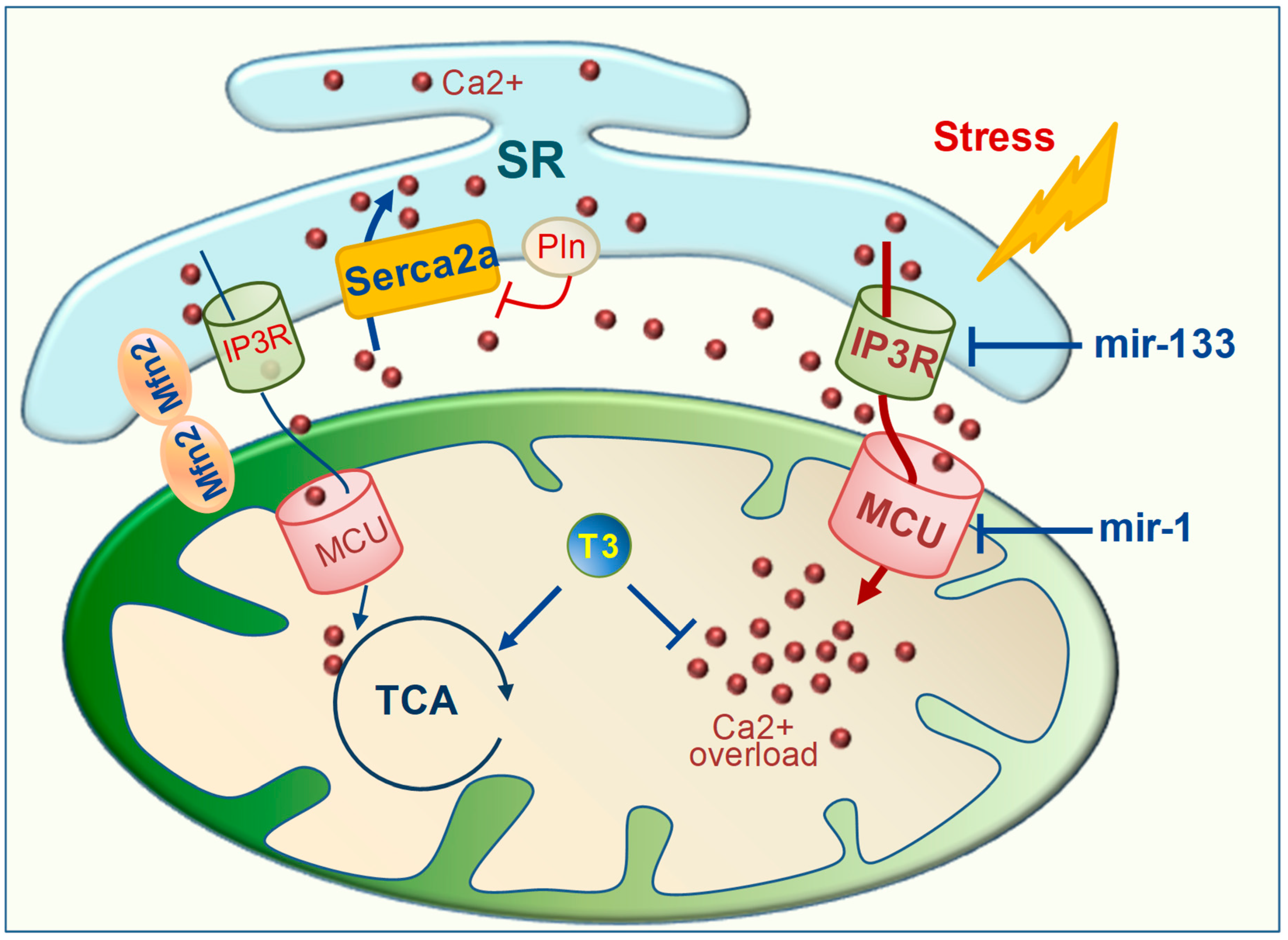{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. TH and Oxidative Stress
3. TH Regulation of Mitochondrial Calcium
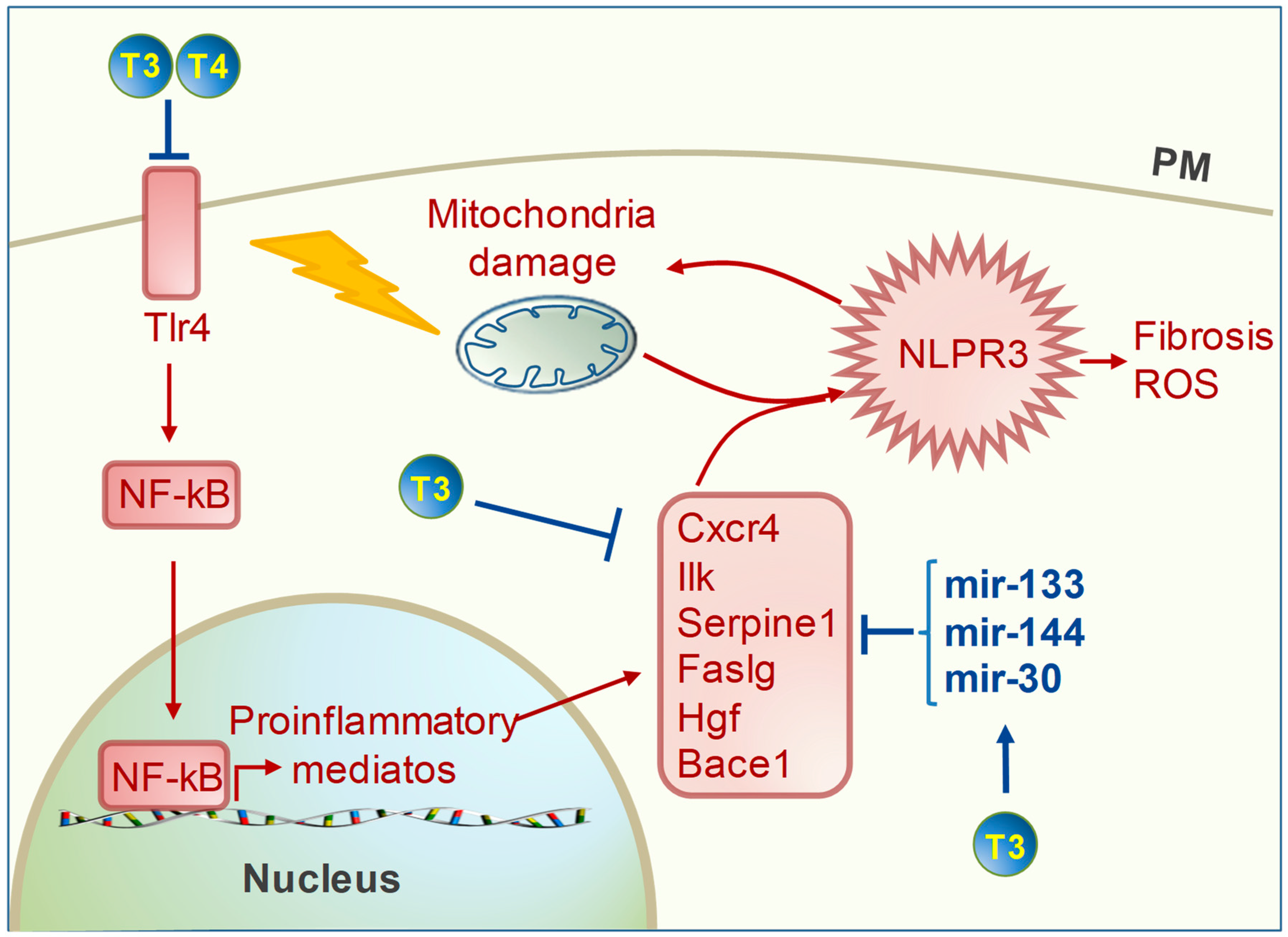
4. TH Regulation of Cardiac Inflammation
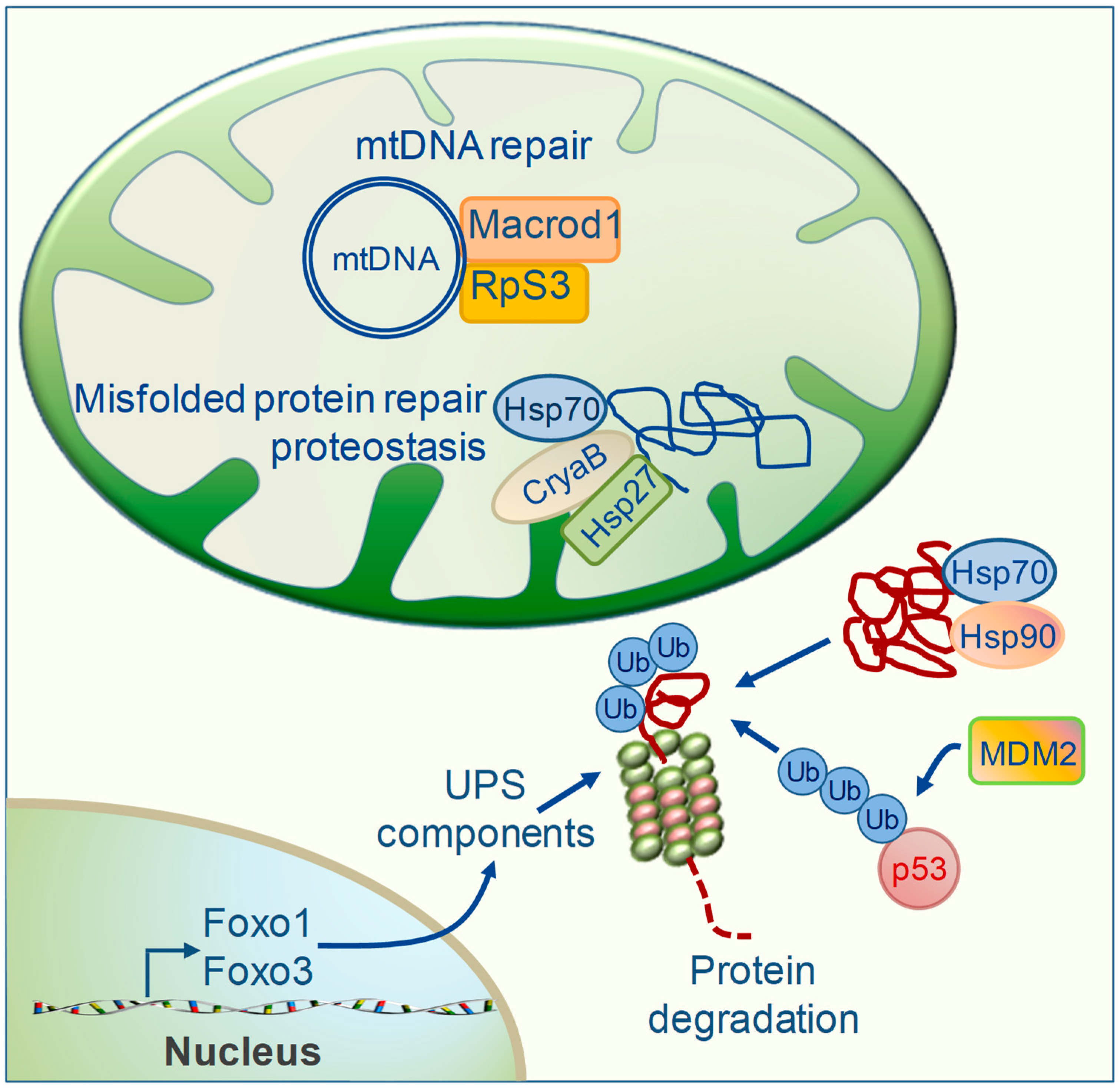
5. TH Regulation of Mitochondrial Repair Systems
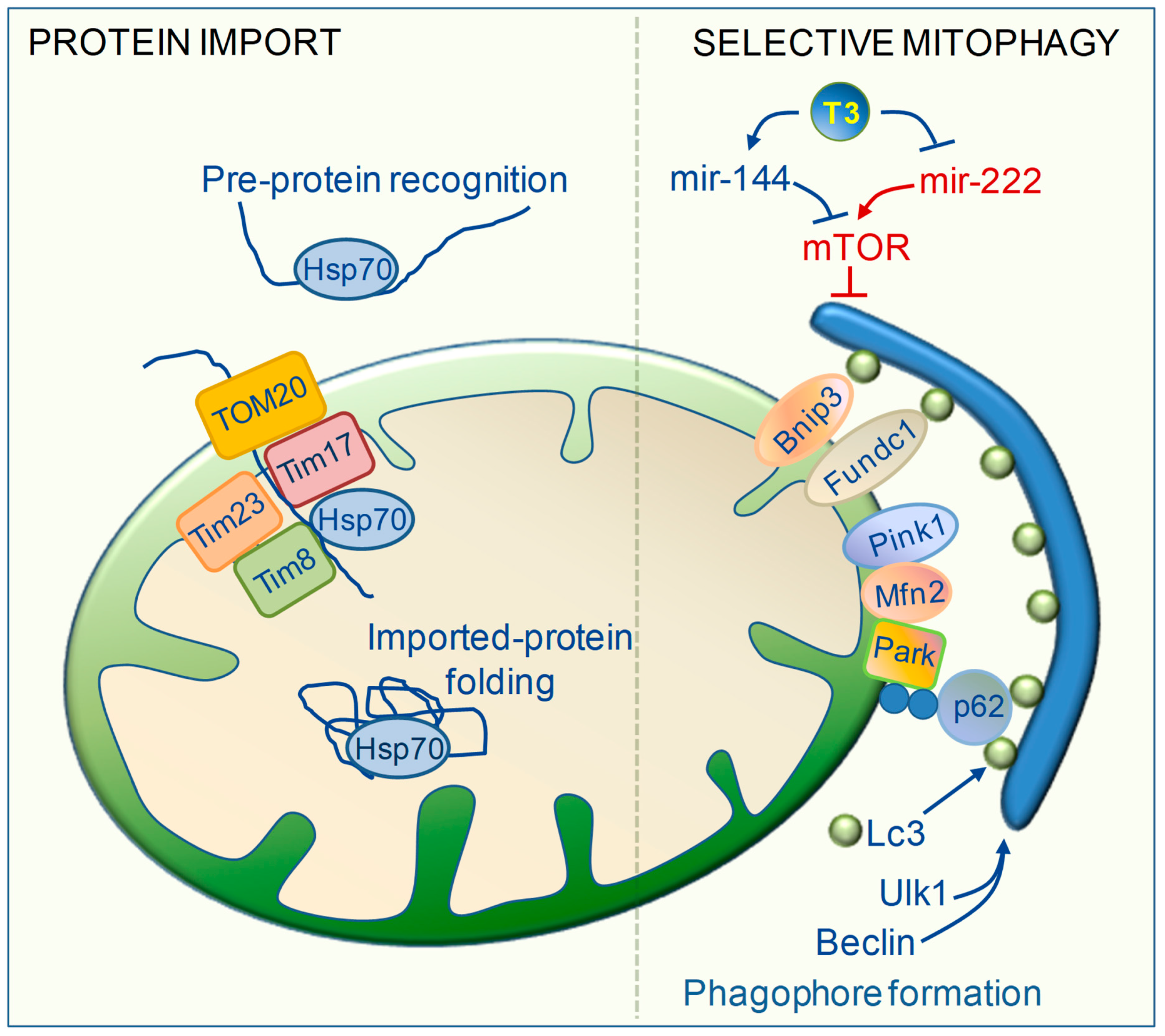
6. TH Regulation of Mitochondrial Clearance
7. TH Regulation of Mitochondrial Biogenesis and Protein Import Machinery
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Aldh2 | aldehyde dehydrogenase 2 |
| Bace 1 | beta secretase |
| Bnip3 | BCL2 interacting protein 3 |
| CryaB | α-crystallin B |
| Cxcr4 | C-X-C motif chemokin receptor 4 |
| eNOS | nitric oxide synthase |
| Faslg | fas ligand |
| Foxo1/3 | forkhead box O 1/3 |
| Fundc1 | FUN14 domain-containing protein 1 |
| Hgf | hepatocyte growth factor |
| Ilk | integrin-linked kinase |
| IMM | inner mitochondrial membrane |
| IP3R | inositol 1,4,5-trisphosphate receptors |
| Lc3 | light chain3 |
| MCU | mitochondrial calcium uniporter |
| Mdm2 | mouse double minute 2 homolog |
| Mfn2 | mitofusin2 |
| mTOR | mammalian target of rapamycin |
| NF-kB | nuclear factor kappa B |
| Tlr4 | toll like receptor 4 |
| OMM | outer mitochondrial membrane |
| Park | parkin |
| Pgc1 | PPARG coactivator 1 alpha |
| Pink1 | PTEN induced kinase 1 |
| Pln | phospholambam |
| PM | plasma membrane |
| Prdx2/5 | peroxiredoxin 2/5 |
| RpS3 | 40s ribosomal protein S3 |
| Serca2a | SR Ca2+ ATPase 2a |
| Sod1/2 | superoxide dismutase1/2 |
| SR | sarcoplasmic reticulum |
| Tfam | mitochondrial transcription factor A |
| Ulk1 | unc-51 like autophagy activating kinase1 |
References
- Moyzis, A.G.; Sadoshima, J.; Gustafsson, A.B. Mending a broken heart: The role of mitophagy in cardioprotection. Am. J. Physiol. Heart. Circ. Physiol. 2015, 308, H183–H192. [Google Scholar] [CrossRef] [PubMed]
- Suliman, H.B.; Piantadosi, C.A. Mitochondrial Quality Control as a Therapeutic Target. Pharmacol. Rev. 2016, 68, 20–48. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, A.B.; Gottlieb, R.A. Heart mitochondria: Gates of life and death. Cardiovasc. Res. 2008, 77, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Yoboue, E.D.; Devin, A. Reactive Oxygen Species-Mediated Control of Mitochondrial Biogenesis. Int. J. Cell. Biol. 2012, 2012, 1–8. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1–44. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, k.; Siddiqi, N.; Singh, S.; Neil, C.J.; Dawson, D.K.; Frenneaux, M.P. The breathing heart. Mitochondrial respiratory chain dysfunction in cardiac disease. Int. J. Cardiol. 2014, 171, 134–143. [Google Scholar] [CrossRef]
- Ide, T.; Tsutsui, H.; Hayashidani, S.; Kang, D.; Suematsu, N.; Nakamura, K.; Utsumi, H.; Hamasaki, N.; Takeshita, A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ. Res. 2001, 88, 529–535. [Google Scholar] [CrossRef]
- Marín-García, J. Mitochondrial DNA repair: A novel therapeutic target for heart failure. Heart Fail. Rev. 2016, 21, 475–487. [Google Scholar] [CrossRef]
- Jain, M.; Rivera, S.; Monclus, E.A.; Synenki, L.; Zirk, A.; Eisenbart, J.; Feghali-Bostwick, C.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial Reactive Oxygen Species Regulate Transforming Growth Factor-β Signaling. J. Biol. Chem. 2013, 288, 770–777. [Google Scholar] [CrossRef]
- Liu, R.M.; Desai, L.P. Reciprocal regulation of TGF-β and reactive oxygen species A perverse cycle for fibrosis. Redox Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef]
- Cao, J.L.; Adaniya, S.M.; Cypress, M.W.; Suzuki, Y.; Kusakari, Y.; Jhun, B.S.; Jin, O. Role of mitochondrial Ca2+ homeostasis in cardiac muscles. Arch. Biochem. Biophys. 2019, 663, 276–287. [Google Scholar] [CrossRef] [PubMed]
- López-Armada, M.J.; Riveiro-Naveira, R.R.; Vaamonde-García, C.; Valcárcel-Ares, M.N. Mitochondrial dysfunction and the inflammatory response. Mitochondrion 2013, 13, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Missiroli, S.; Patergnani, S.; Caroccia, N.; Pedriali, G.; Perrone, M.; Previati, M.; Wieckowski, M.R.; Giorgi, C. Mitochondria-associated membranes (MAMs) and inflammation. Cell Death Dis. 2018, 9, 329. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wang, S.; Hu, S.; Chen, Y.; Ren, J. ER–Mitochondria Microdomains in Cardiac Ischemia–Reperfusion Injury: A Fresh Perspective. Front. Physiol. 2018, 9, 755. [Google Scholar] [CrossRef] [PubMed]
- Forini, F.; Ucciferri, N.; Kusmic, C.; Nicolini, G.; Cecchettini, A.; Rocchiccioli, S.; Citti, L.; Iervasi, G. Low T3 State Is Correlated with Cardiac Mitochondrial Impairments after Ischemia Reperfusion Injury: Evidence from a Proteomic Approach. Int. J. Mol. Sci. 2015, 16, 26687–26705. [Google Scholar] [CrossRef]
- Forini, F.; Nicolini, G.; Kusmic, C.; D’Aurizio, R.; Rizzo, M.; Baumgart, M.; Groth, M.; Doccini, S.; Iervasi, G.; Pitto, L. Integrative analysis of differentially expressed genes and miRNAs predicts complex T3-mediated protective circuits in a rat model of cardiac ischemia reperfusion. Sci. Rep. 2018, 8, 13870. [Google Scholar] [CrossRef]
- Razvi, S.; Jabbar, A.; Pingitore, A.; Danzi, S.; Biondi, B.; Klein, I.; Peeters, R.; Zaman, A.; Iervasi, G. Thyroid Hormones and Cardiovascular Function and Diseases. J. Am. Coll. Cardiol. 2018, 71, 1781–1796. [Google Scholar] [CrossRef]
- Kahali, G.J.; Dillmann, W.H. Thyroid Hormone Action in the Heart. Endocr. Rev. 2005, 26, 704–728. [Google Scholar] [CrossRef]
- Iervasi, G.; Pingitore, A.; Landi, P.; Raciti, M.; Ripoli, A.; Scarlattini, M.; L’Abbate, A.; Donato, L. Low-T3 syndrome: A strong prognostic predictor of death in patients with heart disease. Circulation 2003, 107, 708–713. [Google Scholar] [CrossRef]
- Marwali, E.M.; Boom, C.E.; Budiwardhana, N.; Fakhri, D.; Roebiono, P.S.; Santoso, A.; Sastroasmoro, S.; Slee, A.; Portman, M.A. Oral triiodothyronine normalizes triiodothyronine levels after surgery for pediatric congenital heart disease. Pediatr. Crit. Care Med. 2013, 14, 701–708. [Google Scholar] [CrossRef]
- Pingitore, A.; Mastorci, F.; Piaggi, P.; Aquaro, G.D.; Molinaro, S.; Ravani, M.; De Caterina, A.; Trianni, G.; Ndreu, R.; Berti, S.; et al. Usefulness of Triiodothyronine Replacement Therapy in Patients With ST Elevation Myocardial Infarction and Borderline/Reduced Triiodothyronine Levels. Am. J. Cardiol. 2019, 123, 905–912. [Google Scholar] [CrossRef]
- Pingitore, A.; Nicolini, G.; Kusmic, C.; Iervasi, G.; Grigolini, P.; Forini, F. Cardioprotection and thyroid hormones. Heart Fail. Rev. 2016, 21, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, V.; Zhang, Y.; Pol, C.; Costello, C.; Seitter, S.; Lehto, A.; Savinova, O.V.; Chen, Y.F.; Gerdes, A.M. Modified Low-Dose Triiodo-L-thyronine Therapy Safely Improves Function Following Myocardial Ischemia-Reperfusion Injury. Front. Physiol. 2017, 8, 225. [Google Scholar] [CrossRef] [PubMed]
- Pantos, C.; Mourouzis, I.; Markakis, K.; Tsagoulis, N.; Panagiotou, M.; Cokkinos, D.V. Long-term thyroid hormone administration re-shapes left ventricular chamber and improves cardiac function after myocardial infarction in rats. Basic. Res. Cardiol. 2008, 103, 308–318. [Google Scholar] [CrossRef]
- Pantos, C.; Mourouzis, I.; Cokkinos, D.V. Thyroid hormone and cardiac repair/regeneration: From Prometheus myth to reality? Can. J. Physiol. Pharmacol. 2012, 90, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Pingitore, A.; Galli, E.; Barison, A.; Iervasi, A.; Scarlattini, M.; Nucci, D.; L’abbate, A.; Mariotti, R.; Iervasi, G. Acute effects of triiodothyronine (T3) replacement therapy in patients with chronic heart failure and low-T3 syndrome: A randomized, placebo-controlled study. J. Clin. Endocrinol. Metab. 2008, 93, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Forini, F.; Nicolini, G.; Iervasi, G. Mitochondria as key targets of cardioprotection in cardiac ischemic disease: Role of thyroid hormone triiodothyronine. Int. J. Mol. Sci. 2015, 16, 6312–6336. [Google Scholar] [CrossRef]
- Goldenthal, M.J.; Weiss, H.R.; Marín-García, J. Bioenergetic remodeling of heart mitochondria by thyroid hormone. Mol. Cell. Biochem. 2004, 265, 97–106. [Google Scholar] [CrossRef]
- Portman, M.A. Thyroid hormone regulation of heart metabolism. Thyroid 2008, 18, 217–225. [Google Scholar] [CrossRef]
- Forini, F.; Lionetti, V.; Ardehali, H.; Pucci, A.; Cecchetti, F.; Ghanefar, M.; Nicolini, G.; Yoshihiko, I.; Nannipieri, M.; Recchia, F.A.; et al. Early long-term L-T3 replacement rescues mitochondria and prevents ischemic cardiac remodelling in rats. J. Cell. Mol. Med. 2011, 15, 514–524. [Google Scholar] [CrossRef]
- Forini, F.; Kusmic, C.; Nicolini, G.; Mariani, L.; Zucchi, R.; Matteucci, M.; Pitto, L.; Iervasi, G. Triiodothyronine prevents cardiac ischemia/reperfusion mitochondrial impairment and cell loss by regulating miR30a/p53 axis. Endocrinology 2014, 155, 4581–4590. [Google Scholar] [CrossRef] [PubMed]
- Tavares, A.M.; da Rosa Araujo, A.S.; Llesuy, S.; Khaper, N.; Rohde, L.E.; Clausell, N.; Belló-Klein, A. Early loss of cardiac function in acute myocardial infarction is associated with redox imbalance. Exp. Clin. Cardiol. 2012, 17, 263–267. [Google Scholar] [PubMed]
- Dey, S.; DeMazumder, D.; Sidor, A.; Foster, D.B.; O’Rourke, B. Mitochondrial ROS Drive Sudden Cardiac Death and Chronic Proteome Remodeling in Heart Failure. Circ. Res. 2018, 123, 356–371. [Google Scholar] [CrossRef] [PubMed]
- De Castro, A.L.; Tavares, A.V.; Campos, C.; Fernandes, R.O.; Siqueira, R.; Conzatti, A.; Bicca, A.M.; Fernandes, T.R.; Sartório, C.L.; Schenkel, P.C.; et al. Cardioprotective effects of thyroid hormones in a rat model of myocardial infarction are associated with oxidative stress reduction. Mol. Cell. Endocrinol. 2014, 391, 22–29. [Google Scholar] [CrossRef] [PubMed]
- De Castro, A.L.; Tavares, A.V.; Fernandes, R.O.; Campos, C.; Conzatti, A.; Siqueira, R.; Fernandes, T.R.; Schenkel, P.C.; Sartório, C.L.; Llesuy, S.; et al. T3 and T4 decrease ROS levels and increase endothelial nitric oxide synthase expression in the myocardium of infarcted rats. Mol. Cell. Biochem. 2015, 408, 235–243. [Google Scholar] [CrossRef]
- Corssac, G.B.; de Castro, A.L.; Tavares, A.V.; Campos, C.; Fernandes, R.O.; Ortiz, V.D.; Siqueira, R.; Fernandes, T.R.; Belló-Klein, A.; Araujo, A.S. Thyroid hormones effects on oxidative stress and cardiac remodeling in the right ventricle of infarcted rats. Life Sci. 2016, 146, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Qi, X.; Jia, W. 3,3′,5-triiodothyroxine inhibits apoptosis and oxidative stress by the PKM2/PKM1 ratio during oxygen-glucose deprivation/reperfusion AC16 and HCM-a cells: T3 inhibits apoptosis and oxidative stress by PKM2/PKM1 ratioBiochem. Biophys. Res. Commun. 2016, 17, 51–56. [Google Scholar] [CrossRef]
- Abdolghaffari, A.H.; Baghaei, A.; Solgi, R.; Gooshe, M.; Baeeri, M.; Navaei-Nigjeh, M.; Hassani, S.; Jafari, A.; Rezayat, S.M.; Dehpour, A.R.; et al. Molecular and biochemical evidences on the protective effects of triiodothyronine against phosphine-induced cardiac and mitochondrial toxicity. Life Sci. 2015, 139, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, R.O.; Bonetto, J.H.; Baregzay, B.; de Castro, A.L.; Puukila, S.; Forsyth, H.; Schenkel, P.C.; Llesuy, S.F.; Brum, I.S.; Araujo, A.S.; et al. Modulation of apoptosis by sulforaphane is associated with PGC-1α stimulation and decreased oxidative stress in cardiac myoblasts. Mol. Cell. Biochem. 2015, 401, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Xu, X.; Hu, X.; Fassett, J.; Zhu, G.; Tao, Y.; Li, J.; Huang, Y.; Zhang, P.; Zhao, B.; et al. PGC-1α regulates expression of myocardial mitochondrial antioxidants and myocardial oxidative stress after chronic systolic overload. Antioxid. Redox Signal. 2010, 13, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Men, M.; Yang, W.; Zheng, H.; Xue, S. MiR-31 downregulation protects against cardiac ischemia/reperfusion injury by targeting protein kinase C epsilon (PKCε) directly. Cell. Physiol. Biochem. 2015, 36, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Martinez, E.C.; Lilyanna, S.; Wang, P.; Vardy, L.A.; Jiang, X.; Armugam, A.; Jeyaseelan, K.; Richards, A.M. MicroRNA-31 promotes adverse cardiac remodeling and dysfunction in ischemic heart disease. J. Mol. Cell. Cardiol. 2017, 112, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Chen, Z.; Wang, C.; Song, L.; Zou, Y.; Zhang, L.; Hui, R.; Wang, J. Cardiac-Specific Overexpression of miR-222 Induces Heart Failure and Inhibits Autophagy in Mice. Cell. Physiol. Biochem. 2016, 39, 1503–1511. [Google Scholar] [CrossRef] [PubMed]
- Seok, H.Y.; Chen, J.; Kataoka, M.; Huang, Z.P.; Ding, J.; Yan, J.; Hu, X.; Wang, D.Z. Loss of microRNA-155 Protects the Heart from Pathological Cardiac Hypertrophy. Circ. Res. 2014, 114, 1585–1595. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Yigang, W.; Ahmar, A.; Muhammad, A. Mitochondrial KATP channel activation reduces anoxic injury by restoring mitochondrial membrane potential. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H1295–H1303. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef] [PubMed]
- Contreras, L.; Gomez-Puertas, P.; Iijima, M.; Kobayashi, K.; Saheki, T.; Satrústegui, J. Ca2+ Activation kinetics of the two aspartate-glutamate mitochondrial carriers, aralar and citrin: Role in the heart malate-aspartate NADH shuttle. J. Biol. Chem. 2007, 282, 7098–7106. [Google Scholar] [CrossRef] [PubMed]
- Hopper, R.K.; Carroll, S.; Aponte, A.M.; Johnson, D.T.; French, S.; Shen, R.F.; Witzmann, F.A.; Harris, R.A.; Balaban, R.S. Mitochondrial matrix phosphoproteome: Effect of extra mitochondrial calcium. Biochemistry 2006, 45, 2524–2536. [Google Scholar] [CrossRef] [PubMed]
- Kawase, Y.; Hajjar, R.J. The cardiac sarcoplasmic/endoplasmic reticulum calcium ATPase: A potent target for cardiovascular diseases. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Kawase, Y.; Ly, H.Q.; Prunier, F.; Lebeche, D.; Shi, Y.; Jin, H.; Hadri, L.; Yoneyama, R.; Hoshino, K.; Takewa, Y.; et al. Reversal of cardiac dysfunction after long-term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J. Am. Coll. Cardiol. 2008, 51, 1112–1119. [Google Scholar] [CrossRef]
- Lipskaia, L.; Hadri, L.; Lopez, J.J.; Hajjar, R.J.; Bobe, R. Benefit of SERCA2a gene transfer to vascular endothelial and smooth muscle cells: A new aspect in therapy of cardiovascular diseases. Curr. Vasc. Pharmacol. 2013, 11, 465–479. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, M.I.; del Monte, F.; Schmidt, U.; DiSalvo, T.S.; Kang, Z.B.; Matsui, T.; Guerrero, J.L.; Gwathmey, J.K.; Rosenzweig, A.; Hajjar, R.J. Adenoviral gene transfer of SERCA2a improves left-ventricular function in aortic-banded rats in transition to heart failure. Proc. Natl. Acad. Sci. USA 2000, 97, 793–798. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.M.; Pound, K.; Xu, X.; Lewandowski, E.D. SERCA1 expression enhances the metabolic efficiency of improved contractility in post-ischemic heart. J. Mol. Cell. Cardiol. 2009, 47, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Reed, T.D.; Babu, G.J.; Ji, Y.; Zilberman, A.; Heyen, M.V.; Wuytack, F. The Expression of SR Calcium Transport ATPase and the Na+/Ca2+Exchanger are Antithetically Regulated During Mouse Cardiac Development and in Hypo/hyperthyroidism. J. Mol. Cell. Cardiol. 2000, 32, 453–464. [Google Scholar] [CrossRef]
- Klein, I.; Danzi, S. Thyroid Disease and the Heart. Circulation 2007, 116, 1725–1735. [Google Scholar] [CrossRef]
- Kiss, E.; Jakab, G.; Kranias, E.G.; Edes, I. Thyroid hormone-induced alterations in phospholamban protein expression: Regulatory effects on sarcoplasmic reticulum Ca2 transport and myocardial relaxation. Circ Res. 1994, 75, 245–251. [Google Scholar] [CrossRef]
- Forini, F.; Paolicchi, A.; Pizzorusso, T.; Ratto, G.M.; Saviozzi, M.; Vanini, V.; Iervasi, G. 3,5,3′-Triiodothyronine deprivation affects phenotype and intracellular [Ca2+]i of human cardiomyocytes in culture. Cardiovasc. Res. 2001, 51, 322–330. [Google Scholar] [CrossRef]
- Drawnel, F.M.; Wachten, D.; Molkentin, J.D.; Maillet, M.; Aronsen, J.M.; Swift, F.; Sjaastad, I.; Liu, N.; Catalucci, D.; Mikoshiba, K.; et al. Mutual antagonism between IP3RII and miRNA-133a regulates calcium signals and cardiac hypertrophy. J. Cell Boil. 2012, 199, 783–798. [Google Scholar] [CrossRef]
- Blackshaw, S.; Sawa, A.; Sharp, A.H.; Ross, C.A.; Snyder, S.H.; Khan, A.A. Type 3 inositol 1,4,5-trisphosphate receptor modulates cell death. FASEB J. 2000, 14, 1375–1379. [Google Scholar] [CrossRef]
- Patron, M.; Raffaello, A.; Granatiero, V.; Tosatto, A.; Merli, G.; De Stefani, D.; Wright, L.; Pallafacchina, G.; Terrin, A.; Mammucari, C.; et al. The Mitochondrial Calcium Uniporter (MCU): Molecular Identity and Physiological Roles. J. Boil. Chem. 2013, 288, 10750–10758. [Google Scholar] [CrossRef]
- Kwong, J.Q.; Lu, X.; Correll, R.N.; Schwanekamp, J.A.; Vagnozzi, R.J.; Sargent, M.A.; York, A.J.; Zhang, J.; Bers, D.M.; Molkentin, J.D. The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart. Cell Rep. 2015, 12, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Chen, R.; Li, M.; Yu, Y.; Liang, Y.; Han, F.; Qin, S.; Chen, X.; Su, Y.; Ge, J. Mitochondrial calcium uniporter inhibition provides cardioprotection in pressure overload-induced heart failure through autophagy enhancement. Int. J. Cardiol. 2018, 271, 161–168. [Google Scholar] [CrossRef]
- Zaglia, T.; Ceriotti, P.; Campo, A.; Borile, G.; Armani, A.; Carullo, P.; Prando, V.; Coppini, R.; Vida, V.; Stølen, T.O.; et al. Content of mitochondrial calcium uniporter (MCU) in cardiomyocytes is regulated by microRNA-1 in physiologic and pathologic hypertrophy. Proc. Natl. Acad. Sci. USA 2017, 114, E9006–E9015. [Google Scholar] [CrossRef]
- Nicolini, G.; Forini, F.; Kusmic, C.; Pitto, L.; Mariani, L.; Iervasi, G. Early and Short-term Triiodothyronine Supplementation Prevents Adverse Postischemic Cardiac Remodeling: Role of Transforming Growth Factor-β1 and Antifibrotic miRNA Signaling. Mol. Med. 2016, 21, 900–911. [Google Scholar] [CrossRef] [PubMed]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Csordás, G.; Jowdy, C.; Schneider, T.G.; Csordás, N.; Wang, W.; Liu, Y.; Kohlhaas, M.; Meiser, M.; Bergem, S.; et al. Mitofusin 2 containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca2+ crosstalk. Circ. Res. 2012, 111, 863–875. [Google Scholar] [CrossRef]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef]
- De Castro, A.L.; Fernandes, R.O.; Ortiz, V.D.; Campos, C.; Bonetto, J.H.; Fernandes, T.R.G.; Conzatti, A.; Siqueira, R.; Tavares, A.V.; Belló-Klein, A.; et al. Thyroid hormones decrease the proinflammatory TLR4/NF-κβ pathway and improve functional parameters of the left ventricle of infarcted rats. Mol. Cell. Endocrinol. 2018, 461, 132–142. [Google Scholar] [CrossRef]
- Lu, L.; Chen, S.S.; Zhang, J.Q.; Ramires, F.J.; Sun, Y. Activation of nuclear factor-kappaB and its proinflammatory mediator cascade in the infarcted rat heart. Biochem. Biophys. Res. Commun. 2004, 321, 879–885. [Google Scholar] [CrossRef]
- Onai, Y.; Suzuki, J.; Maejima, Y.; Haraguchi, G.; Muto, S.; Itai, A.; Isobe, M.; Onai, Y. Inhibition of NF-kappa B improves left ventricular remodeling and cardiac dysfunction after myocardial infarction. Am. J. Physiol. Heart. Circ. Physiol. 2007, 292, H530–H538. [Google Scholar] [CrossRef]
- Rajagopalan, V.; Zhang, Y.; Ojamaa, K.; Chen, Y.F.; Pingitore, A.; Pol, C.J.; Balasubramanian, K.; Towner, R.A.; Gerdes, A.M. Safe oral triiodo-l-thyronine therapy protects from post-infarct cardiac dysfunction and arrhythmias without cardiovascular adverse effects. PLoS ONE 2016, 11, e0151413. [Google Scholar] [CrossRef] [PubMed]
- Nural-Guvener, H.F.; Mutlu, N.; Gaballa, M.A. BACE1 levels are elevated in congestive heart failure. Neurosci. Lett. 2013, 532, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Thackeray, J.T.; Hupe, H.C.; Wang, Y.; Bankstahl, J.P.; Berding, G.; Ross, T.L.; Bauersachs, J.; Wollert, K.C.; Bengel, F.M. Myocardial Inflammation Predicts Remodeling and Neuroinflammation After Myocardial Infarction. J. Am. Coll. Cardiol. 2018, 71, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.S.; Benjamin, I.J. Protective responses in the ischemic myocardium. HSPs play an important role in the defense mechanism against IR injury. J. Clin. Investig. 2000, 106, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Willis, M.S.; Patterson, C. Hold me tight: The role of the HSP family of chaperones in cardiac disease. Circulation 2010, 122, 1740–1751. [Google Scholar] [CrossRef] [PubMed]
- Henning, R.H.; Brundel, B.J.J.M. Proteostasis in cardiac health and disease. Nat. Rev. Cardiol. 2017, 14, 637–653. [Google Scholar] [CrossRef] [PubMed]
- Heide, R.S.V. Increased expression of HSP27 protects canine myocytes from simulated ischemia reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H935–H941. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Benjamin, I.J. Stress response proteins in cardiovascular disease. Am. J. Hum. Genet. 1999, 64, 685–690. [Google Scholar] [CrossRef]
- Brundel, B.J.; Henning, R.H.; Ke, L.; van Gelder, I.C.; Crijns, H.J.; Kampinga, H.H. Heat shock protein upregulation protects against pacing-induced myolysis in HL-1 atrial myocytes and in human atrial fibrillation. J. Mol. Cell. Cardiol. 2006, 41, 555–562. [Google Scholar] [CrossRef]
- Young, J.C.; Barral, J.M.; Ulrich Hartl, F. More than folding: Localized functions of cytosolic chaperones. Trends Biochem. Sci. 2003, 28, 541–547. [Google Scholar] [CrossRef]
- Webb, A.E.; Brunet, A. FOXO transcription factors: Key regulators of cellular quality control. Trends Biochem. Sci. 2014, 39, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Willis, M.S.; Patterson, C. Into the heart: The emerging role of the ubiquitin-proteasome system. J. Mol. Cell. Cardiol. 2006, 41, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Lesmana, R.; Sinha, R.A.; Singh, B.K.; Zhou, J.; Ohba, K.; Wu, Y.; Yau, W.W.; Bay, B.H.; Yen, P.M. Thyroid Hormone Stimulation of Autophagy Is Essential for Mitochondrial Biogenesis and Activity in Skeletal Muscle. Endocrinology 2016, 157, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.C.; Tsai, C.Y.; Tsai, M.M.; Yeh, C.T.; Lin, K.H. Molecular functions and clinical impact of thyroid hormone-triggered autophagy in liver-related diseases. J. Biomed. Sci. 2019, 26, 24. [Google Scholar] [CrossRef] [PubMed]
- Lino, C.A.; Barreto-Chaves, M.L.M. Modulation of the ubiquitin proteasome system (UPS) in thyroid hormone-induced cardiac hypertrophy. FASEB J. 2012, 26. [Google Scholar] [CrossRef]
- Hauck, L.; Stanley-Hasnain, S.; Fung, A.; Grothe, D.; Rao, V.; Mak, T.W.; Billia, F. Cardiac-specific ablation of the E3 ubiquitin ligase Mdm2 leads to oxidative stress, broad mitochondrial deficiency and early death. PLoS ONE 2017, 12, e0189861. [Google Scholar] [CrossRef] [PubMed]
- Ramos, G.V.; Cruz, A.; Silva, W.J.; Rozanski, A.; Baptista, I.L.; Silvestre, J.G.; Moriscot, A.S. Thyroid hormone upregulates MDM2 in rat type I fibre: Implications for skeletal muscle mass regulation. Acta Physiol. 2018, 222, e13003. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, H.D.; Kim, J. Cytoplasmic ribosomal protein S3 (rpS3) plays a pivotal role in mitochondrial DNA damage surveillance. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2013, 1833, 2943–2952. [Google Scholar] [CrossRef]
- Agnew, T.; Munnur, D.; Crawford, K.; Palazzo, L.; Mikoč, A.; Ahel, I. MacroD1 Is a Promiscuous ADP-Ribosyl Hydrolase Localized to Mitochondria. Front. Microbiol. 2018, 9, 20. [Google Scholar] [CrossRef]
- Chen, X.J.; Wang, X.; Kaufman, B.A.; Butow, R.A. Aconitase Couples Metabolic Regulation to Mitochondrial DNA Maintenance. Science 2005, 307, 714–717. [Google Scholar] [CrossRef]
- Kim, S.J.; Cheresh, P.; Williams, D.; Cheng, Y.; Ridge, K.; Schumacker, P.T.; Weitzman, S.; Bohr, V.A.; Kamp, D.W. Mitochondria-targeted Ogg1 and Aconitase-2 Prevent Oxidant-induced Mitochondrial DNA Damage in Alveolar Epithelial Cells. J. Boil. Chem. 2014, 289, 6165–6176. [Google Scholar] [CrossRef] [PubMed]
- Kloner, R.A.; Brown, D.A.; Csete, M.; Dai, W.; Downey, J.M.; Gottlieb, R.A.; Hale, S.L.; Shi, J. New and revisited approaches to preserving the reperfused myocardium. Nat. Rev. Cardiol. 2017, 14, 679–693. [Google Scholar] [CrossRef] [PubMed]
- Bravo-San Pedro, JM.; Kroemer, G.; Galluzzi, L. Autophagy and Mitophagy in Cardiovascular Disease. Circ. Res. 2017, 120, 1812–1824. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Sadoshima, J. The Molecular Mechanisms of Mitochondrial Autophagy Mitophagy in the heart. Circ. Res. 2015, 116, 1477–1490. [Google Scholar] [CrossRef] [PubMed]
- Yau, W.W.; Singh, B.K.; Lesmana, R.; Zhou, J.; Sinha, R.A.; Wong, K.A.; Wu, Y.; Bay, B.H.; Sugii, S.; Sun, L.; et al. Thyroid hormone (T3) stimulates brown adipose tissue activation via mitochondrial biogenesis and MTOR-mediated mitophagy. Autophagy 2019, 15, 131–150. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.A.; You, S.H.; Zhou, J.; Siddique, M.M.; Bay, B.H.; Zhu, X.; Privalsky, M.L.; Cheng, S.Y.; Stevens, R.D.; Summers, S.A.; et al. Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. J. Clin. Investig. 2012, 122, 2428–2438. [Google Scholar] [CrossRef]
- Sinha, R.A.; Singh, B.K.; Zhou, J.; Wu, Y.; Farah, B.L.; Ohba, K.; Lesmana, R.; Gooding, J.; Bay, B.H.; Yen, P.M. Thyroid hormone induction of mitochondrial activity is coupled to mitophagy via ROS-AMPK-ULK1 signaling. Autophagy 2015, 11, 1341–1357. [Google Scholar] [CrossRef]
- Singh, B.K.; Sinha, R.A.; Tripathi, M.; Mendoza, A.; Ohba, K.; Sy, J.A.C.; Xie, S.Y.; Zhou, J.; Ho, J.P.; Chang, C.Y.; et al. Thyroid hormone receptor and ERRα coordinately regulate mitochondrial fission, mitophagy, biogenesis, and function. Sci. Signal. 2018, 11, eaam5855. [Google Scholar] [CrossRef]
- Chi, H.C.; Chen, S.L.; Lin, S.L.; Tsai, C.Y.; Chuang, W.Y.; Lin, Y.H.; Huang, Y.H.; Tsai, M.M.; Yeh, C.T.; Lin, K.H. Thyroid hormone protects hepatocytes from HBx-induced carcinogenesis by enhancing mitochondrial turnover. Oncogene 2017, 36, 5274–5284. [Google Scholar] [CrossRef]
- Yang, J.; Wang, Y.; Sui, M.; Liu, F.; Fu, Z.; Wang, Q.X. Tri-iodothyronine preconditioning protects against liver ischemia reperfusion injury through the regulation of autophagy by the MEK ERK mTORC1 axis. Biochem. Biophys. Res. Commun. 2015, 467, 704–710. [Google Scholar] [CrossRef]
- Xiong, W.; Ma, Z.; An, D.; Liu, Z.; Cai, W.; Bai, Y.; Zhan, Q.; Lai, W.; Zeng, Q.; Ren, H.; et al. Mitofusin 2 Participates in Mitophagy and Mitochondrial Fusion Against Angiotensin II-Induced Cardiomyocyte Injury. Front. Physiol. 2019, 10, 411. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Rohailla, S.; Gelber, N.; Rutka, J.; Sabah, N.; Gladstone, R.A.; Wei, C.; Hu, P.; Kharbanda, R.K.; Redington, A.N. MicroRNA-144 is a circulating effector of remote ischemic preconditioning. Basic Res. Cardiol. 2014, 109, 423. [Google Scholar] [CrossRef] [PubMed]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V. Transcriptional control of mitochondrial biogenesis: the central role of PGC-1. Cardiovasc. Res. 2008, 79, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, G.H.; Chaturvedi, P.; Tyagi, S.C. Mitochondrial pathways to cardiac recovery: TFAM. Heart Fail. Rev. 2016, 21, 499–517. [Google Scholar] [CrossRef] [PubMed]
- Wrutniak-Cabello, C.; Casas, F.; Cabello, G. Thyroid hormone action in mitochondria. J. Mol. Endocrinol. 2001, 26, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Goldenthal, M.J.; Ananthakrishnan, R.; Marín-García, J. Nuclear-mitochondrial cross-talk in cardiomyocyte T3 signaling: A time-course analysis. J. Mol. Cell. Cardiol. 2005, 39, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Marín-García, J. Thyroid hormone and myocardial mitochondrial biogenesis. Vascul Pharmacol. 2010, 52, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Weitzel, J.M.; Iwen, K.A. Coordination of mitochondrial biogenesis by thyroid hormone. Mol. Cell. Endocrinol. 2011, 342, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wulf, A.; Harneit, A.; Kröger, M.; Kebenko, M.; Wetzel, M.G.; Weitzel, J.M. T3-mediated expression of PGC-1α via a far upstream located thyroid hormone response element. Mol. Cell. Endocrinol. 2008, 287, 90–95. [Google Scholar] [CrossRef]
- Kang, P.J.; Ostermann, J.; Shilling, J.; Neupert, W.; Craig, E.A.; Pfanner, N. Requirement for hsp70 in the mitochondrial matrix for translocation and folding of precursor proteins. Nature 1990, 348, 137–143. [Google Scholar] [CrossRef]
- Colavecchia, M.; Christie, L.N.; Kanwar, Y.S.; Hood, D.A. Functional consequences of thyroid hormone-induced changes in the mitochondrial protein import pathway. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E29–E35. [Google Scholar] [CrossRef] [PubMed]
- Craig, E.E.; Chesley, A.; Hood, D.A. Thyroid hormone modifies mitochondrial phenotype by increasing protein import without altering degradation. Am. J. Physiol. 1998, 275, C1508–C1515. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.J.; Hood, D.A. Effect of thyroid hormone on mthsp70 expression, mitochondrial import, and processing in cardiac muscle. J. Endocrinol. 2000, 165, 9–17. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Abdul, K.M.; Terada, K.; Yano, M.; Ryan, M.T.; Streimann, I.; Hoogenraad, N.J.; Mori, M. Functional analysis of human metaxin in mitochondrial protein import in cultured cells and its relationship with the Tom complex. Biochem. Biophys. Res. Commun. 2000, 276, 1028–1034. [Google Scholar] [CrossRef] [PubMed]
- Paschen, S.A.; Rothbauer, U.; Káldi, K.; Bauer, M.F.; Neupert, W.; Brunner, M. The role of the TIM8–13 complex in the import of Tim23 into mitochondria. EMBO J. 2000, 19, 6392–6400. [Google Scholar] [CrossRef] [PubMed]
- Picou, F.; Fauquier, T.; Chatonnet, F.; Richard, S.; Flamant, F. Deciphering direct and indirect influence of thyroid hormone with mouse genetics. Mol. Endocrinol. 2014, 4, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Henderson, K.K.; Danzi, S.; Paul, J.T.; Leya, G.; Klein, I.; Samarel, A.M. Physiological Replacement of T 3 Improves Left Ventricular Function in an Animal Model of Myocardial Infarction-Induced Congestive Heart Failure. Circ. Heart Fail. 2009, 2, 243–252. [Google Scholar] [CrossRef]
- Lymvaios, I.; Mourouzis, I.; Cokkinos, D.V.; A Dimopoulos, M.; Toumanidis, S.T.; Pantos, C.; Dimopoulos, M. Thyroid hormone and recovery of cardiac function in patients with acute myocardial infarction: A strong association? Eur. J. Endocrinol. 2011, 165, 107–114. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forini, F.; Nicolini, G.; Kusmic, C.; Iervasi, G. Protective Effects of Euthyroidism Restoration on Mitochondria Function and Quality Control in Cardiac Pathophysiology. Int. J. Mol. Sci. 2019, 20, 3377. https://doi.org/10.3390/ijms20143377
Forini F, Nicolini G, Kusmic C, Iervasi G. Protective Effects of Euthyroidism Restoration on Mitochondria Function and Quality Control in Cardiac Pathophysiology. International Journal of Molecular Sciences. 2019; 20(14):3377. https://doi.org/10.3390/ijms20143377
Chicago/Turabian StyleForini, Francesca, Giuseppina Nicolini, Claudia Kusmic, and Giorgio Iervasi. 2019. "Protective Effects of Euthyroidism Restoration on Mitochondria Function and Quality Control in Cardiac Pathophysiology" International Journal of Molecular Sciences 20, no. 14: 3377. https://doi.org/10.3390/ijms20143377
APA StyleForini, F., Nicolini, G., Kusmic, C., & Iervasi, G. (2019). Protective Effects of Euthyroidism Restoration on Mitochondria Function and Quality Control in Cardiac Pathophysiology. International Journal of Molecular Sciences, 20(14), 3377. https://doi.org/10.3390/ijms20143377