Relevance of Receptor for Advanced Glycation end Products (RAGE) in Murine Antibody-Mediated Autoimmune Diseases

 ,
,
Abstract
1. Introduction
2. Results
2.1. Influence of RAGE Deficiency in Pristane-Induced Lupus
2.2. Phenotype of Splenocytes in WT and RAGE−/− Mice after Pristane Injection
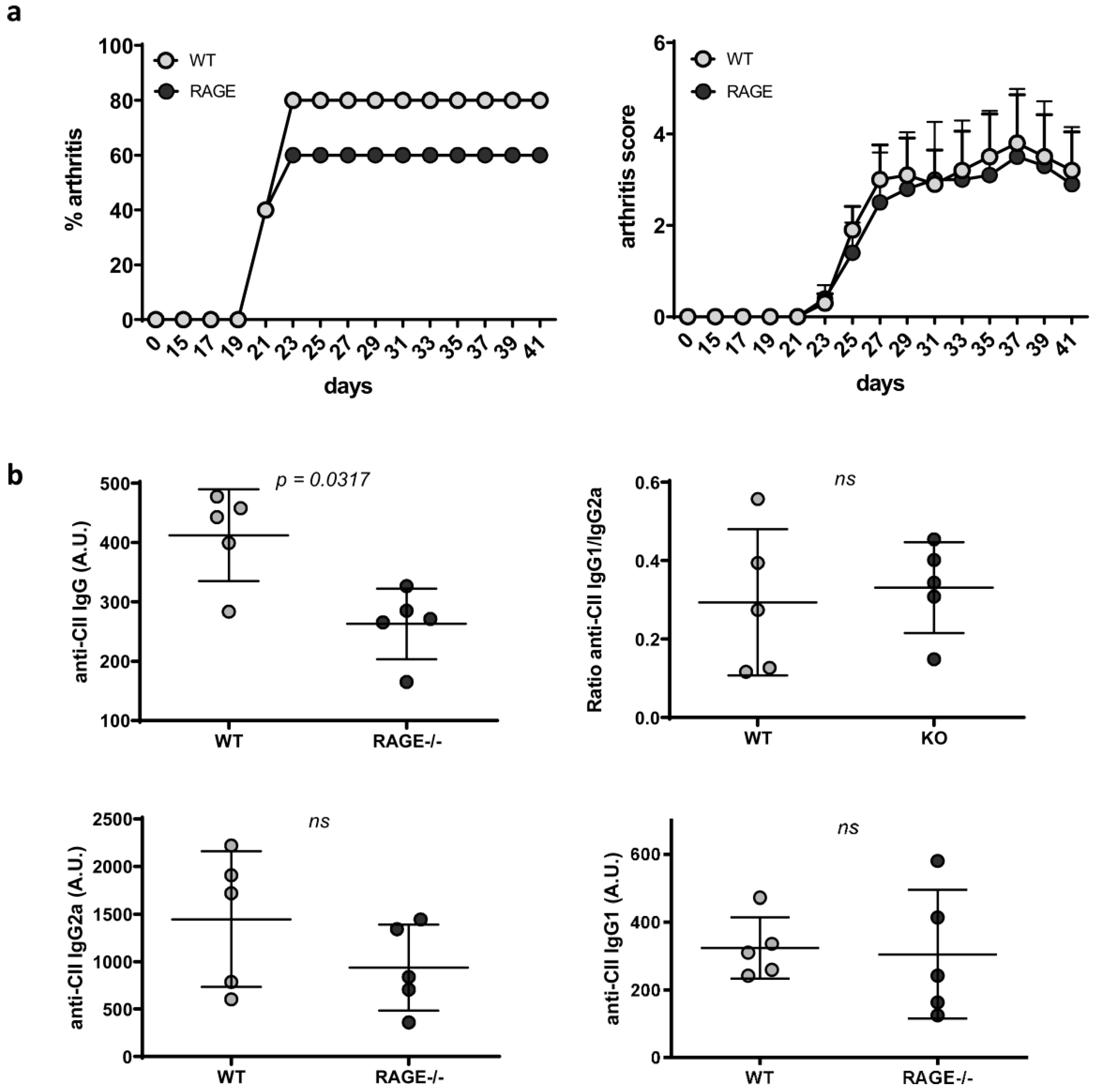
2.3. Influence of RAGE Deficiency on Development of Arthritis
3. Discussion
4. Materials and Methods
4.1. Animals and Treatments
4.2. Arthritis Scoring
4.3. Assessment of Kidney Disease
4.4. Determination of Serum Autoantibodies
4.4.1. Anti-CII IgG Antibodies and Its Subclasses
4.4.2. Anti-dsDNA Autoantibodies
4.5. Flow Cytometry
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| RAGE | receptor for advanced glycation end products |
| CIA | collagen-induced |
| ELISA | enzyme-linked immunosorbent assay |
| SLE | systemic lupus erythematosus |
| RA | rheumatoid arthritis |
| dsDNA | double-stranded DNA |
| PAMPs | pathogen-associated molecular patterns |
| DAMPs | danger-associated molecular patterns |
| PRRs | pattern recognition receptors |
| DCs | dendritic cells |
| AGEs | advanced glycation end products |
| HMGB1 | high mobility group box 1 |
| sRAGE | soluble receptor for advanced glycation end products |
| ALPS | autoimmune lymphoproliferative syndrome |
| Lpr | lymphoproliferation |
| PAS | periodic acid Schiff |
| OD | optical density |
| FCS | fetal calf serum |
| A.U. | arbitrary unit |
References
- Harley, I.T.; Kaufman, K.M.; Langefeld, C.D.; Harley, J.B.; Kelly, J.A. Genetic susceptibility to SLE: New insights from fine mapping and genome-wide association studies. Nat. Rev. Genet. 2009, 10, 285–290. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [PubMed]
- Ehrenstein, M.R.; Katz, D.R.; Griffiths, M.H.; Papadaki, L.; Winkler, T.H.; Kalden, J.R.; Isenberg, D.A. Human IgG anti-DNA antibodies deposit in kidneys and induce proteinuria in SCID mice. Kidney Int. 1995, 48, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Winfield, J.B.; Faiferman, I.; Koffler, D. Avidity of anti-DNA antibodies in serum and IgG glomerular eluates from patients with systemic lupus erythematosus. Association of high avidity antinative DNA antibody with glomerulonephritis. J. Clin. Invest. 1977, 59, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Derksen, V.; Huizinga, T.W.J.; van der Woude, D. The role of autoantibodies in the pathophysiology of rheumatoid arthritis. Semin. Immunopathol. 2017, 39, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.M.; Schur, P.H.; Carr, R.I.; Kunkel, H.G. Deoxybonucleic acid (DNA) and antibodies to DNA in the serum of patients with systemic lupus erythematosus. J. Clin. Invest. 1966, 45, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Suurmond, J.; Diamond, B. Autoantibodies in systemic autoimmune diseases: Specificity and pathogenicity. J. Clin. Invest. 2015, 125, 2194–2202. [Google Scholar] [CrossRef]
- Kierdorf, K.; Fritz, G. RAGE regulation and signaling in inflammation and beyond. J. Leukoc. Biol. 2013, 94, 55–68. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Hori, O.; Cao, R.; Yan, S.D.; Brett, J.; Wautier, J.L.; Ogawa, S.; Kuwabara, K.; Matsumoto, M.; Stern, D. RAGE: A novel cellular receptor for advanced glycation end products. Diabetes 1996, 45 (Suppl. 3), S77–S80. [Google Scholar] [CrossRef]
- Hofmann, M.A.; Drury, S.; Fu, C.; Qu, W.; Taguchi, A.; Lu, Y.; Avila, C.; Kambham, N.; Bierhaus, A.; Nawroth, P.; et al. RAGE mediates a novel proinflammatory axis: A central cell surface receptor for S100/calgranulin polypeptides. Cell 1999, 97, 889–901. [Google Scholar] [CrossRef]
- Leclerc, E.; Fritz, G.; Vetter, S.W.; Heizmann, C.W. Binding of S100 proteins to RAGE: An update. Biochim. Biophys. Acta 2009, 1793, 993–1007. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.M.; Yan, S.D.; Yan, S.F.; Stern, D.M. The biology of the receptor for advanced glycation end products and its ligands. Biochim. Biophys. Acta 2000, 1498, 99–111. [Google Scholar] [CrossRef]
- Lander, H.M.; Tauras, J.M.; Ogiste, J.S.; Hori, O.; Moss, R.A.; Schmidt, A.M. Activation of the receptor for advanced glycation end products triggers a p21(ras)-dependent mitogen-activated protein kinase pathway regulated by oxidant stress. J. Biol. Chem. 1997, 272, 17810–17814. [Google Scholar] [CrossRef] [PubMed]
- Kislinger, T.; Fu, C.; Huber, B.; Qu, W.; Taguchi, A.; Du Yan, S.; Hofmann, M.; Yan, S.F.; Pischetsrieder, M.; Stern, D.; et al. N(epsilon)-(carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J. Biol. Chem. 1999, 274, 31740–31749. [Google Scholar] [CrossRef] [PubMed]
- Basta, G.; Lazzerini, G.; Massaro, M.; Simoncini, T.; Tanganelli, P.; Fu, C.; Kislinger, T.; Stern, D.M.; Schmidt, A.M.; De Caterina, R. Advanced glycation end products activate endothelium through signal-transduction receptor RAGE: A mechanism for amplification of inflammatory responses. Circulation 2002, 105, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, R.; Yan, S.F.; Schmidt, A.M. Receptor for AGE (RAGE): Signaling mechanisms in the pathogenesis of diabetes and its complications. Ann. N. Y. Acad Sci 2011, 1243, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.I.; Lippman, M.E. Targeting RAGE Signaling in Inflammatory Disease. Annu. Rev. Med. 2018, 69, 349–364. [Google Scholar] [CrossRef]
- Brett, J.; Schmidt, A.M.; Yan, S.D.; Zou, Y.S.; Weidman, E.; Pinsky, D.; Nowygrod, R.; Neeper, M.; Przysiecki, C.; Shaw, A.; et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am. J. Pathol 1993, 143, 1699–1712. [Google Scholar]
- Tanji, N.; Markowitz, G.S.; Fu, C.; Kislinger, T.; Taguchi, A.; Pischetsrieder, M.; Stern, D.; Schmidt, A.M.; D’Agati, V.D. Expression of advanced glycation end products and their cellular receptor RAGE in diabetic nephropathy and nondiabetic renal disease. J. Am. Soc. Nephrol. 2000, 11, 1656–1666. [Google Scholar]
- Reynaert, N.L.; Gopal, P.; Rutten, E.P.A.; Wouters, E.F.M.; Schalkwijk, C.G. Advanced glycation end products and their receptor in age-related, non-communicable chronic inflammatory diseases; Overview of clinical evidence and potential contributions to disease. Int. J. Biochem. Cell Biol. 2016, 81, 403–418. [Google Scholar] [CrossRef]
- Bierhaus, A.; Stern, D.M.; Nawroth, P.P. RAGE in inflammation: A new therapeutic target? Curr. Opin. Investig. Drugs 2006, 7, 985–991. [Google Scholar] [PubMed]
- Kalea, A.Z.; Schmidt, A.M.; Hudson, B.I. RAGE: A novel biological and genetic marker for vascular disease. Clin. Sci. (Lond) 2009, 116, 621–637. [Google Scholar] [CrossRef] [PubMed]
- Piperi, C.; Adamopoulos, C.; Dalagiorgou, G.; Diamanti-Kandarakis, E.; Papavassiliou, A.G. Crosstalk between advanced glycation and endoplasmic reticulum stress: Emerging therapeutic targeting for metabolic diseases. J. Clin. Endocrinol. Metab. 2012, 97, 2231–2242. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Taneda, S.; Richey, P.L.; Miyata, S.; Yan, S.D.; Stern, D.; Sayre, L.M.; Monnier, V.M.; Perry, G. Advanced Maillard reaction end products are associated with Alzheimer disease pathology. Proc. Natl. Acad. Sci. USA 1994, 91, 5710–5714. [Google Scholar] [CrossRef] [PubMed]
- Raucci, A.; Cugusi, S.; Antonelli, A.; Barabino, S.M.; Monti, L.; Bierhaus, A.; Reiss, K.; Saftig, P.; Bianchi, M.E. A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). FASEB J. 2008, 22, 3716–3727. [Google Scholar] [CrossRef] [PubMed]
- Geroldi, D.; Falcone, C.; Emanuele, E. Soluble receptor for advanced glycation end products: From disease marker to potential therapeutic target. Curr. Med. Chem. 2006, 13, 1971–1978. [Google Scholar] [CrossRef]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. (Berl) 2005, 83, 876–886. [Google Scholar] [CrossRef]
- Sims, G.P.; Rowe, D.C.; Rietdijk, S.T.; Herbst, R.; Coyle, A.J. HMGB1 and RAGE in inflammation and cancer. Annu. Rev. Immunol. 2010, 28, 367–388. [Google Scholar] [CrossRef]
- Harris, H.E.; Andersson, U.; Pisetsky, D.S. HMGB1: A multifunctional alarmin driving autoimmune and inflammatory disease. Nat. Rev. Rheumatol. 2012, 8, 195–202. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Urbonaviciute, V.; Furnrohr, B.G.; Meister, S.; Munoz, L.; Heyder, P.; de Marchis, F.; Bianchi, M.E.; Kirschning, C.; Wagner, H.; Manfredi, A.A.; et al. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: Implications for the pathogenesis of SLE. J. Exp. Med. 2008, 205, 3007–3018. [Google Scholar] [CrossRef] [PubMed]
- Perry, D.; Sang, A.; Yin, Y.; Zheng, Y.Y.; Morel, L. Murine models of systemic lupus erythematosus. J. Biomed. Biotechnol. 2011, 2011, 271694. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, N.; Macia, L.; Tan, J.K.; Mason, L.J.; Robert, R.; Thorburn, A.N.; Wong, C.H.; Tsai, L.M.; Bourne, K.; Brink, R.; et al. The Role of Follicular Helper T Cell Molecules and Environmental Influences in Autoantibody Production and Progression to Inflammatory Arthritis in Mice. Arthritis Rheumatol. 2016, 68, 1026–1038. [Google Scholar] [CrossRef] [PubMed]
- Pietrosimone, K.M.; Jin, M.; Poston, B.; Liu, P. Collagen-Induced Arthritis: A model for Murine Autoimmune Arthritis. Bio. Protoc. 2015, 5, e1612. [Google Scholar] [CrossRef]
- Korganow, A.S.; Ji, H.; Mangialaio, S.; Duchatelle, V.; Pelanda, R.; Martin, T.; Degott, C.; Kikutani, H.; Rajewsky, K.; Pasquali, J.L.; et al. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity 1999, 10, 451–461. [Google Scholar] [CrossRef]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune. Netw. 2018, 18, e27. [Google Scholar] [CrossRef]
- Sirois, C.M.; Jin, T.; Miller, A.L.; Bertheloot, D.; Nakamura, H.; Horvath, G.L.; Mian, A.; Jiang, J.; Schrum, J.; Bossaller, L.; et al. RAGE is a nucleic acid receptor that promotes inflammatory responses to DNA. J. Exp. Med. 2013, 210, 2447–2463. [Google Scholar] [CrossRef]
- Lahoud, M.H.; Ahmet, F.; Zhang, J.G.; Meuter, S.; Policheni, A.N.; Kitsoulis, S.; Lee, C.N.; O’Keeffe, M.; Sullivan, L.C.; Brooks, A.G.; et al. DEC-205 is a cell surface receptor for CpG oligonucleotides. Proc. Natl. Acad. Sci. USA 2012, 109, 16270–16275. [Google Scholar] [CrossRef]
- Zhou, H.; Liao, J.; Aloor, J.; Nie, H.; Wilson, B.C.; Fessler, M.B.; Gao, H.M.; Hong, J.S. CD11b/CD18 (Mac-1) is a novel surface receptor for extracellular double-stranded RNA to mediate cellular inflammatory responses. J. Immunol. 2013, 190, 115–125. [Google Scholar] [CrossRef]
- Baid, K.; Nellimarla, S.; Huynh, A.; Boulton, S.; Guarne, A.; Melacini, G.; Collins, S.E.; Mossman, K.L. Direct binding and internalization of diverse extracellular nucleic acid species through the collagenous domain of class A scavenger receptors. Immunol. Cell Biol. 2018, 96, 922–934. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Lin, L. RAGE on the Toll Road? Cell Mol. Immunol. 2006, 3, 351–358. [Google Scholar] [PubMed]
- Urbonaviciute, V.; Starke, C.; Pirschel, W.; Pohle, S.; Frey, S.; Daniel, C.; Amann, K.; Schett, G.; Herrmann, M.; Voll, R.E. Toll-like receptor 2 is required for autoantibody production and development of renal disease in pristane-induced lupus. Arthritis Rheum. 2013, 65, 1612–1623. [Google Scholar] [CrossRef] [PubMed]
- Lartigue, A.; Colliou, N.; Calbo, S.; Francois, A.; Jacquot, S.; Arnoult, C.; Tron, F.; Gilbert, D.; Musette, P. Critical role of TLR2 and TLR4 in autoantibody production and glomerulonephritis in lpr mutation-induced mouse lupus. J. Immunol. 2009, 183, 6207–6216. [Google Scholar] [CrossRef] [PubMed]
- Moreth, K.; Brodbeck, R.; Babelova, A.; Gretz, N.; Spieker, T.; Zeng-Brouwers, J.; Pfeilschifter, J.; Young, M.F.; Schaefer, R.M.; Schaefer, L. The proteoglycan biglycan regulates expression of the B cell chemoattractant CXCL13 and aggravates murine lupus nephritis. J. Clin. Invest. 2010, 120, 4251–4272. [Google Scholar] [CrossRef] [PubMed]
- Loser, K.; Vogl, T.; Voskort, M.; Lueken, A.; Kupas, V.; Nacken, W.; Klenner, L.; Kuhn, A.; Foell, D.; Sorokin, L.; et al. The Toll-like receptor 4 ligands Mrp8 and Mrp14 are crucial in the development of autoreactive CD8+ T cells. Nat. Med. 2010, 16, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yin, H.; Zhao, M.; Lu, Q. TLR2 and TLR4 in autoimmune diseases: A comprehensive review. Clin. Rev. Allergy Immunol. 2014, 47, 136–147. [Google Scholar] [CrossRef]
- Abdollahi-Roodsaz, S.; Joosten, L.A.; Koenders, M.I.; Devesa, I.; Roelofs, M.F.; Radstake, T.R.; Heuvelmans-Jacobs, M.; Akira, S.; Nicklin, M.J.; Ribeiro-Dias, F.; et al. Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J. Clin. Invest. 2008, 118, 205–216. [Google Scholar] [CrossRef]
- Pierer, M.; Wagner, U.; Rossol, M.; Ibrahim, S. Toll-like receptor 4 is involved in inflammatory and joint destructive pathways in collagen-induced arthritis in DBA1J mice. PLoS ONE 2011, 6, e23539. [Google Scholar] [CrossRef]
- Ospelt, C.; Brentano, F.; Rengel, Y.; Stanczyk, J.; Kolling, C.; Tak, P.P.; Gay, R.E.; Gay, S.; Kyburz, D. Overexpression of toll-like receptors 3 and 4 in synovial tissue from patients with early rheumatoid arthritis: Toll-like receptor expression in early and longstanding arthritis. Arthritis Rheum 2008, 58, 3684–3692. [Google Scholar] [CrossRef] [PubMed]
- Radstake, T.R.; Roelofs, M.F.; Jenniskens, Y.M.; Oppers-Walgreen, B.; van Riel, P.L.; Barrera, P.; Joosten, L.A.; van den Berg, W.B. Expression of toll-like receptors 2 and 4 in rheumatoid synovial tissue and regulation by proinflammatory cytokines interleukin-12 and interleukin-18 via interferon-gamma. Arthritis Rheum. 2004, 50, 3856–3865. [Google Scholar] [CrossRef] [PubMed]
- Komatsuda, A.; Wakui, H.; Iwamoto, K.; Ozawa, M.; Togashi, M.; Masai, R.; Maki, N.; Hatakeyama, T.; Sawada, K. Up-regulated expression of Toll-like receptors mRNAs in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Clin. Exp. Immunol 2008, 152, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, M.; Sonnenschein, A.; Schoofs, S.; Schmidtke, P.; Umlauf, V.N.; Mannhardt-Laakmann, W. Surface expression and genotypes of Toll-like receptors 2 and 4 in patients with juvenile idiopathic arthritis and systemic lupus erythematosus. Pediatr. Rheumatol. Online J. 2013, 11, 9. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, M.L.; Wolska, A.; Grzegorczyk, J.; Hilt, J.; Jarzebska, M.; Drobniewski, M.; Synder, M.; Kurowski, M. Increased responsiveness to toll-like receptor 4 stimulation in peripheral blood mononuclear cells from patients with recent onset rheumatoid arthritis. Mediators Inflamm. 2008, 2008, 132732. [Google Scholar] [CrossRef] [PubMed]
- Chovanova, L.; Vlcek, M.; Krskova, K.; Penesova, A.; Radikova, Z.; Rovensky, J.; Cholujova, D.; Sedlak, J.; Imrich, R. Increased production of IL-6 and IL-17 in lipopolysaccharide-stimulated peripheral mononuclears from patients with rheumatoid arthritis. Gen. Physiol. Biophys. 2013, 32, 395–404. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tsao, J.T.; Hsieh, S.C.; Chiang, B.L.; Yu, C.L.; Lin, S.C. Altered IL-10 and TNF-alpha production in peripheral blood mononuclear cells of systemic lupus erythematosus patients after Toll-like receptor 2, 4, or 9 activation. Clin. Exp. Med. 2012, 12, 153–158. [Google Scholar] [CrossRef]
- Bangert, A.; Andrassy, M.; Muller, A.M.; Bockstahler, M.; Fischer, A.; Volz, C.H.; Leib, C.; Goser, S.; Korkmaz-Icoz, S.; Zittrich, S.; et al. Critical role of RAGE and HMGB1 in inflammatory heart disease. Proc. Natl. Acad. Sci. USA 2016, 113, E155–E164. [Google Scholar] [CrossRef]
- D’Agati, V.; Schmidt, A.M. RAGE and the pathogenesis of chronic kidney disease. Nat. Rev. Nephrol. 2010, 6, 352–360. [Google Scholar] [CrossRef]
- Andersson, U.; Tracey, K.J. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu. Rev. Immunol. 2011, 29, 139–162. [Google Scholar] [CrossRef]
- Magna, M.; Pisetsky, D.S. The role of HMGB1 in the pathogenesis of inflammatory and autoimmune diseases. Mol. Med. 2014, 20, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.S.; Yan, W.; Geczy, C.L.; Brown, M.A.; Thomas, R. Serum levels of soluble receptor for advanced glycation end products and of S100 proteins are associated with inflammatory, autoantibody, and classical risk markers of joint and vascular damage in rheumatoid arthritis. Arthritis Res. Ther. 2009, 11, R39. [Google Scholar] [CrossRef] [PubMed]
- De Leeuw, K.; Graaff, R.; de Vries, R.; Dullaart, R.P.; Smit, A.J.; Kallenberg, C.G.; Bijl, M. Accumulation of advanced glycation endproducts in patients with systemic lupus erythematosus. Rheumatology (Oxford) 2007, 46, 1551–1556. [Google Scholar] [CrossRef] [PubMed]
- Pullerits, R.; Bokarewa, M.; Dahlberg, L.; Tarkowski, A. Decreased levels of soluble receptor for advanced glycation end products in patients with rheumatoid arthritis indicating deficient inflammatory control. Arthritis Res. Ther. 2005, 7, R817-24. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.Y.; Ma, J.L.; Jiao, Y.L.; Li, J.F.; Wang, L.C.; Yang, Q.R.; You, L.; Cui, B.; Chen, Z.J.; Zhao, Y.R. The plasma level of soluble receptor for advanced glycation end products is decreased in patients with systemic lupus erythematosus. Scand. J. Immunol. 2012, 75, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Myles, A.; Viswanath, V.; Singh, Y.P.; Aggarwal, A. Soluble receptor for advanced glycation endproducts is decreased in patients with juvenile idiopathic arthritis (ERA category) and inversely correlates with disease activity and S100A12 levels. J. Rheumatol. 2011, 38, 1994–1999. [Google Scholar] [CrossRef] [PubMed]
- Martens, H.A.; Nienhuis, H.L.; Gross, S.; van der Steege, G.; Brouwer, E.; Berden, J.H.; de Sevaux, R.G.; Derksen, R.H.; Voskuyl, A.E.; Berger, S.P.; et al. Receptor for advanced glycation end products (RAGE) polymorphisms are associated with systemic lupus erythematosus and disease severity in lupus nephritis. Lupus 2012, 21, 959–968. [Google Scholar] [CrossRef]
- Lee, S.W.; Park, K.H.; Park, S.; Kim, J.H.; Hong, S.Y.; Lee, S.K.; Choi, D.; Park, Y.B. Soluble receptor for advanced glycation end products alleviates nephritis in (NZB/NZW)F1 mice. Arthritis Rheum. 2013, 65, 1902–1912. [Google Scholar] [CrossRef]
- Goury, A.; Meghraoui-Kheddar, A.; Belmokhtar, K.; Vuiblet, V.; Ortillon, J.; Jaisson, S.; Devy, J.; Le Naour, R.; Tabary, T.; Cohen, J.H.; et al. Deletion of receptor for advanced glycation end products exacerbates lymphoproliferative syndrome and lupus nephritis in B6-MRL Fas lpr/j mice. J. Immunol. 2015, 194, 3612–3622. [Google Scholar] [CrossRef]
- Reap, E.A.; Leslie, D.; Abrahams, M.; Eisenberg, R.A.; Cohen, P.L. Apoptosis abnormalities of splenic lymphocytes in autoimmune lpr and gld mice. J. Immunol. 1995, 154, 936–943. [Google Scholar]
- Teachey, D.T.; Seif, A.E.; Grupp, S.A. Advances in the management and understanding of autoimmune lymphoproliferative syndrome (ALPS). Br. J. Haematol. 2010, 148, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.; Morales, M.; Gonzalez, I.; Araya, P. Inhibition of RAGE Axis Signaling: A Pharmacological Challenge. Curr. Drug Targets 2019, 20, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Zhu, X.; Kwiecinski, J.; Gjertsson, I.; Lindholm, C.; Iwakura, Y.; Wang, X.; Lycke, N.; Josefsson, E.; Pullerits, R.; et al. Antibiotic-killed Staphylococcus aureus induces destructive arthritis in mice. Arthritis Rheumatol. 2015, 67, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, M.; Na, M.; Welin, A.; Svensson, M.N.; Ali, A.; Jin, T.; Pullerits, R. RAGE Deficiency Impairs Bacterial Clearance in Murine Staphylococcal Sepsis, but Has No Significant Impact on Staphylococcal Septic Arthritis. PLoS One 2016, 11, e0167287. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Grevers, L.C.; de Vries, T.J.; Vogl, T.; Abdollahi-Roodsaz, S.; Sloetjes, A.W.; Leenen, P.J.; Roth, J.; Everts, V.; van den Berg, W.B.; van Lent, P.L. S100A8 enhances osteoclastic bone resorption in vitro through activation of Toll-like receptor 4: Implications for bone destruction in murine antigen-induced arthritis. Arthritis Rheum. 2011, 63, 1365–1375. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Katsuta, S.; Tamura, Y.; Nagase, N.; Suzuki, K.; Nomura, M.; Tomatsu, S.; Miyamoto, K.; Kobayashi, S. Bone-targeting endogenous secretory receptor for advanced glycation end products rescues rheumatoid arthritis. Mol. Med. 2013, 19, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.; Delgado-Lopez, F.; Gonzalez, I.; Perez-Castro, R.; Romero, J.; Rojas, I. The receptor for advanced glycation end-products: A complex signaling scenario for a promiscuous receptor. Cell Signal. 2013, 25, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Liliensiek, B.; Weigand, M.A.; Bierhaus, A.; Nicklas, W.; Kasper, M.; Hofer, S.; Plachky, J.; Grone, H.J.; Kurschus, F.C.; Schmidt, A.M.; et al. Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J. Clin. Invest. 2004, 113, 1641–1650. [Google Scholar] [CrossRef]
- Englert, J.M.; Hanford, L.E.; Kaminski, N.; Tobolewski, J.M.; Tan, R.J.; Fattman, C.L.; Ramsgaard, L.; Richards, T.J.; Loutaev, I.; Nawroth, P.P.; et al. A role for the receptor for advanced glycation end products in idiopathic pulmonary fibrosis. Am. J. Pathol. 2008, 172, 583–591. [Google Scholar] [CrossRef]
- Kai, H.; Shibuya, K.; Wang, Y.; Kameta, H.; Kameyama, T.; Tahara-Hanaoka, S.; Miyamoto, A.; Honda, S.; Matsumoto, I.; Koyama, A.; et al. Critical role of M. tuberculosis for dendritic cell maturation to induce collagen-induced arthritis in H-2b background of C57BL/6 mice. Immunology 2006, 118, 233–239. [Google Scholar] [CrossRef]
- Chevalier, N.; Tan, J.K.; Mason, L.J.; Robert, R.; McKenzie, C.I.; Lim, F.; Wong, C.H.; Macia, L.; Thorburn, A.N.; Russ, B.E.; et al. Avenues to autoimmune arthritis triggered by diverse remote inflammatory challenges. J. Autoimmun. 2016, 73, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, N.; Thorburn, A.N.; Macia, L.; Tan, J.; Juglair, L.; Yagita, H.; Yu, D.; Hansbro, P.M.; Mackay, C.R. Inflammation and lymphopenia trigger autoimmunity by suppression of IL-2-controlled regulatory T cell and increase of IL-21-mediated effector T cell expansion. J. Immunol. 2014, 193, 4845–4858. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Splenic Cell Subsets | WT | RAGE−/− | |
|---|---|---|---|
| CD11chi | / | 1.448 (0.226) | 1.208 (0.222) |
| CD11b+ | / | 6.369 (1.127) | 5.973 (1.697) |
| Ly6Ghi | 3.351 (0.838) | 2.736 (1.199) | |
| Ly6Chi | 0.918 (0.376) | 1.090 (0.356) | |
| Ly6Clo | 1.917 (0.454) | 1.980 (0.387) | |
| CD4+ | / | 13.740 (1.225) | 14.686 (2.554) |
| CD44hi | 54.810 (4.043) | 54.486 (7.926) | |
| CD69+ | 24.690 (2.591) | 26.766 (5.135) | |
| CXCR5hiPD1hi | 11.119 (2.381) | 10.789 (2.735) | |
| IFNγ | 10.666 (3.198) | 10.064 (1.686) | |
| IL17 | 0.365 (0.232) | 0.403 (0.134) | |
| FoxP3 | 26.944 (6.147) | 27.206 (4.679) | |
| B220+ | / | 50.620 (3.572) | 57.600 (4.227) |
| FashiGL7hi (*) | 6.554 (4.515) | 4.515 (1.618) | |
| CD21loCD23hi (**) | 45.530 (8.893) | 52.186 (6.125) | |
| CD21hiCD23lo | 6.360 (3.437) | 6.892 (2.670) | |
| IgD+IgM+ | 24.550 (7.334) | 27.773 (6.501) | |
| IgM+CD5+ | 1.117 (0.288) | 1.106 (0.249) | |
| LC+CD138hi (***) | 1.785 (0.324) | 1.453 (0.397) | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eichhorst, A.; Daniel, C.; Rzepka, R.; Sehnert, B.; Nimmerjahn, F.; Voll, R.E.; Chevalier, N. Relevance of Receptor for Advanced Glycation end Products (RAGE) in Murine Antibody-Mediated Autoimmune Diseases. Int. J. Mol. Sci. 2019, 20, 3234. https://doi.org/10.3390/ijms20133234
Eichhorst A, Daniel C, Rzepka R, Sehnert B, Nimmerjahn F, Voll RE, Chevalier N. Relevance of Receptor for Advanced Glycation end Products (RAGE) in Murine Antibody-Mediated Autoimmune Diseases. International Journal of Molecular Sciences. 2019; 20(13):3234. https://doi.org/10.3390/ijms20133234
Chicago/Turabian StyleEichhorst, Alexandra, Christoph Daniel, Rita Rzepka, Bettina Sehnert, Falk Nimmerjahn, Reinhard E. Voll, and Nina Chevalier. 2019. "Relevance of Receptor for Advanced Glycation end Products (RAGE) in Murine Antibody-Mediated Autoimmune Diseases" International Journal of Molecular Sciences 20, no. 13: 3234. https://doi.org/10.3390/ijms20133234
APA StyleEichhorst, A., Daniel, C., Rzepka, R., Sehnert, B., Nimmerjahn, F., Voll, R. E., & Chevalier, N. (2019). Relevance of Receptor for Advanced Glycation end Products (RAGE) in Murine Antibody-Mediated Autoimmune Diseases. International Journal of Molecular Sciences, 20(13), 3234. https://doi.org/10.3390/ijms20133234