In Silico Prediction of PAMPA Effective Permeability Using a Two-QSAR Approach



Abstract
1. Introduction
2. Results
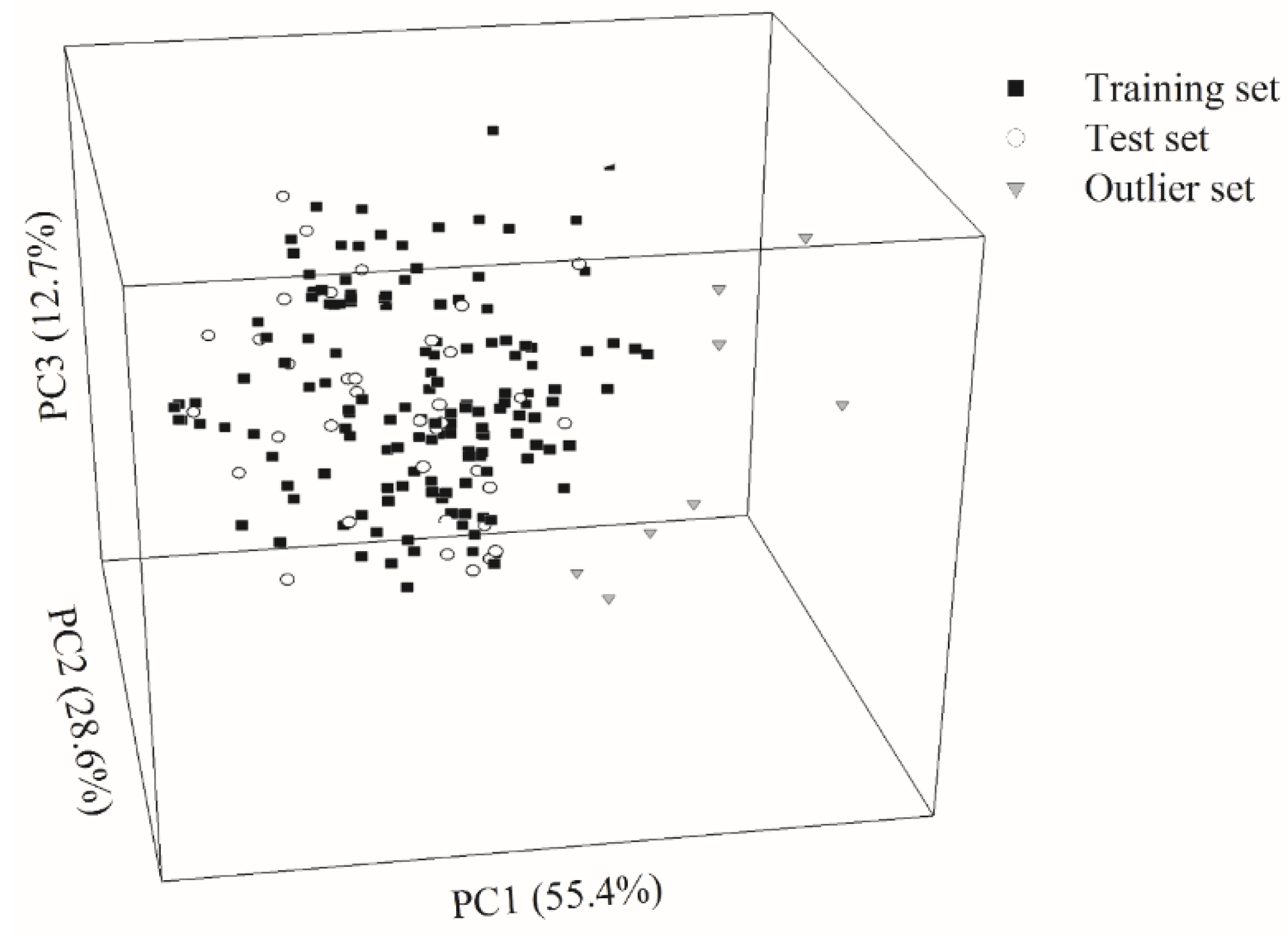
2.1. Data Partition
2.2. HSVR
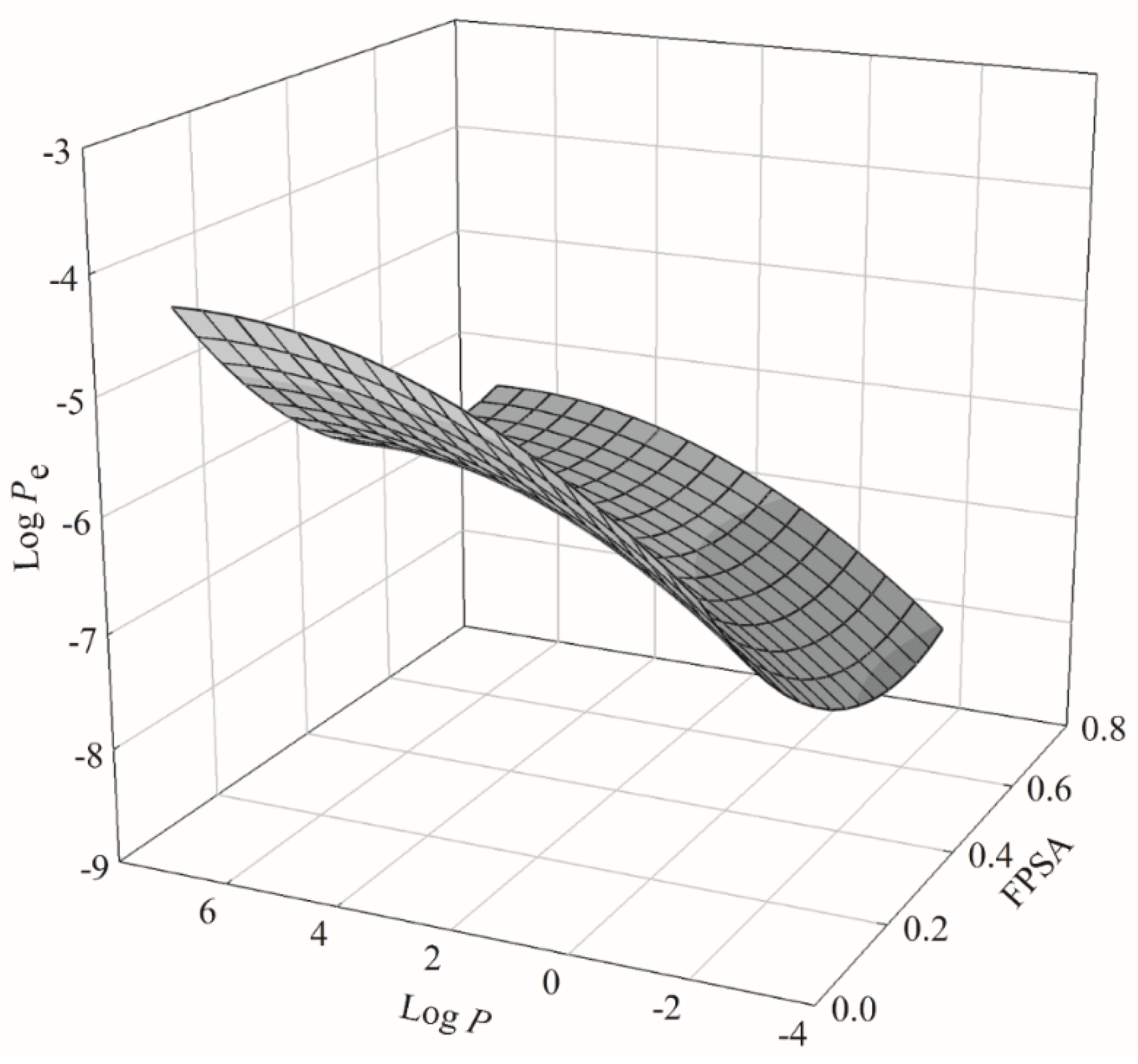
2.3. PLS
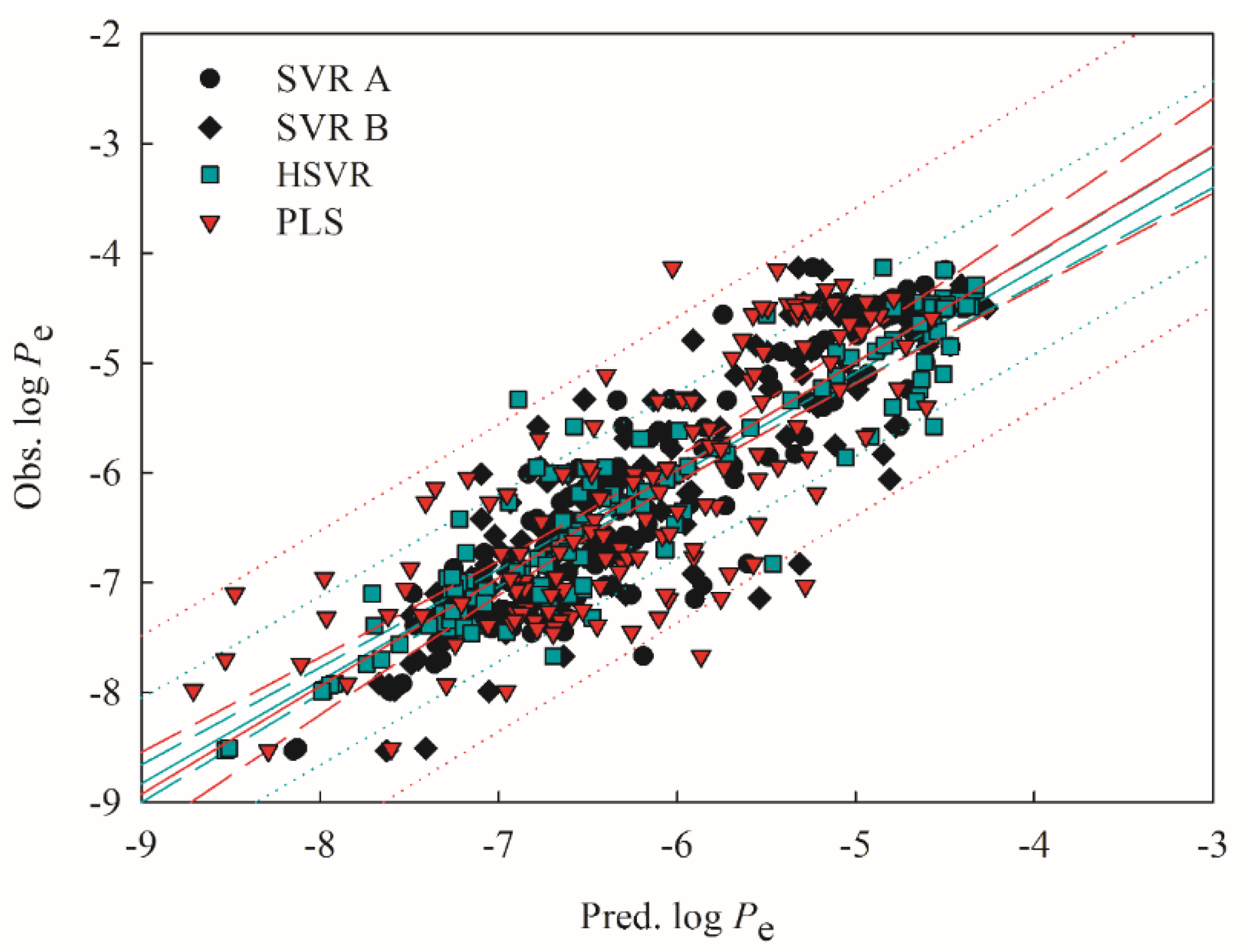
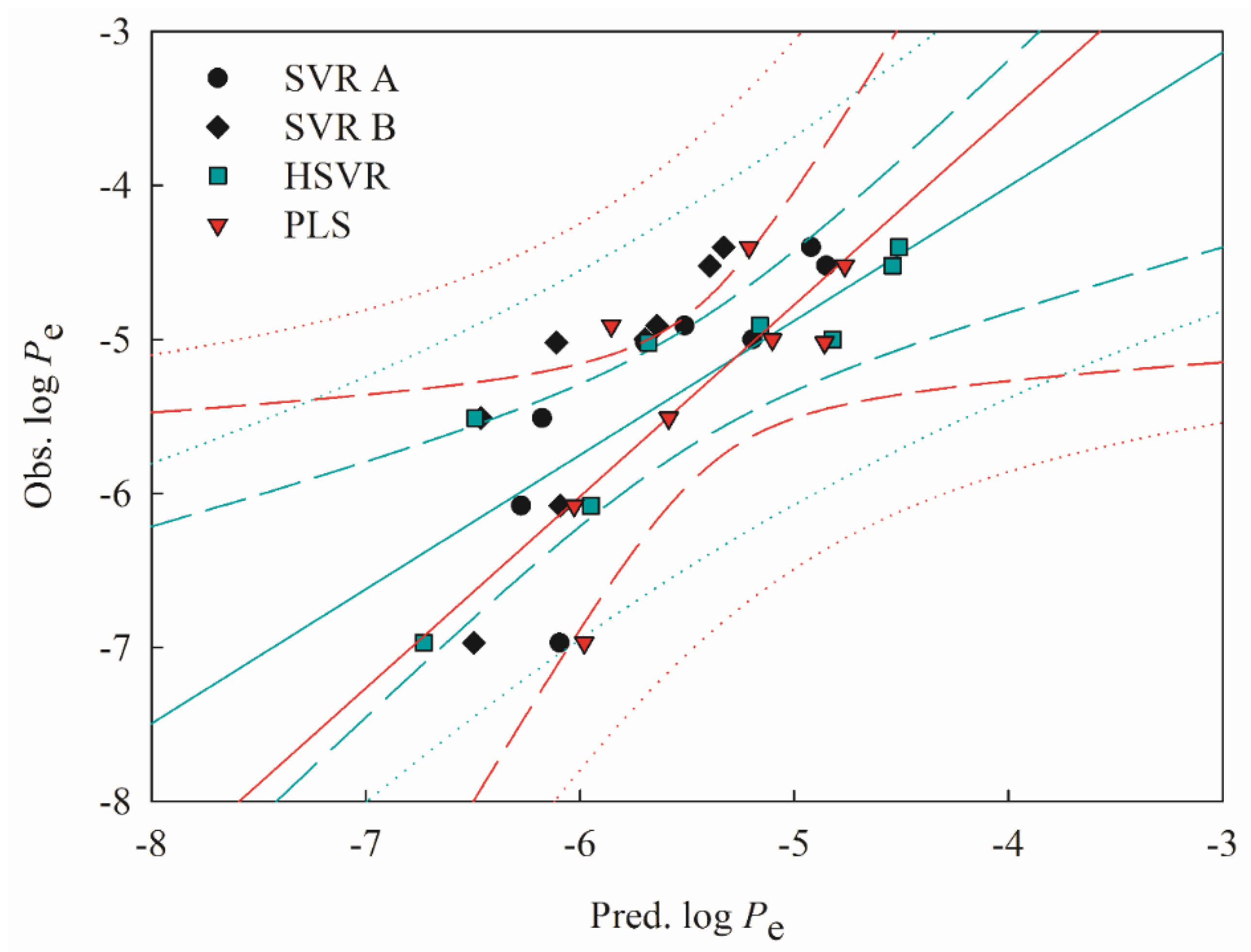
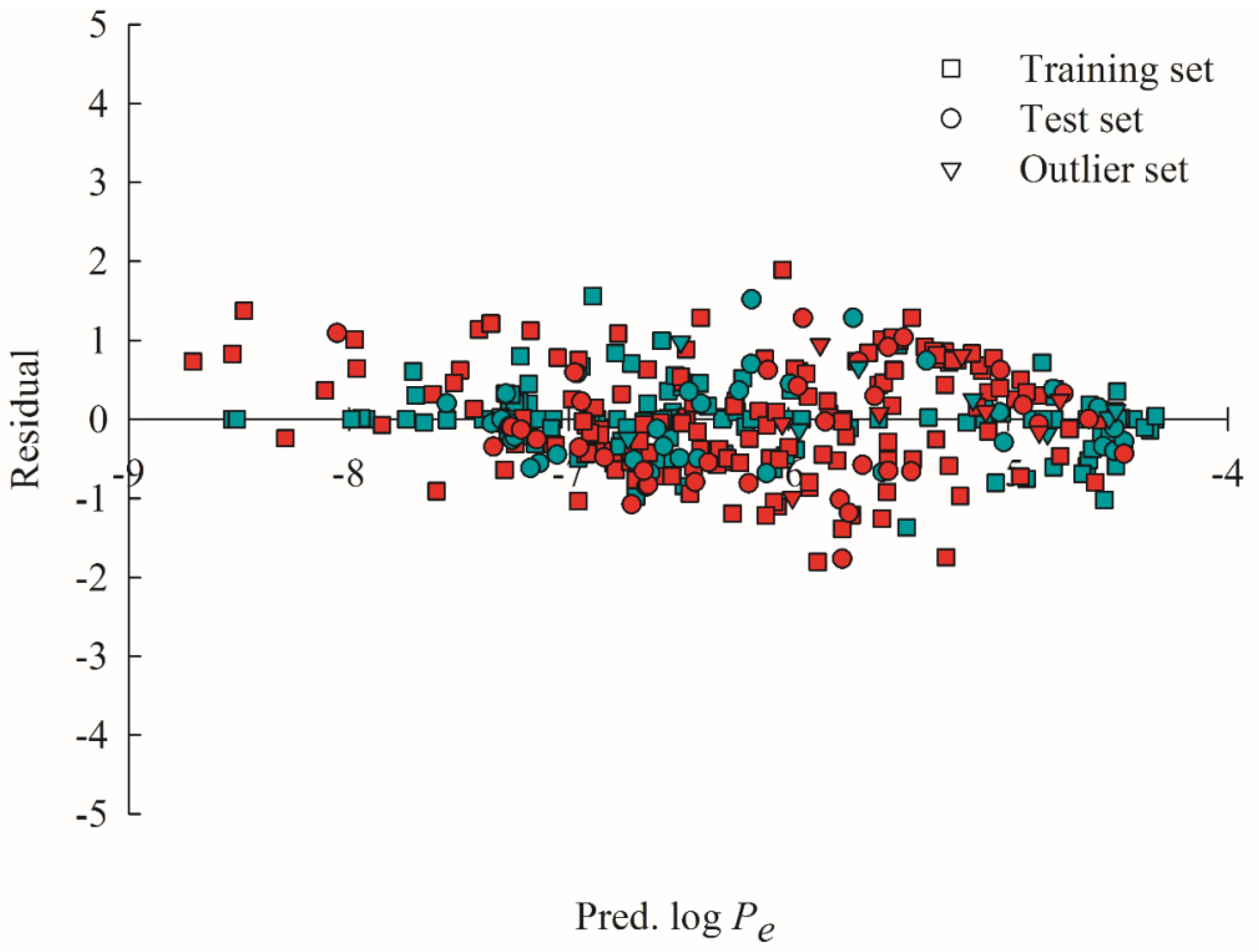
2.4. Predictive Evaluations
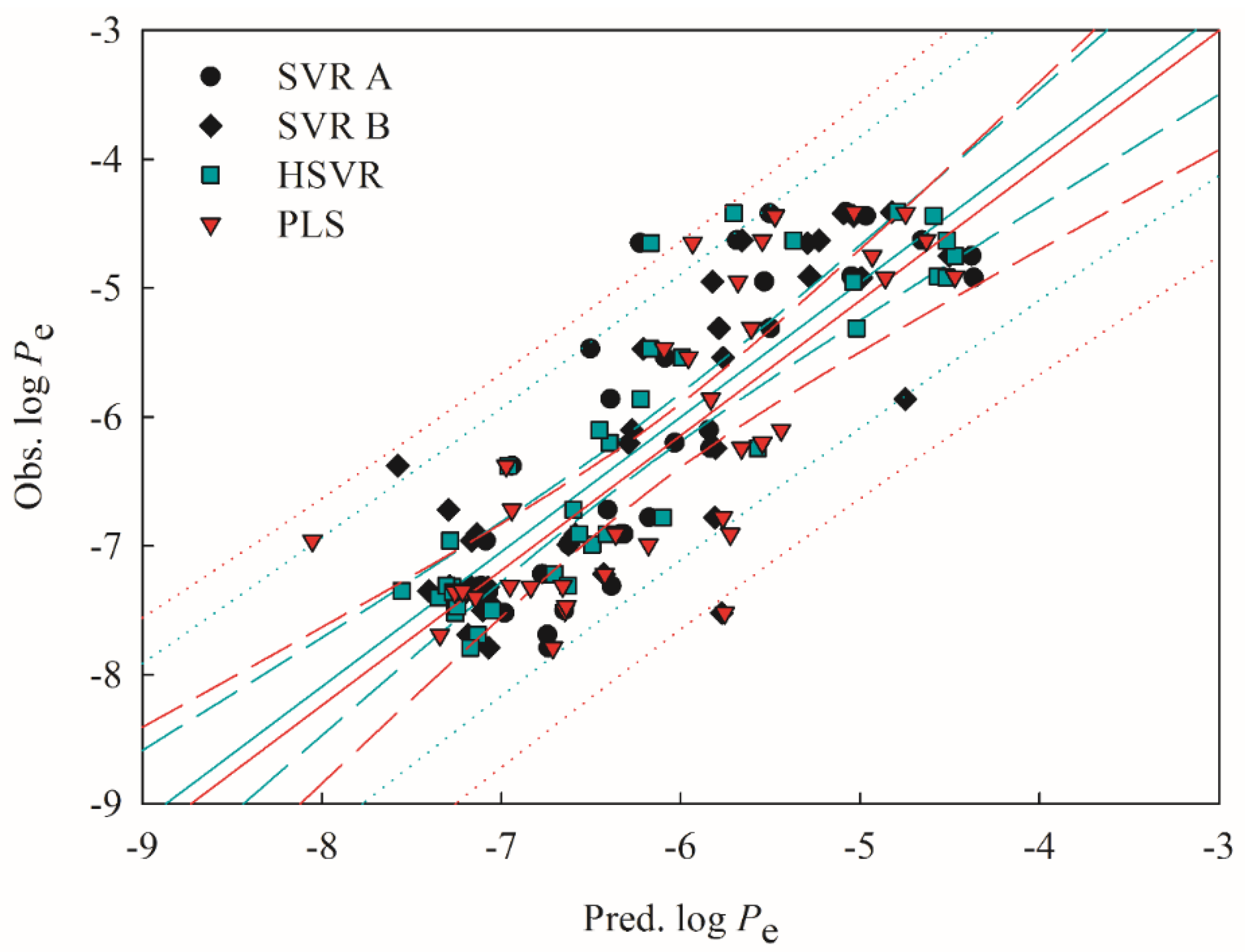
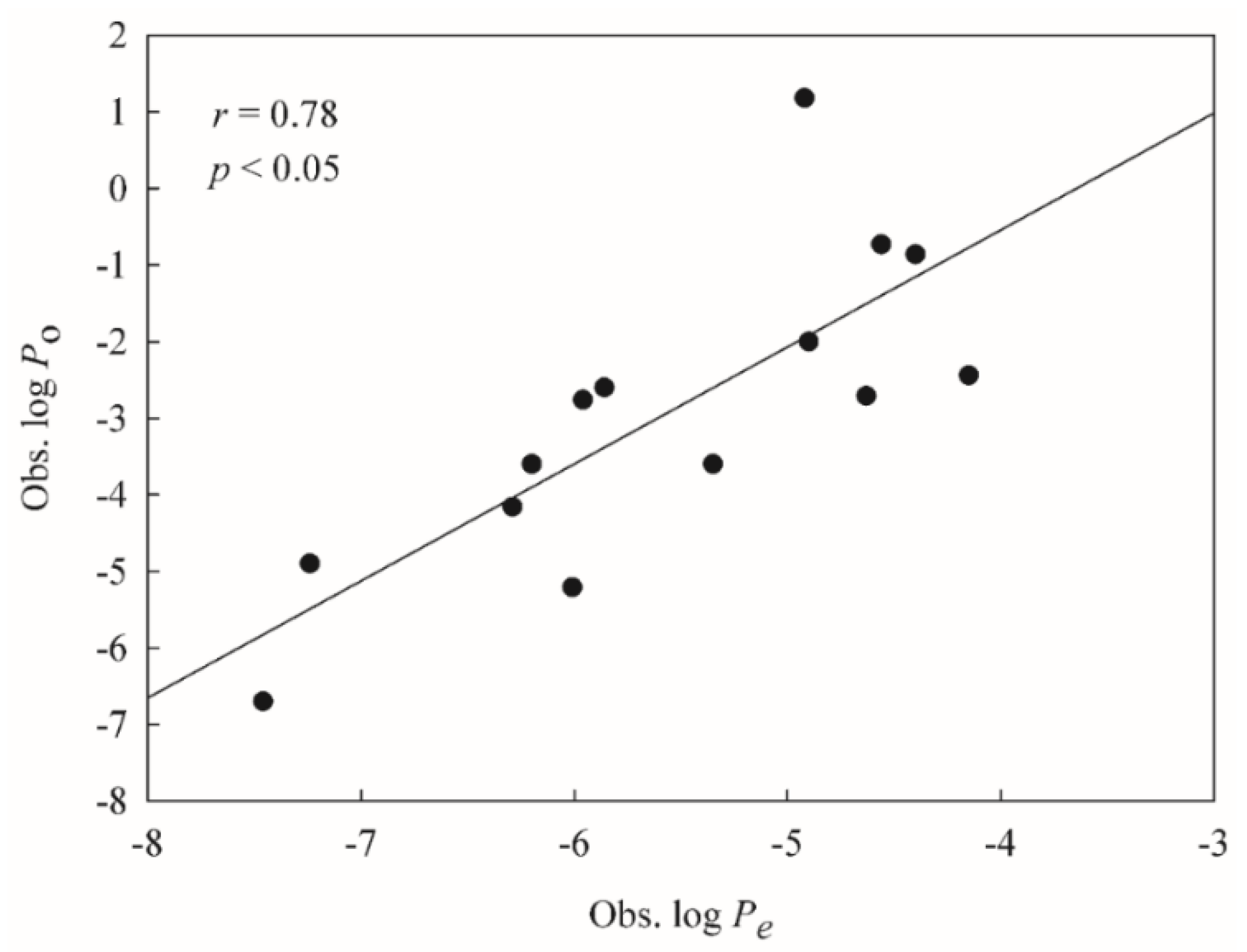
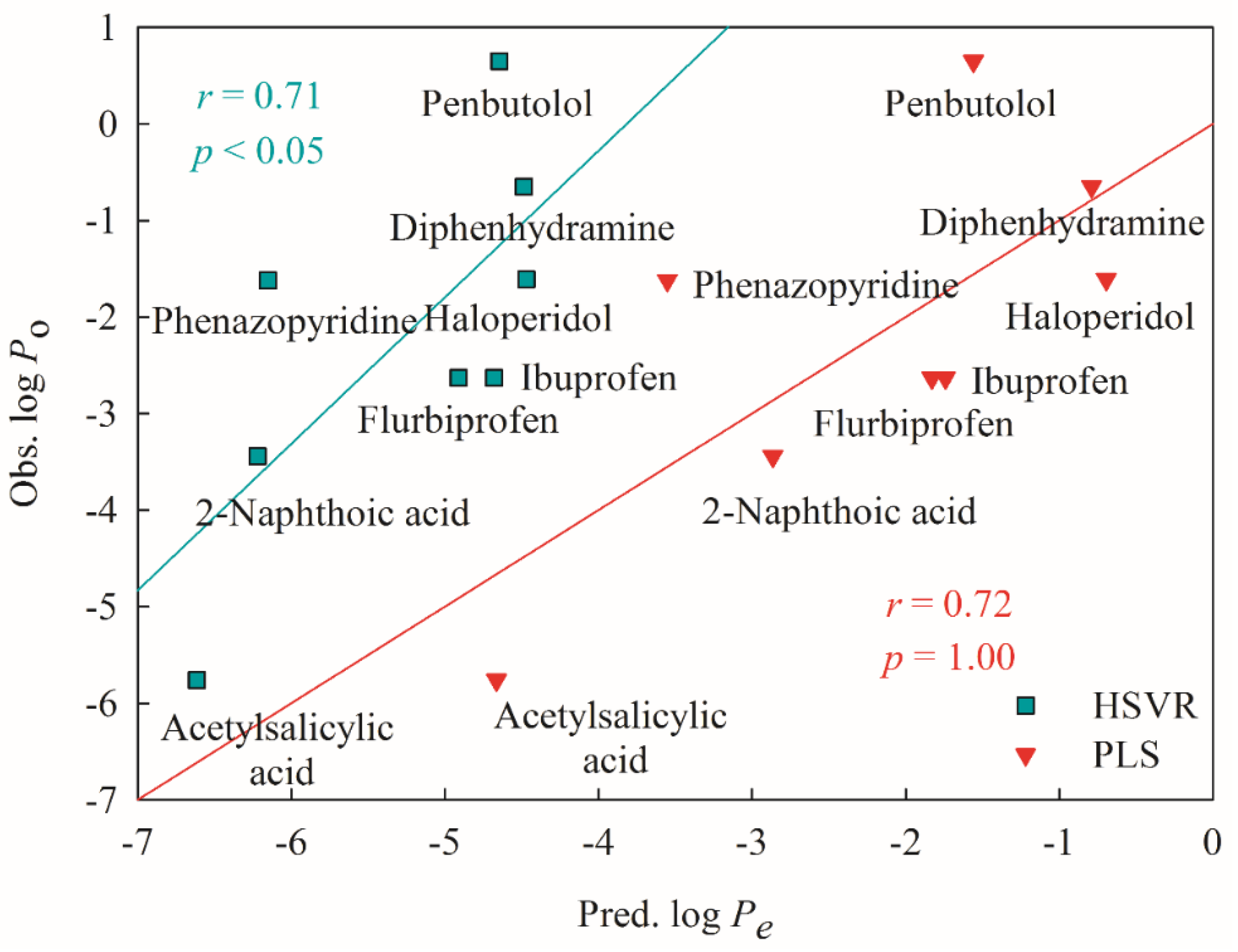
2.5. Mock Test
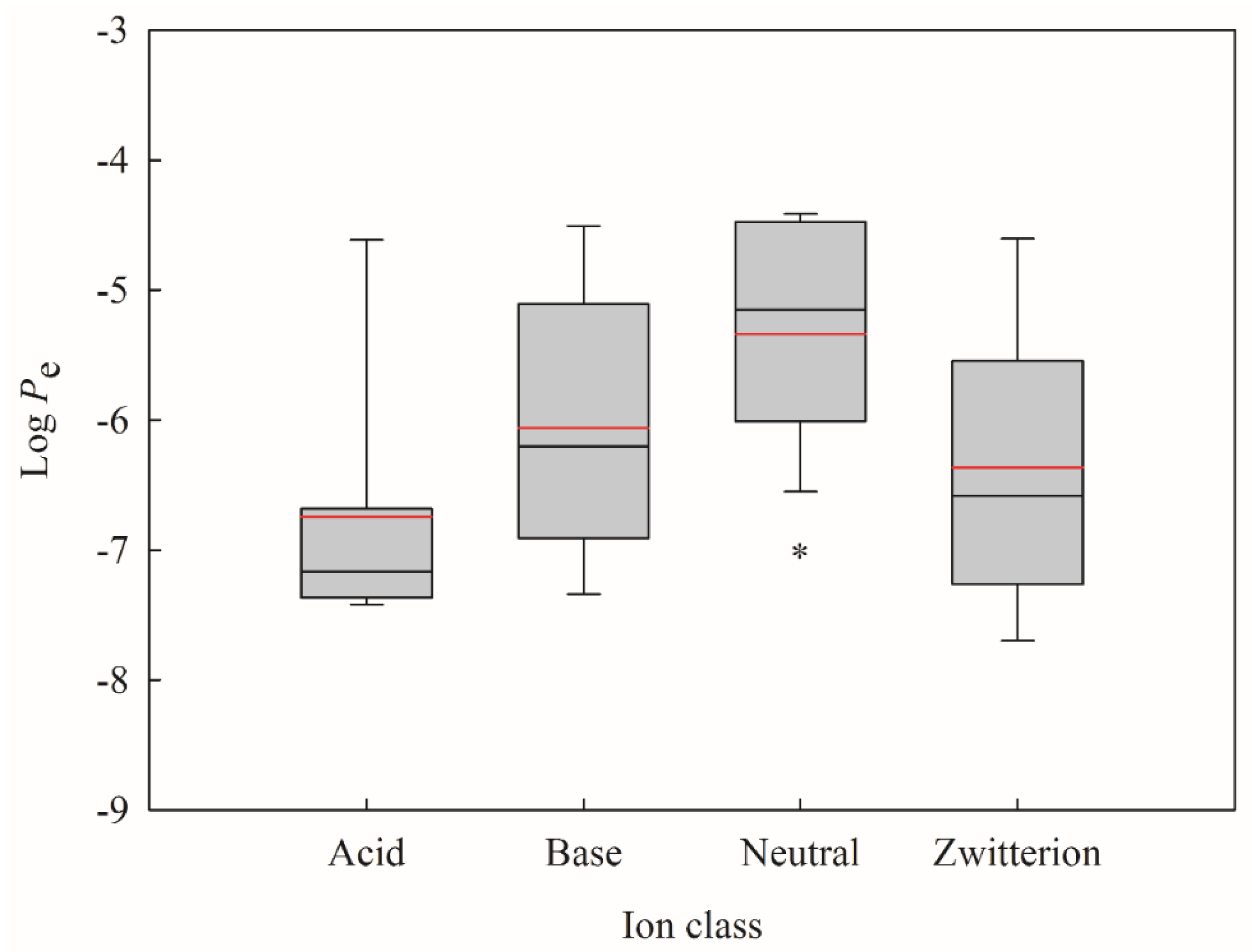
3. Discussion
4. Materials and Methods
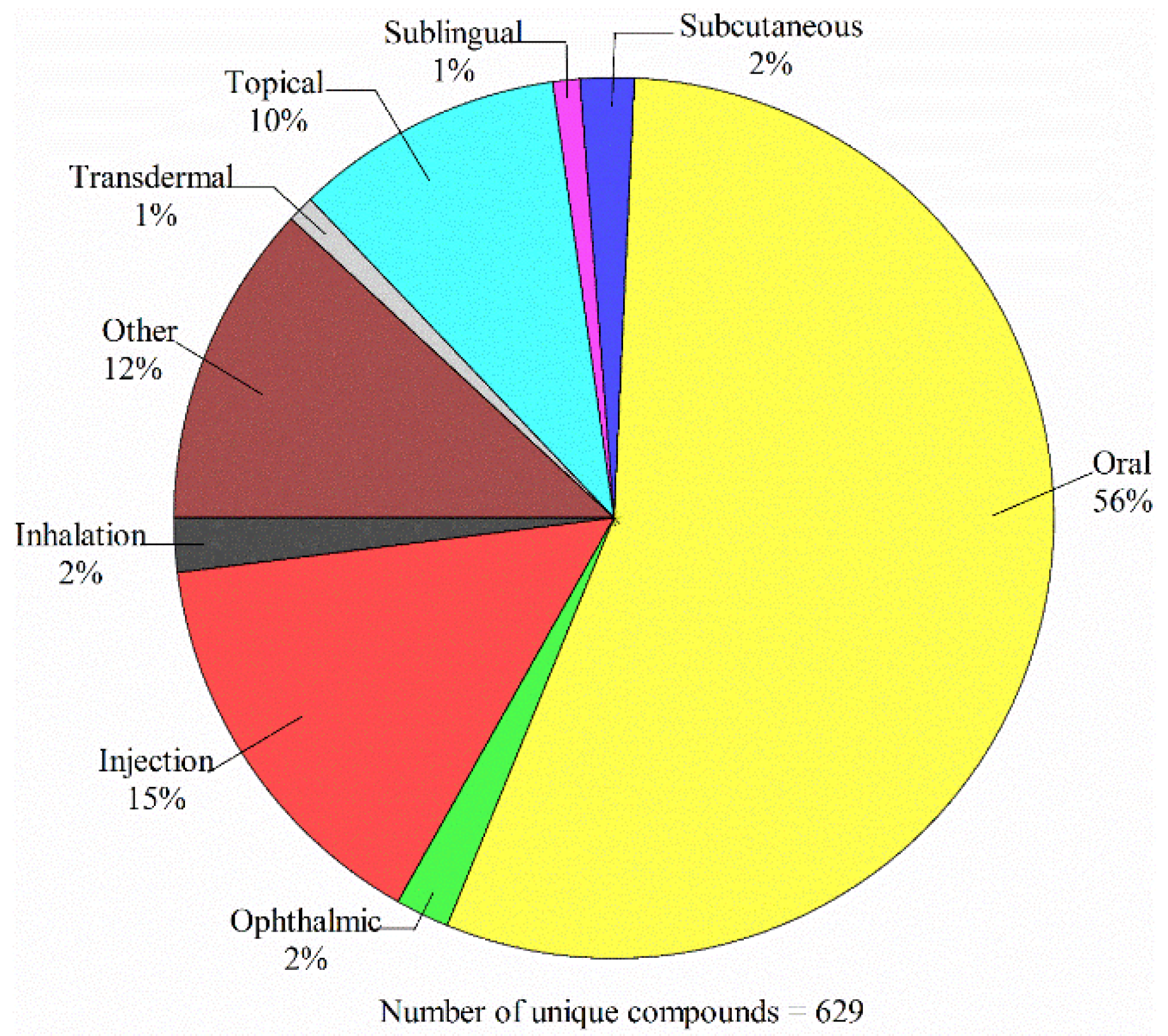
4.1. Data Compilation
4.2. Molecular Descriptors
4.3. Data Partition
4.4. Partial Least Square
4.5. Hierarchical Support Vector Regression
4.6. Predictive Evaluation
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Gad, S.C. Oral Drug Formulation Development in Pharmaceutical Lead Selection Stage. In Oral Formulation Roadmap from Early Drug Discovery to Development; Kwong, E., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017; pp. 39–88. [Google Scholar]
- Avdeef, A. The rise of PAMPA. Expert Opin. Drug Metab. Toxicol. 2005, 1, 325–342. [Google Scholar] [CrossRef] [PubMed]
- van de Waterbeemd, H.; Smith, D.A.; Beaumont, K.; Walker, D.K. Property-based design: Optimization of drug absorption and pharmacokinetics. J. Med. Chem. 2001, 44, 1313–1333. [Google Scholar] [CrossRef] [PubMed]
- Shehzad, A.; Qureshi, M.; Anwar, M.N.; Lee, Y.S. Multifunctional Curcumin Mediate Multitherapeutic Effects. J. Food Sci. 2017, 82, 2006–2015. [Google Scholar] [CrossRef] [PubMed]
- Eke-Okoro, U.J.; Raffa, R.B.; Pergolizzi, J.V., Jr.; Breve, F.; Taylor, R., Jr. Curcumin in turmeric: Basic and clinical evidence for a potential role in analgesia. J. Clin. Pharm. Ther. 2018, 43, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. Molecular Mechanisms of Action of Curcumin and Its Relevance to Some Clinical Conditions. In Curcumin for Neurological and Psychiatric Disorders: Neurochemical and Pharmacological Properties: Neurochemical and Pharmacological Properties; Farooqui, T., Farooqui, A.A., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 325–332. [Google Scholar]
- Sun, H.; Nguyen, K.; Kerns, E.; Yan, Z.; Yu, K.R.; Shah, P.; Jadhav, A.; Xu, X. Highly predictive and interpretable models for PAMPA permeability. Bioorg. Med. Chem. 2017, 25, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Lennernäs, H. Regional intestinal drug permeation: Biopharmaceutics and drug development. Eur. J. Pharm. Sci. 2014, 57, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Parasrampuria, D.A.; Benet, L.Z.; Sharma, A. Why Drugs Fail in Late Stages of Development: Case Study Analyses from the Last Decade and Recommendations. AAPS J. 2018, 20, 46. [Google Scholar] [CrossRef] [PubMed]
- Billat, P.-A.; Roger, E.; Faure, S.; Lagarce, F. Models for drug absorption from the small intestine: Where are we and where are we going? Drug Discov. Today 2017, 22, 761–775. [Google Scholar] [CrossRef] [PubMed]
- Kansy, M.; Senner, F.; Gubernator, K. Physicochemical High Throughput Screening: Parallel Artificial Membrane Permeation Assay in the Description of Passive Absorption Processes. J. Med. Chem. 1998, 41, 1007–1010. [Google Scholar] [CrossRef]
- Linnankoski, J.; Mäkelä, J.; Palmgren, J.; Mauriala, T.; Vedin, C.; Ungell, A.-L.; Lazorova, L.; Artursson, P.; Urtti, A.; Yliperttula, M. Paracellular porosity and pore size of the human intestinal epithelium in tissue and cell culture models. J. Pharm. Sci. 2010, 99, 2166–2175. [Google Scholar] [CrossRef]
- Cabrera-Pérez, M.Á.; Pham-The, H.; Cervera, M.F.; Hernández-Armengol, R.; Miranda-Pérez de Alejo, C.; Brito-Ferrer, Y. Integrating theoretical and experimental permeability estimations for provisional biopharmaceutical classification: Application to the WHO essential medicines. Biopharm. Drug Dispos. 2018, 39, 354–368. [Google Scholar] [CrossRef] [PubMed]
- Berben, P.; Bauer-Brandl, A.; Brandl, M.; Faller, B.; Flaten, G.E.; Jacobsen, A.-C.; Brouwers, J.; Augustijns, P. Drug permeability profiling using cell-free permeation tools: Overview and applications. Eur. J. Pharm. Sci. 2018, 119, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, M.; Avdeef, A.; Ruiz, A.; Nalda, R.; Ruell, J.A.; Tsinman, O.; González, I.; Fernández, C.; Sánchez, G.; Garrigues, T.M.; et al. PAMPA—a drug absorption in vitro model: 7. Comparing rat in situ, Caco-2, and PAMPA permeability of fluoroquinolones. Eur. J. Pharm. Sci. 2004, 21, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Sugano, K.; Kansy, M.; Artursson, P.; Avdeef, A.; Bendels, S.; Di, L.; Ecker, G.F.; Faller, B.; Fischer, H.; Gerebtzoff, G.; et al. Coexistence of passive and carrier-mediated processes in drug transport. Nat. Rev. Drug Discov. 2010, 9, 597–614. [Google Scholar] [CrossRef] [PubMed]
- Diukendjieva, A.; Tsakovska, I.; Alov, P.; Pencheva, T.; Pajeva, I.; Worth, A.P.; Madden, J.C.; Cronin, M.T.D. Advances in the prediction of gastrointestinal absorption: Quantitative Structure-Activity Relationship (QSAR) modelling of PAMPA permeability. Comput. Toxicol. 2019, 10, 51–59. [Google Scholar] [CrossRef]
- Faller, B. Artificial Membrane Assays to Assess Permeability. Curr. Drug Metab. 2008, 9, 886–892. [Google Scholar] [CrossRef]
- Mensch, J.; L, L.J.; Sanderson, W.; Melis, A.; Mackie, C.; Verreck, G.; Brewster, M.E.; Augustijns, P. Application of PAMPA-models to predict BBB permeability including efflux ratio, plasma protein binding and physicochemical parameters. Int. J. Pharm. 2010, 395, 182–197. [Google Scholar] [CrossRef]
- Sinkó, B.; Garrigues, T.M.; Balogh, G.T.; Nagy, Z.K.; Tsinman, O.; Avdeef, A.; Takács-Novák, K. Skin–PAMPA: A new method for fast prediction of skin penetration. Eur. J. Pharm. Sci. 2012, 45, 698–707. [Google Scholar] [CrossRef]
- Shibayama, T.; Morales, M.; Zhang, X.; Martínez-Guerrero, L.J.; Berteloot, A.; Secomb, T.W.; Wright, S.H. Unstirred Water Layers and the Kinetics of Organic Cation Transport. Pharm. Res. 2015, 32, 2937–2949. [Google Scholar] [CrossRef]
- Nielsen, P.E.; Avdeef, A. PAMPA—a drug absorption in vitro model: 8. Apparent filter porosity and the unstirred water layer. Eur. J. Pharm. Sci. 2004, 22, 33–41. [Google Scholar] [CrossRef]
- Akamatsu, M.; Fujikawa, M.; Nakao, K.; Shimizu, R. In silico Prediction of Human Oral Absorption Based on QSAR Analyses of PAMPA Permeability. Chem. Biodivers. 2009, 6, 1845–1866. [Google Scholar] [CrossRef] [PubMed]
- Velický, M.; Bradley, D.F.; Tam, K.Y.; Dryfe, R.A.W. In Situ Artificial Membrane Permeation Assay under Hydrodynamic Control: Permeability-pH Profiles of Warfarin and Verapamil. Pharm. Res. 2010, 27, 1644–1658. [Google Scholar] [CrossRef] [PubMed]
- Mayr, L.M.; Bojanic, D. Novel trends in high-throughput screening. Curr. Opin. Pharmacol. 2009, 9, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.W.; Glick, M.; Jenkins, J.L. Streamlining lead discovery by aligning in silico and high-throughput screening. Curr. Opin. Chem. Biol. 2006, 10, 343–351. [Google Scholar] [CrossRef] [PubMed]
- van de Waterbeemd, H.; Gifford, E. ADMET In Silico Modelling: Towards Prediction Paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Ruell, J.A.; Tsinman, O.; Avdeef, A. Acid-Base Cosolvent Method for Determining Aqueous Permeability of Amiodarone, Itraconazole, Tamoxifen, Terfenadine and Other Very Insoluble Molecules. Chem. Pharm. Bull. 2004, 52, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Ano, R.; Kimura, Y.; Shima, M.; Matsuno, R.; Ueno, T.; Akamatsu, M. Relationships between structure and high-throughput screening permeability of peptide derivatives and related compounds with artificial membranes: Application to prediction of Caco-2 cell permeability. Bioorg. Med. Chem. 2004, 12, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, M.; Ano, R.; Nakao, K.; Shimizu, R.; Akamatsu, M. Relationships between structure and high-throughput screening permeability of diverse drugs with artificial membranes: Application to prediction of Caco-2 cell permeability. Bioorg. Med. Chem. 2005, 13, 4721–4732. [Google Scholar] [CrossRef] [PubMed]
- Avdeef, A.; Tsinman, O. PAMPA—A drug absorption in vitro model: 13. Chemical selectivity due to membrane hydrogen bonding: In combo comparisons of HDM-, DOPC-, and DS-PAMPA models. Eur. J. Pharm. Sci. 2006, 28, 43–50. [Google Scholar] [CrossRef]
- Verma, R.; Hansch, C.; Selassie, C. Comparative QSAR studies on PAMPA/modified PAMPA for high throughput profiling of drug absorption potential with respect to Caco-2 cells and human intestinal absorption. J. Comput.-Aided Mol. Des. 2007, 21, 3–22. [Google Scholar] [CrossRef]
- Fischer, H.; Kansy, M.; Avdeef, A.; Senner, F. Permeation of permanently positive charged molecules through artificial membranes—Influence of physico-chemical properties. Eur. J. Pharm. Sci. 2007, 31, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, M.; Nakao, K.; Shimizu, R.; Akamatsu, M. QSAR study on permeability of hydrophobic compounds with artificial membranes. Bioorg. Med. Chem. 2007, 15, 3756–3767. [Google Scholar] [CrossRef] [PubMed]
- Nakao, K.; Fujikawa, M.; Shimizu, R.; Akamatsu, M. QSAR application for the prediction of compound permeability with in silico descriptors in practical use. J. Comput.-Aided Mol. Des. 2009, 23, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Karelson, M.; Karelson, G.; Tamm, T.; Indrek, T.; Jänes, J.; Tämm, K.; Lomaka, A.; Savchenko, D.; Dobcheva, D. QSAR study of pharmacological permeabilities. Arkivoc 2009, 218–238. [Google Scholar]
- Tulp, I.; Sild, S.; Maran, U. Relationship Between Structure and Permeability in Artificial Membranes: Theoretical Whole Molecule Descriptors in Development of QSAR Models. QSAR Comb. Sci. 2009, 28, 811–814. [Google Scholar] [CrossRef]
- Wang, C.K.; Northfield, S.E.; Swedberg, J.E.; Colless, B.; Chaousis, S.; Price, D.A.; Liras, S.; Craik, D.J. Exploring experimental and computational markers of cyclic peptides: Charting islands of permeability. Eur. J. Med. Chem. 2015, 97, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Oja, M.; Maran, U. The Permeability of an Artificial Membrane for Wide Range of pH in Human Gastrointestinal Tract: Experimental Measurements and Quantitative Structure-Activity Relationship. Mol. Inform. 2015, 34, 493–506. [Google Scholar] [CrossRef]
- Oja, M.; Maran, U. Quantitative structure–permeability relationships at various pH values for acidic and basic drugs and drug-like compounds. SAR QSAR Environ. Res. 2015, 26, 701–719. [Google Scholar] [CrossRef]
- Oja, M.; Maran, U. Quantitative structure–permeability relationships at various pH values for neutral and amphoteric drugs and drug-like compounds. SAR QSAR Environ. Res. 2016, 27, 813–832. [Google Scholar] [CrossRef]
- Oja, M.; Maran, U. pH-permeability profiles for drug substances: Experimental detection, comparison with human intestinal absorption and modelling. Eur. J. Pharm. Sci. 2018, 123, 429–440. [Google Scholar] [CrossRef]
- Savić, J.; Dobričić, V.; Nikolic, K.; Vladimirov, S.; Dilber, S.; Brborić, J. In vitro prediction of gastrointestinal absorption of novel β-hydroxy-β-arylalkanoic acids using PAMPA technique. Eur. J. Pharm. Sci. 2017, 100, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Diukendjieva, A.; Alov, P.; Tsakovska, I.; Pencheva, T.; Richarz, A.; Kren, V.; Cronin, M.T.D.; Pajeva, I. In vitro and in silico studies of the membrane permeability of natural flavonoids from Silybum marianum (L.) Gaertn. and their derivatives. Phytomedicine 2019, 53, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.-X.; Shang, Z.-C.; Zou, J.-W.; Yang, G.-M.; Yu, Q.-S. QSAR Study and VolSurf Characterization of Human Intestinal Absorption of Drugs. Chin. J. Chem. 2003, 21, 238–243. [Google Scholar]
- Kalyanaraman, C.; Jacobson, M.P. An atomistic model of passive membrane permeability: Application to a series of FDA approved drugs. J. Comput.-Aided Mol. Des. 2007, 21, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Huque, F.T.T.; Box, K.; Platts, J.A.; Comer, J. Permeability through DOPC/dodecane membranes: Measurement and LFER modelling. Eur. J. Pharm. Sci. 2004, 23, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Vizserálek, G.; Balogh, T.; Takács-Novák, K.; Sinkó, B. PAMPA study of the temperature effect on permeability. Eur. J. Pharm. Sci. 2014, 53, 45–49. [Google Scholar] [CrossRef]
- Avdeef, A.; Nielsen, P.E.; Tsinman, O. PAMPA—a drug absorption in vitro model: 11. Matching the in vivo unstirred water layer thickness by individual-well stirring in microtitre plates. Eur. J. Pharm. Sci. 2004, 22, 365–374. [Google Scholar] [CrossRef]
- Ruell, J.A.; Avdeef, A. Absorption Screening Using the PAMPA Approach. In Optimization in Drug Discovery: In Vitro Methods; Yan, Z., Caldwell, G.W., Eds.; Humana Press: Totowa, NJ, USA, 2004; pp. 37–64. [Google Scholar]
- Sugano, K.; Hamada, H.; Machida, M.; Ushio, H.; Saitoh, K.; Terada, K. Optimized conditions of bio-mimetic artificial membrane permeation assay. Int. J. Pharm. 2001, 228, 181–188. [Google Scholar] [CrossRef]
- Nitsche, J.M.; Kasting, G.B. Permeability of Fluid-Phase Phospholipid Bilayers: Assessment and Useful Correlations for Permeability Screening and Other Applications. J. Pharm. Sci. 2013, 102, 2005–2032. [Google Scholar] [CrossRef]
- Korjamo, T.; Heikkinen, A.T.; Mönkkönen, J. Analysis of Unstirred Water Layer in In Vitro Permeability Experiments. J. Pharm. Sci. 2009, 98, 4469–4479. [Google Scholar] [CrossRef]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR Modeling: Where Have You Been? Where Are You Going To? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef] [PubMed]
- Kansy, M.; Fischer, H.; Kratzat, K.; Senner, F.; Wagner, B.; Parrilla, I. High-Throughput Artificial Membrane Permeability Studies in Early Lead Discovery and Development. In Pharmacokinetic Optimization in Drug Research; Testa, B., Van de Waterbeend, H., Folkers, G., Guy, R., Eds.; Verlag Helvetica Chimica Acta/Wiley/VCH: Zurich, Switzerland, 2001; pp. 447–464. [Google Scholar]
- Hou, T.; Li, Y.; Zhang, W.; Wang, J. Recent Developments of In Silico Predictions of Intestinal Absorption and Oral Bioavailability. Comb. Chem. High Throughput Screen. 2009, 12, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.-L.; Lyu, Y.-C.; Leong, M.K. In Silico Prediction of the Mutagenicity of Nitroaromatic Compounds Using a Novel Two-QSAR Approach. Toxicol. Vitro 2017, 40, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Leong, M.K.; Chen, Y.-M.; Chen, T.-H. Prediction of Human Cytochrome P450 2B6-Substrate Interactions Using Hierarchical Support Vector Regression Approach. J. Comput. Chem. 2009, 30, 1899–1909. [Google Scholar] [CrossRef] [PubMed]
- Reymond, J.-L.; van Deursen, R.; Blum, L.C.; Ruddigkeit, L. Chemical space as a source for new drugs. MedChemComm 2010, 1, 30–38. [Google Scholar] [CrossRef]
- Rücker, C.; Rücker, G.; Meringer, M. y-Randomization and Its Variants in QSPR/QSAR. J. Chem. Inf. Model. 2007, 47, 2345–2357. [Google Scholar] [CrossRef] [PubMed]
- Gnanadesikan, R.; Kettenring, J.R. Robust estimates, residuals, and outlier detection with multiresponse data. Biometrics 1972, 28, 81–124. [Google Scholar] [CrossRef]
- Golbraikh, A.; Shen, M.; Xiao, Z.Y.; Xiao, Y.D.; Lee, K.H.; Tropsha, A. Rational selection of training and test sets for the development of validated QSAR models. J. Comput.-Aided Mol. Des. 2003, 17, 241–253. [Google Scholar] [CrossRef]
- Ojha, P.K.; Mitra, I.; Das, R.N.; Roy, K. Further exploring rm2 metrics for validation of QSPR models. Chemometrics Intell. Lab. Syst. 2011, 107, 194–205. [Google Scholar] [CrossRef]
- Roy, K.; Mitra, I.; Kar, S.; Ojha, P.K.; Das, R.N.; Kabir, H. Comparative Studies on Some Metrics for External Validation of QSPR Models. J. Chem. Inf. Model. 2012, 52, 396–408. [Google Scholar] [CrossRef]
- Chirico, N.; Gramatica, P. Real External Predictivity of QSAR Models. Part 2. New Intercomparable Thresholds for Different Validation Criteria and the Need for Scatter Plot Inspection. J. Chem. Inf. Model. 2012, 52, 2044–2058. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.L.; Kuczera, K. Simulating the free energy of passive membrane permeation for small molecules. Mol. Simul. 2018, 44, 1147–1157. [Google Scholar] [CrossRef]
- Arnott, J.A.; Kumar, R.; Planey, S.L. Lipophilicity Indices for Drug Development. J. Appl. Biopharm. Pharmacokinet. 2013, 1, 31–36. [Google Scholar]
- Topliss, J.G.; Edwards, R.P. Chance factors in studies of quantitative structure-activity relationships. J. Med. Chem. 1979, 22, 1238–1244. [Google Scholar] [CrossRef] [PubMed]
- Kokate, A.; Li, X.; Jasti, B. Effect of Drug Lipophilicity and Ionization on Permeability Across the Buccal Mucosa: A Technical Note. AAPS PharmSciTech 2008, 9, 501–504. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sun, R.; Han, Y.; Swanson, J.M.J.; Tan, J.S.; Rose, J.P.; Voth, G.A. Molecular transport through membranes: Accurate permeability coefficients from multidimensional potentials of mean force and local diffusion constants. J. Chem. Phys. 2018, 149, 072310. [Google Scholar] [CrossRef]
- Iyer, M.; Tseng, Y.J.; Senese, C.L.; Liu, J.; Hopfinger, A.J. Prediction and Mechanistic Interpretation of Human Oral Drug Absorption Using MI-QSAR Analysis. Mol. Pharmaceutics 2007, 4, 218–231. [Google Scholar] [CrossRef]
- Yen, T.E.; Agatonovic-Kustrin, S.; Evans, A.M.; Nation, R.L.; Ryand, J. Prediction of drug absorption based on immobilized artificial membrane (IAM) chromatography separation and calculated molecular descriptors. J. Pharm. Biomed. Anal. 2005, 38, 472–478. [Google Scholar] [CrossRef]
- Ekins, S. Progress in computational toxicology. J. Pharmacol. Toxicol. Methods 2014, 69, 115–140. [Google Scholar] [CrossRef]
- Kelder, J.; Grootenhuis, P.D.J.; Bayada, D.M.; Delbressine, L.P.C.; Ploemen, J.-P. Polar Molecular Surface as a Dominating Determinant for Oral Absorption and Brain Penetration of Drugs. Pharm. Res. 1999, 16, 1514–1519. [Google Scholar] [CrossRef]
- Zhu, C.; Jiang, L.; Chen, T.-M.; Hwang, K.-K. A comparative study of artificial membrane permeability assay for high throughput profiling of drug absorption potential. Eur. J. Med. Chem. 2002, 37, 399–407. [Google Scholar] [CrossRef]
- Flaten, G.E.; Dhanikula, A.B.; Luthman, K.; Brandl, M. Drug permeability across a phospholipid vesicle based barrier: A novel approach for studying passive diffusion. Eur. J. Pharm. Sci. 2006, 27, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Kerns, E.H.; Di, L.; Petusky, S.; Farris, M.; Ley, R.; Jupp, P. Combined application of parallel artificial membrane permeability assay and Caco-2 permeability assays in drug discovery. J. Pharm. Sci. 2004, 93, 1440–1453. [Google Scholar] [CrossRef] [PubMed]
- Galinis-Luciani, D.; Nguyen, L.; Yazdanian, M. Is PAMPA a useful tool for discovery? J. Pharm. Sci. 2007, 96, 2886–2892. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Murawski, A.; Patel, K.; Crespi, C.L.; Balimane, P.V. A Novel Design of Artificial Membrane for Improving the PAMPA Model. Pharm. Res. 2008, 25, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Avdeef, A. Permeability—PAMPA. In Absorption and Drug Development: Solubility, Permeability, and Charge State, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 319–498. [Google Scholar]
- Cammi, R.; Tomasi, J. Remarks on the use of the apparent surface charges (ASC) methods in solvation problems: Iterative versus matrix-inversion procedures and the renormalization of the apparent charges. J. Comput. Chem. 1995, 16, 1449–1458. [Google Scholar] [CrossRef]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Besler, B.H.; Merz, K.M.J.; Kollman, P.A. Atomic charges derived from semiempirical methods. J. Comput. Chem. 1990, 11, 431–439. [Google Scholar] [CrossRef]
- Muehlbacher, M.; Spitzer, G.; Liedl, K.; Kornhuber, J. Qualitative prediction of blood–brain barrier permeability on a large and refined dataset. J. Comput.-Aided Mol. Des. 2011, 25, 1095–1106. [Google Scholar] [CrossRef]
- Bemporad, D.; Luttmann, C.; Essex, J.W. Computer Simulation of Small Molecule Permeation across a Lipid Bilayer: Dependence on Bilayer Properties and Solute Volume, Size, and Cross-Sectional Area. Biophys. J. 2004, 87, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.J.; Hopfinger, A.J.; Esposito, E.X. The great descriptor melting pot: Mixing descriptors for the common good of QSAR models. J. Comput.-Aided Mol. Des. 2012, 26, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Burden, F.R.; Ford, M.G.; Whitley, D.C.; Winkler, D.A. Use of Automatic Relevance Determination in QSAR Studies Using Bayesian Neural Networks. J. Chem. Inf. Comput. Sci. 2000, 40, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.; Hopfinger, A.J. Application of Genetic Function Approximation to Quantitative Structure-Activity Relationships and Quantitative Structure-Property Relationships. J. Chem. Inf. Comput. Sci. 1994, 34, 854–866. [Google Scholar] [CrossRef]
- Guyon, I.; Weston, J.; Barnhill, S.; Vapnik, V. Gene Selection for Cancer Classification using Support Vector Machines. Mach. Learn. 2002, 46, 389–422. [Google Scholar] [CrossRef]
- Tropsha, A.; Gramatica, P.; Gombar, V.K. The Importance of Being Earnest: Validation is the Absolute Essential for Successful Application and Interpretation of QSPR Models. QSAR Comb. Sci. 2003, 22, 69–77. [Google Scholar] [CrossRef]
- Kennard, R.W.; Stone, L.A. Computer Aided Design of Experiments. Technometrics 1969, 11, 137–148. [Google Scholar] [CrossRef]
- Tropsha, A. Recent Trends in Statistical QSAR Modeling of Environmental Chemical Toxicity. In Molecular, Clinical and Environmental Toxicology: Volume 3: Environmental Toxicology; Luch, A., Ed.; Springer Base: New York, NY, USA, 2012; Vol. 101, pp. 381–411. [Google Scholar]
- Wold, S. PLS for Multivariate Linear Modeling. In Chemometric Methods in Molecular Design; van de Waterbeemd, H., Ed.; VCH: Weinheim, Germany, 1995; Vol. 2, pp. 195–218. [Google Scholar]
- Clark, M.; Cramer, R.D. The Probability of Chance Correlation Using Partial Least Squares (PLS). Quant. Struct.-Act. Relat. 1993, 12, 137–145. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-Vector Networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Vapnik, V.; Golowich, S.; Smola, A. Support vector method for function approximation, regression estimation, and signal processing; Mozer, M., Jordan, M.I., Petsche, T., Eds.; Advances in Neural Information Processing Systems 9; MIT Press: Cambridge, MA, USA, 1997; pp. 281–287. [Google Scholar]
- Schölkopf, B.; Smola, A. Learning with Kernels: Support Vector Machines, Regularization, Optimization, and Beyond, 1st ed.; MIT Press: Cambridge, MA, USA, 2002. [Google Scholar]
- Netzeva, T.I.; Worth, A.; Aldenberg, T.; Benigni, R.; Cronin, M.T.D.; Gramatica, P.; Jaworska, J.S.; Kahn, S.; Klopman, G.; Marchant, C.A.; et al. Current status of methods for defining the applicability domain of (Quantitative) structure-activity relationships: The report and recommendations of ECVAM workshop 52. Altern. Lab. Anim. 2005, 33, 1–19. [Google Scholar] [CrossRef]
- Leong, M.K.; Lin, S.-W.; Chen, H.-B.; Tsai, F.-Y. Predicting Mutagenicity of Aromatic Amines by Various Machine Learning Approaches. Toxicol. Sci. 2010, 116, 498–513. [Google Scholar] [CrossRef]
- Kecman, V. Learning and Soft Computing: Support Vector Machines, Neural Networks, and Fuzzy Logic Models; MIT Press: Cambridge, MA, USA, 2001; p. 576. [Google Scholar]
- Dearden, J.C.; Cronin, M.T.D.; Kaiser, K.L.E. How not to develop a quantitative structure–activity or structure–property relationship (QSAR/QSPR). SAR QSAR Environ. Res. 2009, 20, 241–266. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L.; Spector, P. Submodel Selection and Evaluation in Regression. The X-Random Case. Int. Stat. Rev. 1992, 60, 291–319. [Google Scholar] [CrossRef]
- Gramatica, P.; Chirico, N.; Papa, E.; Cassani, S.; Kovarich, S. QSARINS: A new software for the development, analysis, and validation of QSAR MLR models. J. Comput. Chem. 2013, 34, 2121–2132. [Google Scholar] [CrossRef]
- Gramatica, P.; Cassani, S.; Chirico, N. QSARINS-chem: Insubria datasets and new QSAR/QSPR models for environmental pollutants in QSARINS. J. Comput. Chem. 2014, 35, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Lee, M.-H.; Weng, C.-F.; Leong, M.K. Theoretical Prediction of the Complex P-Glycoprotein Substrate Efflux Based on the Novel Hierarchical Support Vector Regression Scheme. Molecules 2018, 23, 1820. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Descriptor | SVR A | SVR B | Description |
|---|---|---|---|
| log P | x † | Logarithm of the n-octanol–water partition coefficient | |
| log D | x | x | Logarithm of the n-octanol–water distribution coefficient at pH 7.4 |
| PSA | x | Polar surface area | |
| FPSA | x | The ratio of total partially positively charged molecular surface area to total molecular surface area | |
| μ | x | Dipole moment for the molecule |
| SVR A | SVR B | HSVR | PLS | |
|---|---|---|---|---|
| r2 | 0.84 | 0.79 | 0.88 | 0.61 |
| ΔMax | 1.48 | 1.60 | 1.56 | 1.90 |
| MAE | 0.38 | 0.39 | 0.24 | 0.58 |
| s | 0.26 | 0.33 | 0.31 | 0.38 |
| RMSE | 0.46 | 0.51 | 0.39 | 0.70 |
| 0.57 | 0.14 | 0.80 | 0.76 | |
| 0.06 | 0.06 | 0.03 | 0.06 |
| SVR A | SVR B | HSVR | PLS | |
|---|---|---|---|---|
| q2 | 0.72 | 0.70 | 0.79 | 0.61 |
| 0.70 | 0.70 | 0.79 | 0.60 | |
| 0.70 | 0.70 | 0.79 | 0.60 | |
| 0.68 | 0.68 | 0.86 | 0.58 | |
| CCC | 0.80 | 0.82 | 0.88 | 0.74 |
| ∆Max | 1.58 | 1.75 | 1.52 | 1.77 |
| MAE | 0.53 | 0.51 | 0.42 | 0.61 |
| s | 0.35 | 0.37 | 0.32 | 1.40 |
| RMSE | 0.63 | 0.57 | 0.52 | 0.73 |
| SVR A | SVR B | HSVR | PLS | |
|---|---|---|---|---|
| q2 | 0.68 | 0.69 | 0.76 | 0.54 |
| 0.78 | 0.56 | 0.86 | 0.76 | |
| 0.52 | 0.04 | 0.70 | 0.49 | |
| 0.75 | 0.50 | 0.84 | 0.74 | |
| CCC | 0.69 | 0.48 | 0.85 | 0.63 |
| ∆Max | 0.87 | 1.09 | 0.98 | 0.99 |
| MAE | 0.51 | 0.72 | 0.32 | 0.42 |
| s | 0.25 | 0.34 | 0.33 | 0.41 |
| RMSE | 0.56 | 0.79 | 0.44 | 0.57 |
| Training Set | Test Set | Outlier Set | ||||
|---|---|---|---|---|---|---|
| HSVR | PLS | HSVR | PLS | HSVR | PLS | |
| 0.88 | 0.61 | 0.79 | 0.61 | 0.76 | 0.51 | |
| k | 1.00 | 1.02 | 1.00 | 1.02 | 0.96 | 0.98 |
| 0.88 | 0.38 | 0.71 | 0.35 | 0.74 | –0.46 | |
| 0.83 | 0.23 | 0.76 | 0.61 | 0.69 | 0.45 | |
| 0.84 | 0.60 | 0.57 | 0.30 | 0.64 | 0.00 | |
| 0.83 | 0.42 | 0.67 | 0.45 | 0.67 | 0.23 | |
| 0.01 | 0.29 | 0.19 | 0.30 | 0.05 | 0.45 | |
| Equation (15) | X | X | X | |||
| Equation (16) | X | X | X | |||
| Equation (17) | X | X | X | X | X | X |
| Equation (18) | X | X | X | X | X | X |
| Equation (19) | X | X | X | |||
| Equation (20) | X | X | X | |||
| Equation (21) | X | X | X | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chi, C.-T.; Lee, M.-H.; Weng, C.-F.; Leong, M.K. In Silico Prediction of PAMPA Effective Permeability Using a Two-QSAR Approach. Int. J. Mol. Sci. 2019, 20, 3170. https://doi.org/10.3390/ijms20133170
Chi C-T, Lee M-H, Weng C-F, Leong MK. In Silico Prediction of PAMPA Effective Permeability Using a Two-QSAR Approach. International Journal of Molecular Sciences. 2019; 20(13):3170. https://doi.org/10.3390/ijms20133170
Chicago/Turabian StyleChi, Cheng-Ting, Ming-Han Lee, Ching-Feng Weng, and Max K. Leong. 2019. "In Silico Prediction of PAMPA Effective Permeability Using a Two-QSAR Approach" International Journal of Molecular Sciences 20, no. 13: 3170. https://doi.org/10.3390/ijms20133170
APA StyleChi, C.-T., Lee, M.-H., Weng, C.-F., & Leong, M. K. (2019). In Silico Prediction of PAMPA Effective Permeability Using a Two-QSAR Approach. International Journal of Molecular Sciences, 20(13), 3170. https://doi.org/10.3390/ijms20133170